
The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications

 , , ,
, , , 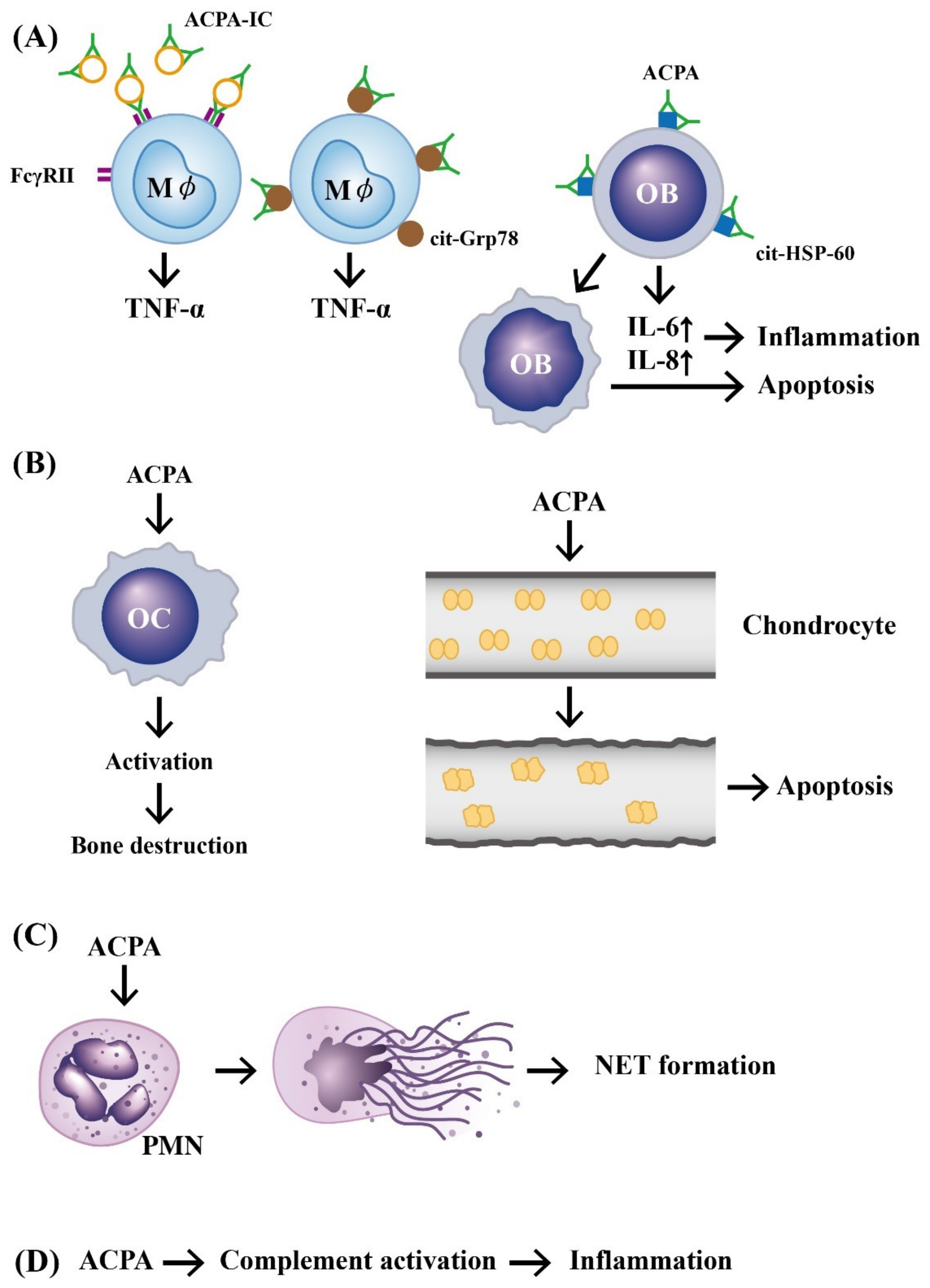{kind=link}
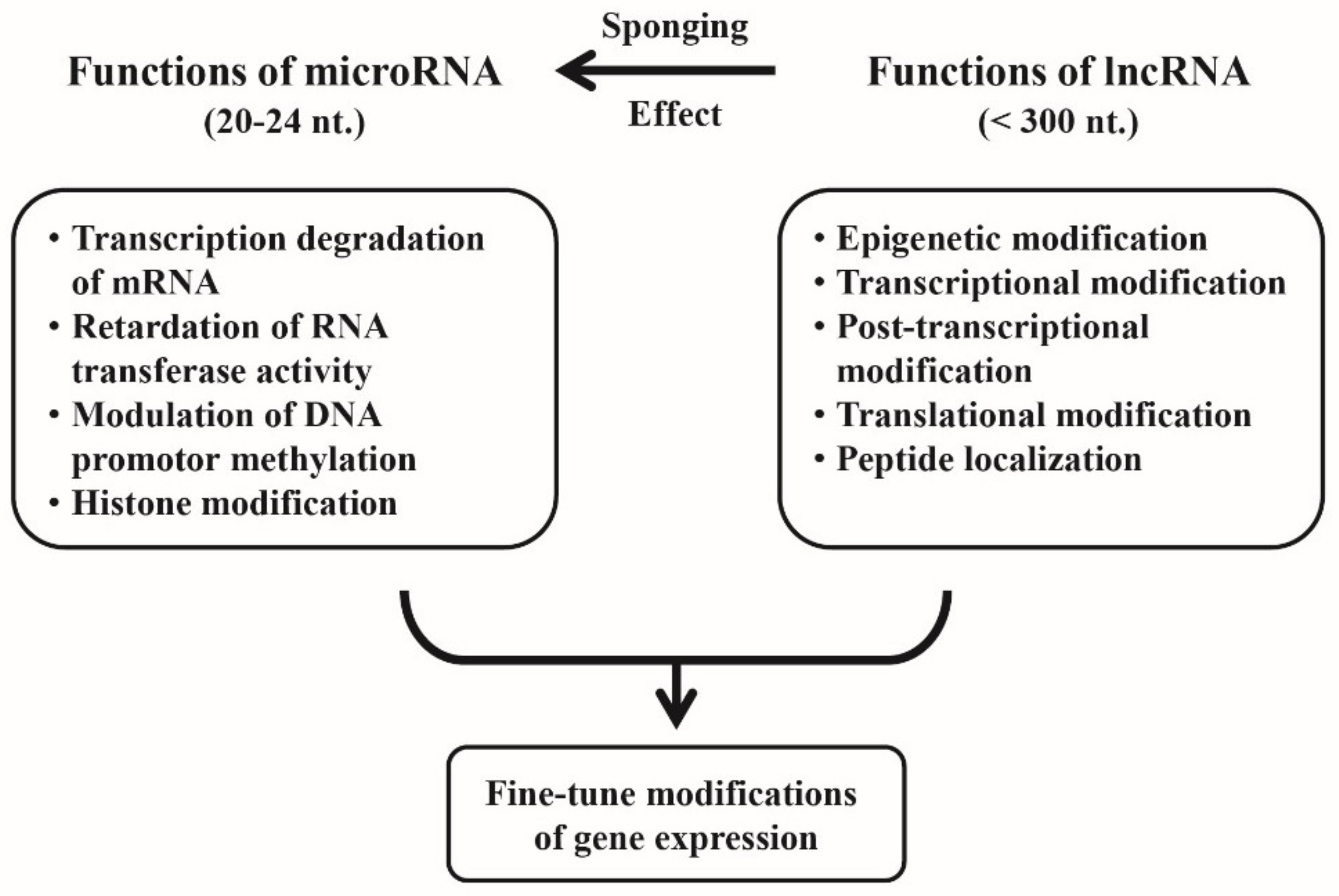{kind=link}
{kind=link}
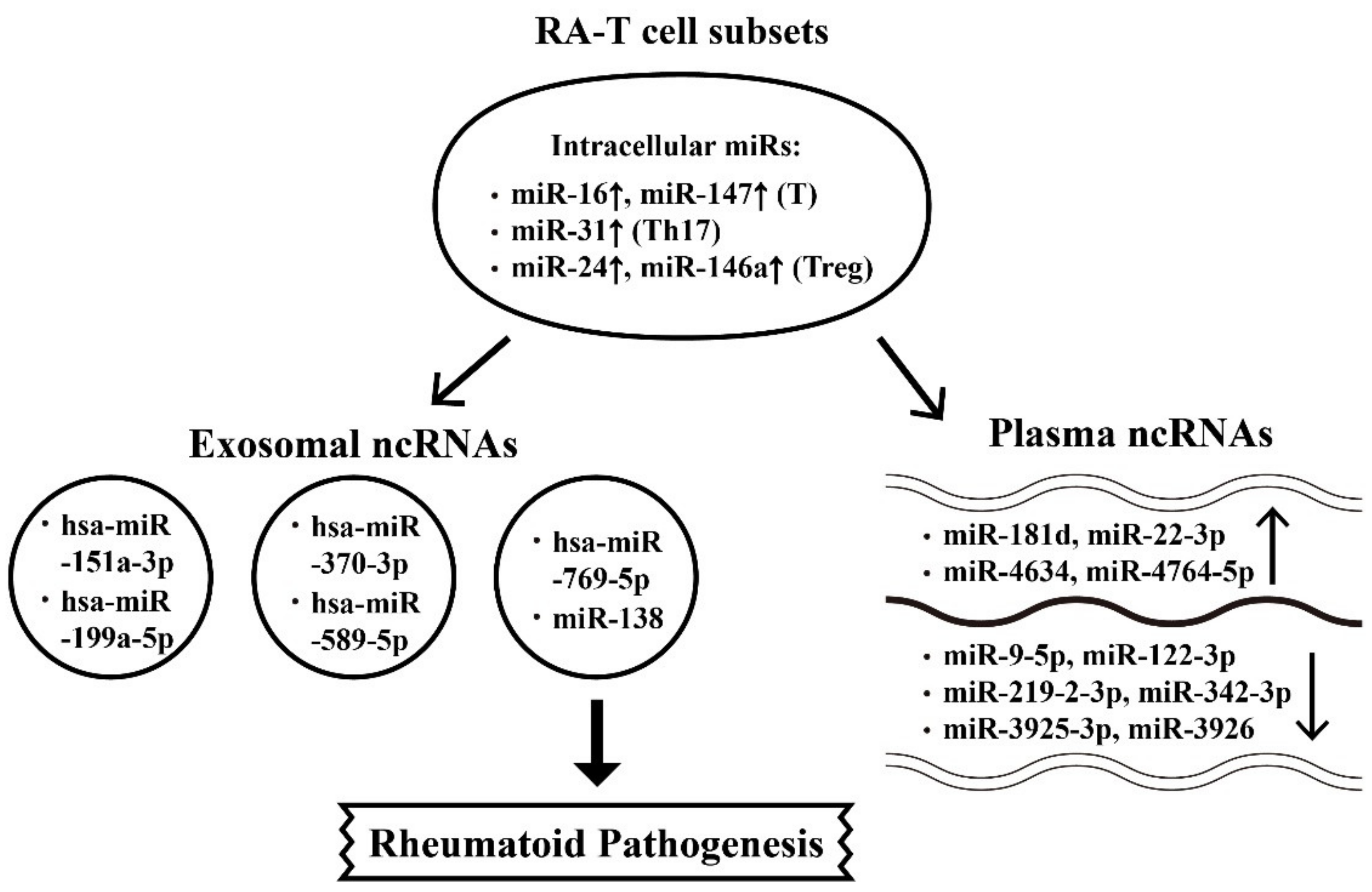{kind=link}
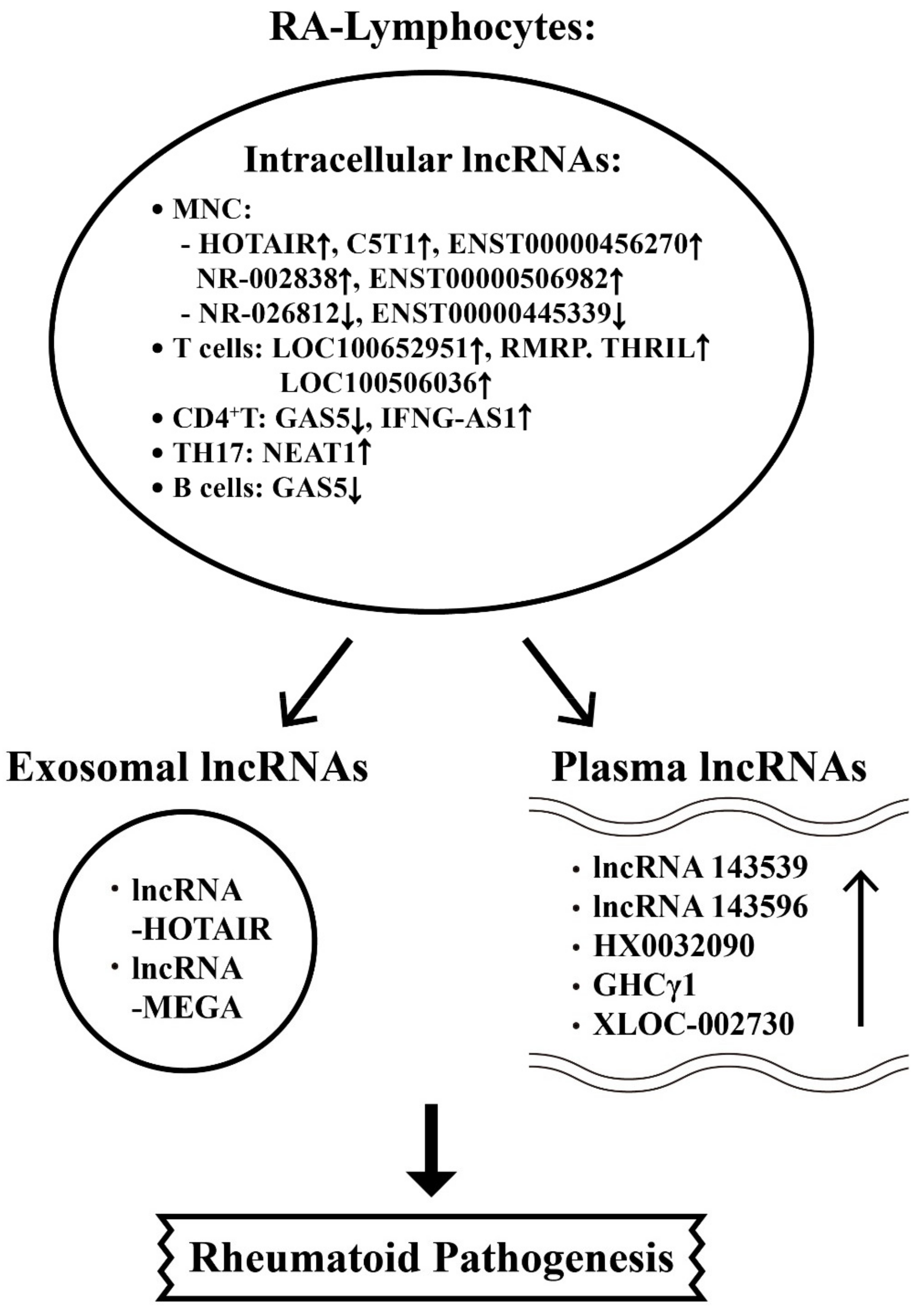{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immunopathologic Roles of RFs and ACPAs in Patients with RA
2.1. Immunopathologic Roles of RFs in RA
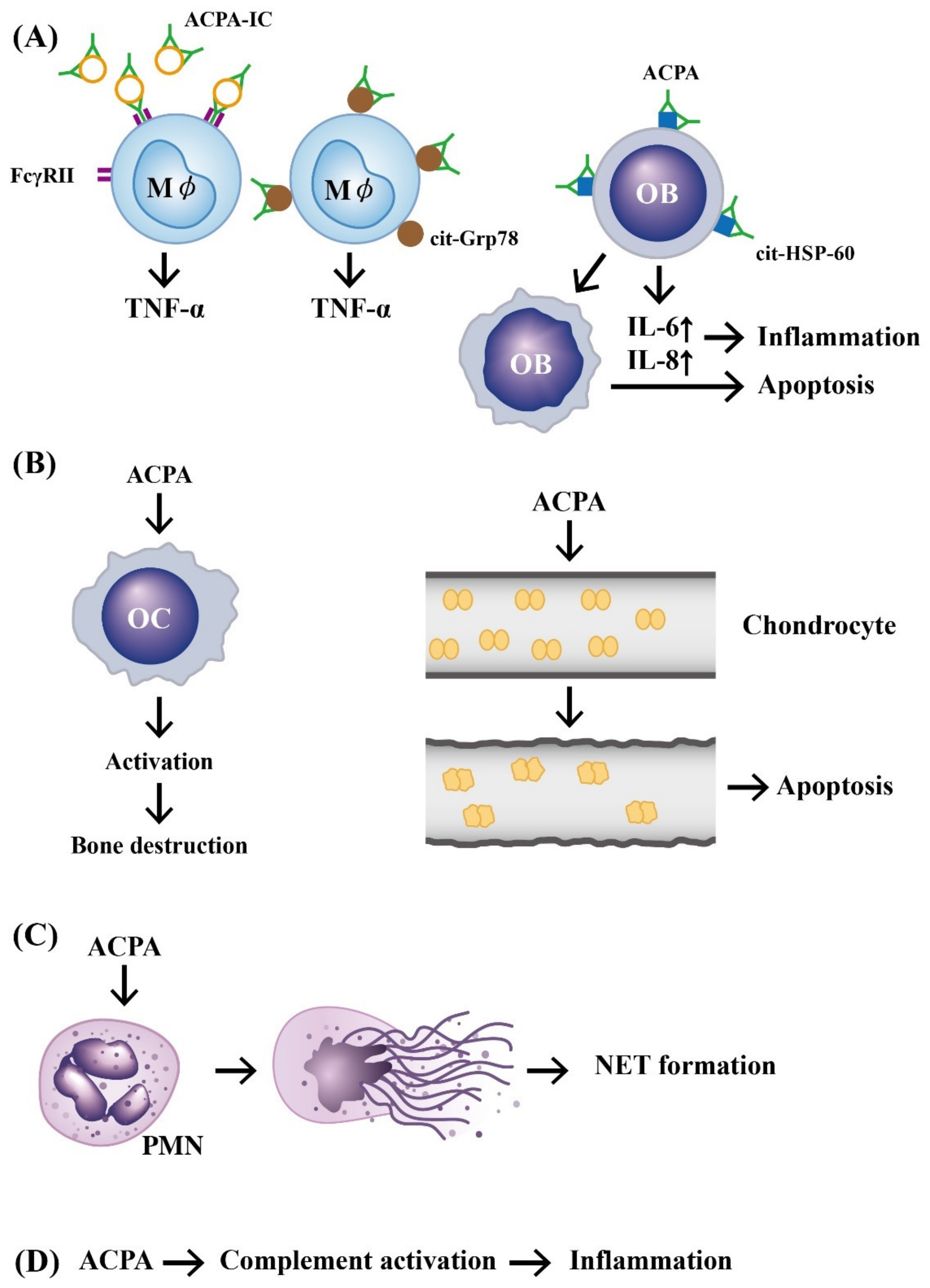
2.2. Immunopathologic Roles of ACPAs in RA
3. The Abnormal Expression and Functions of Peptidylarginine Deiminase 2 (PAD2) and 4 (PAD4) in Generating APCAs and Anti-PAD Autoantibodies in Patients with RA
3.1. The Roles of Excessive Enzymatic Activity of PAD2 and PAD4 in RA Pathogenesis
3.2. The Causes of Anti-PAD4 Autoantibody Production in RA Patients
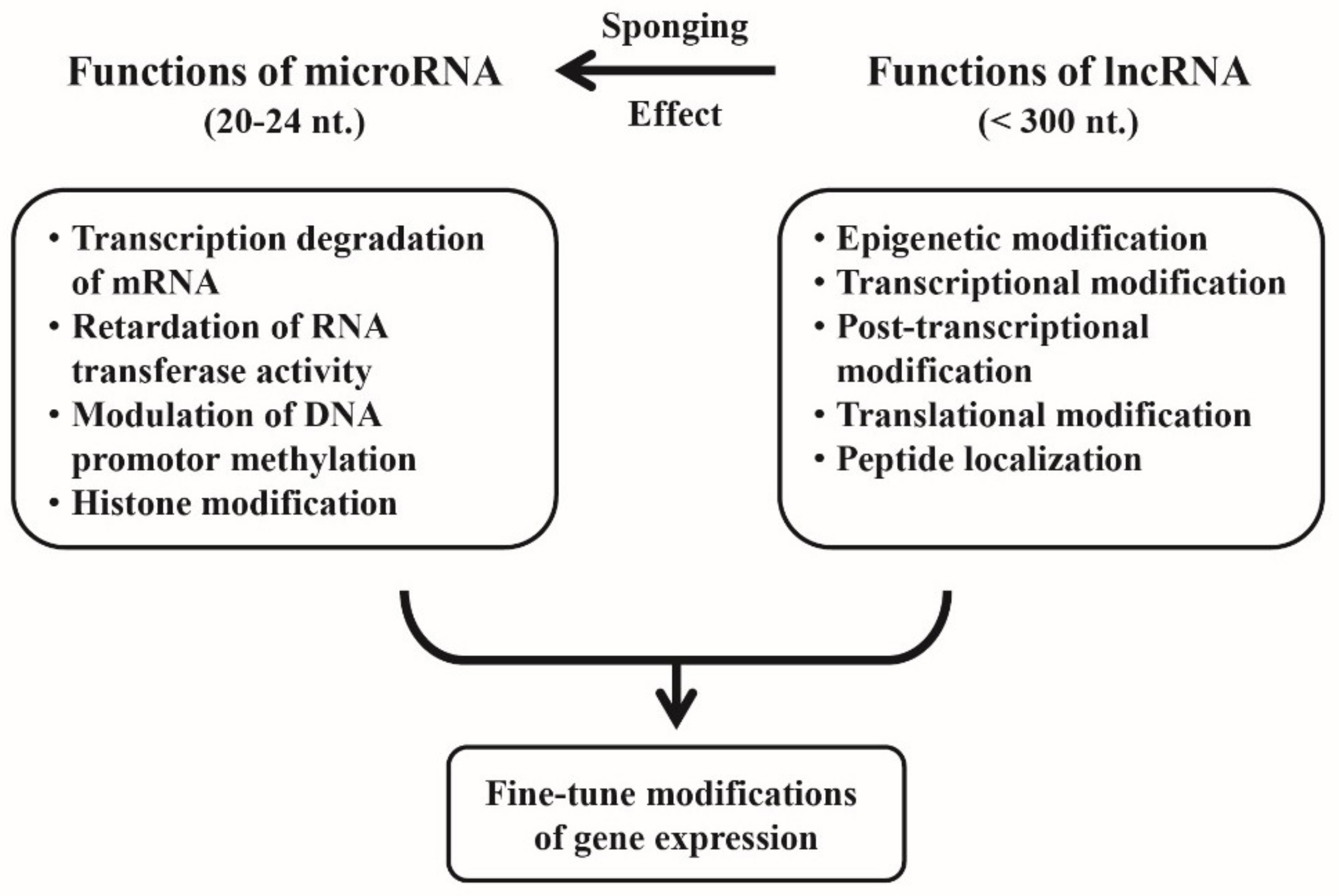
4. Cross-Talk between miRs and lncRNAs for Fine-Tuning of the Gene Expression
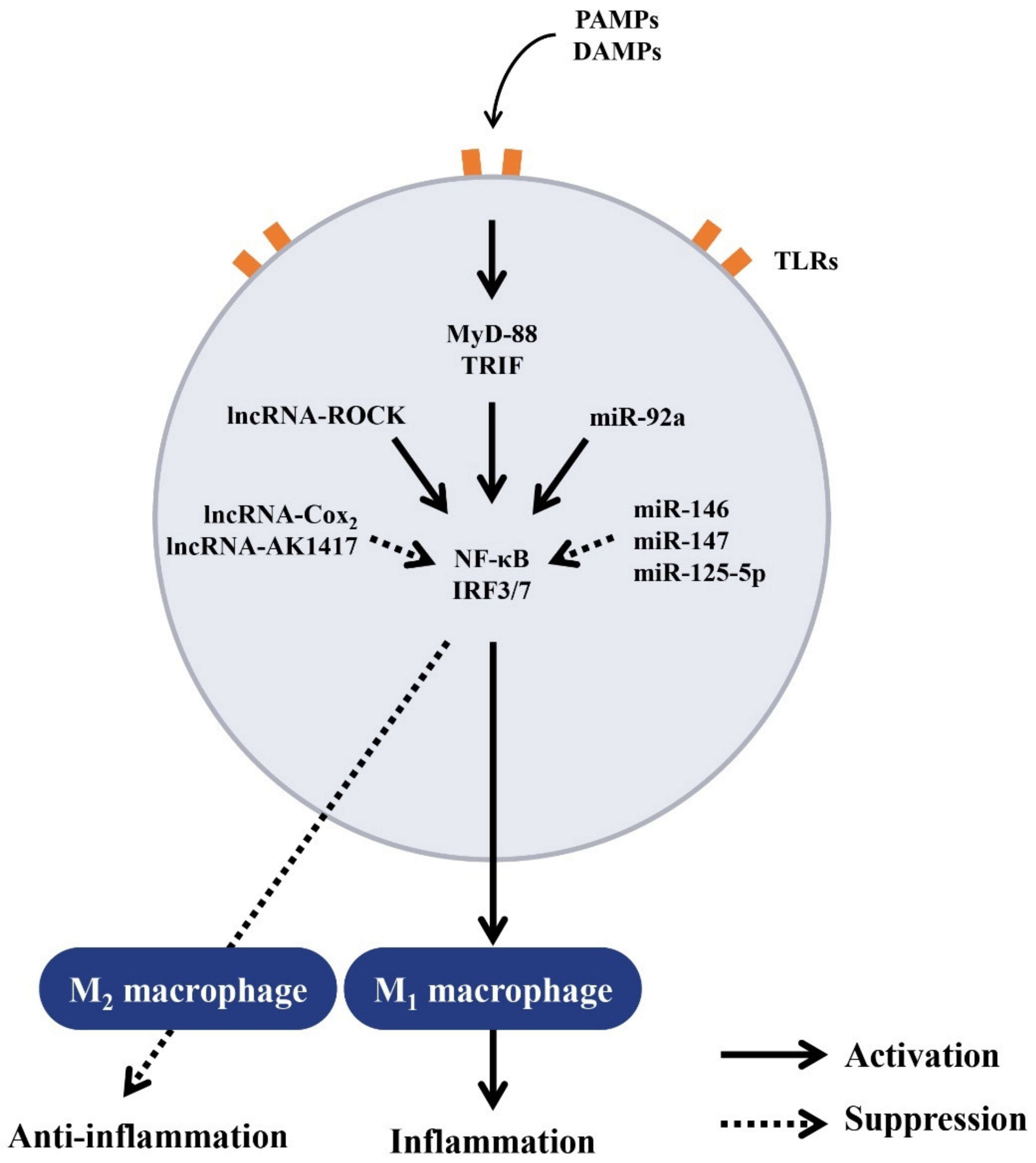
5. Regulation of Macrophage Polarization by ncRNAs
5.1. Regulation of Macrophage Polarization by ncRNAs for Balanced Inflammatory and Anti-Inflammatory Responses
5.2. Regulation of Balanced Inflammatory and Anti-Inflammatory Responses by lncRNAs
6. The Involvement of Aberrant ncRNA Expression in RA Pathogenesis
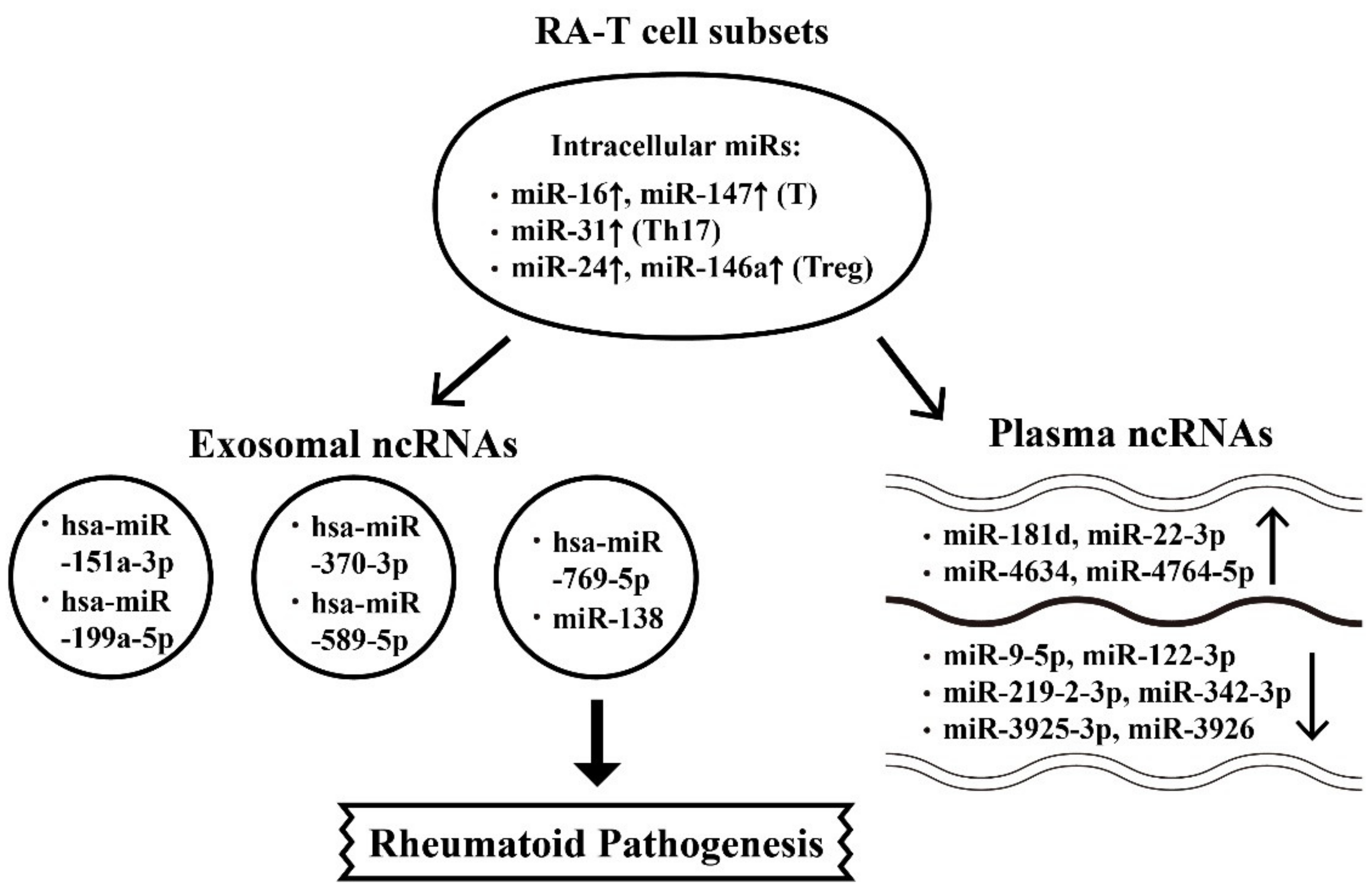
6.1. Abnormal Intracellular, Plasma, and Exosome-Derived miRs and Their Targets in RA Patients
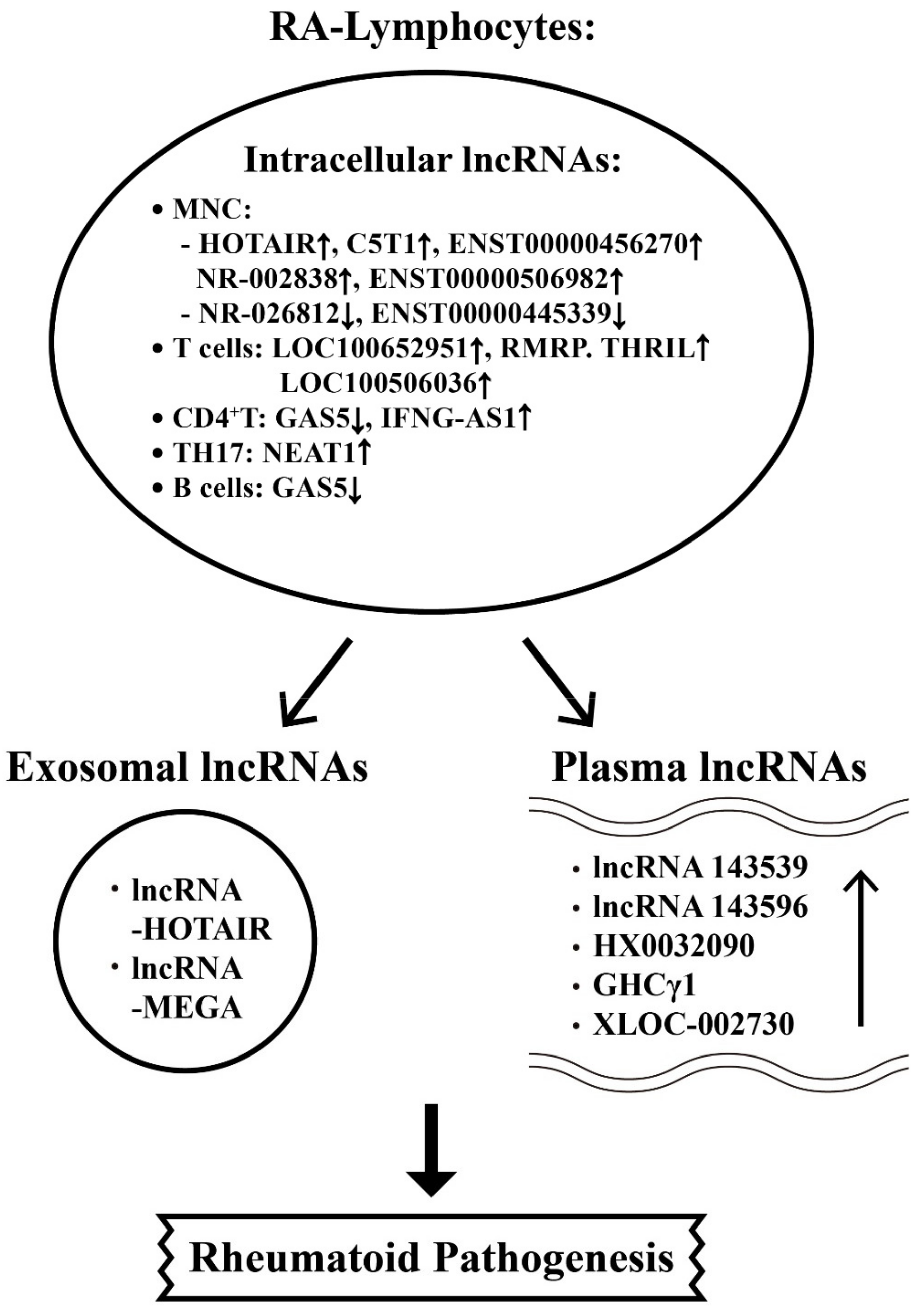
6.2. The Aberrant Expression of Intracellular, Plasma and Exosome-Derived lncRNAs from Different T-cell Subpopulations with Their Targets in RA Patients
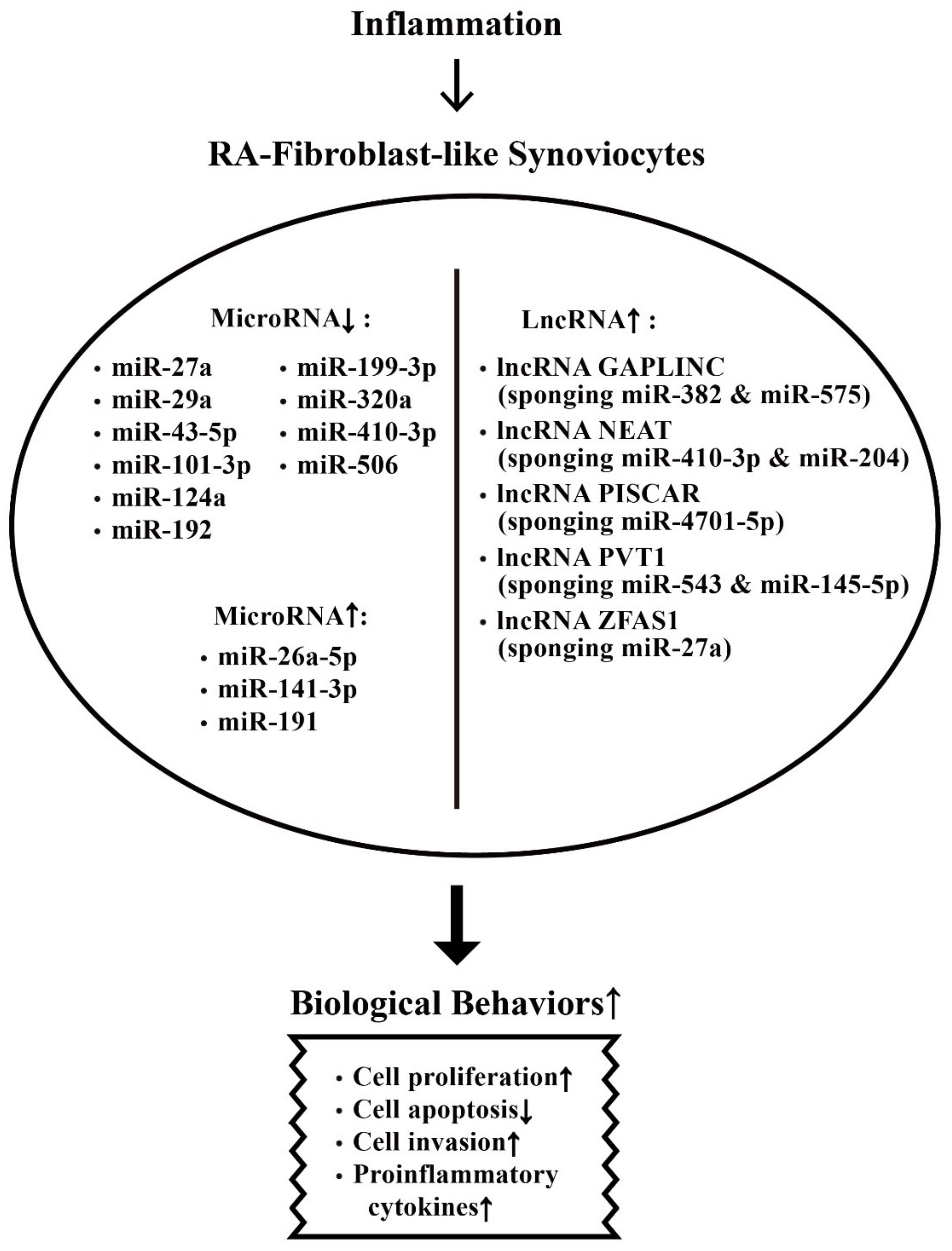
7. The Effects of Rheumatoid Inflammation-Induced Aberrant Non-Coding RNA Expression on the Modification of Biological Behavior in Fibroblast-Like Synoviocytes (FLSs) of RA Patients (RA-FLSs)
7.1. Derangement of the Biological Behaviors of RA-FLSs by Aberrantly Expressed miRs in the Milieu of Chronic Rheumatoid Inflammation
7.2. Inhibition on Biological Behaviors of RA-FLS by Aberrantly Expressed-miRs in Chronic Rheumatoid Inflammation
7.3. Enhancement on Biological Behaviors of RA-FLS by Aberrantly Expressed-lncRNAs in Chronic Rheumatoid Inflammation
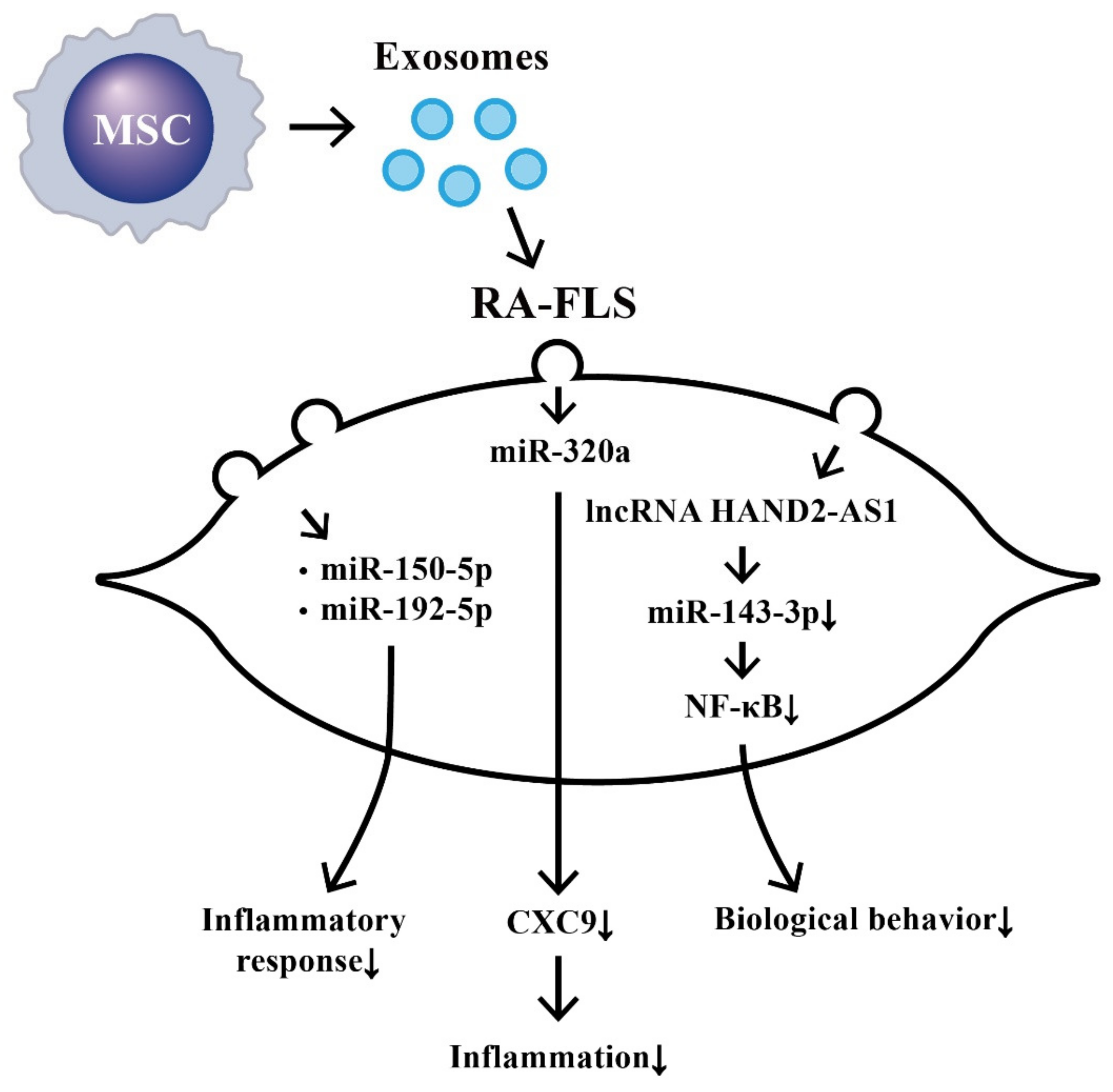
8. Suppression on Biological Behaviors of RA-FLS by Exosome-Derived ncRNAs from Bone Marrow Derived Mesenchymal Stem Cells (BM-MSCs)
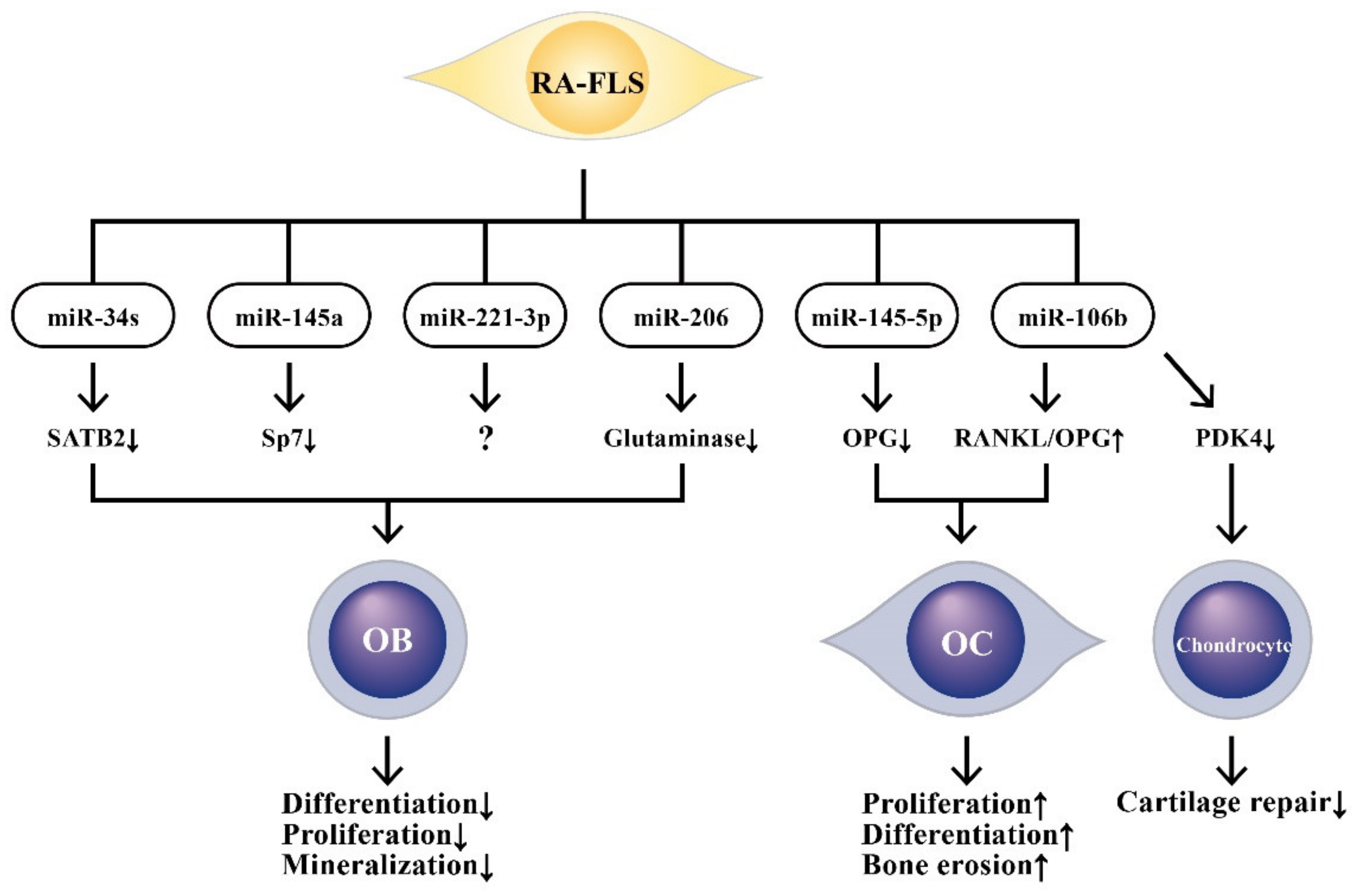
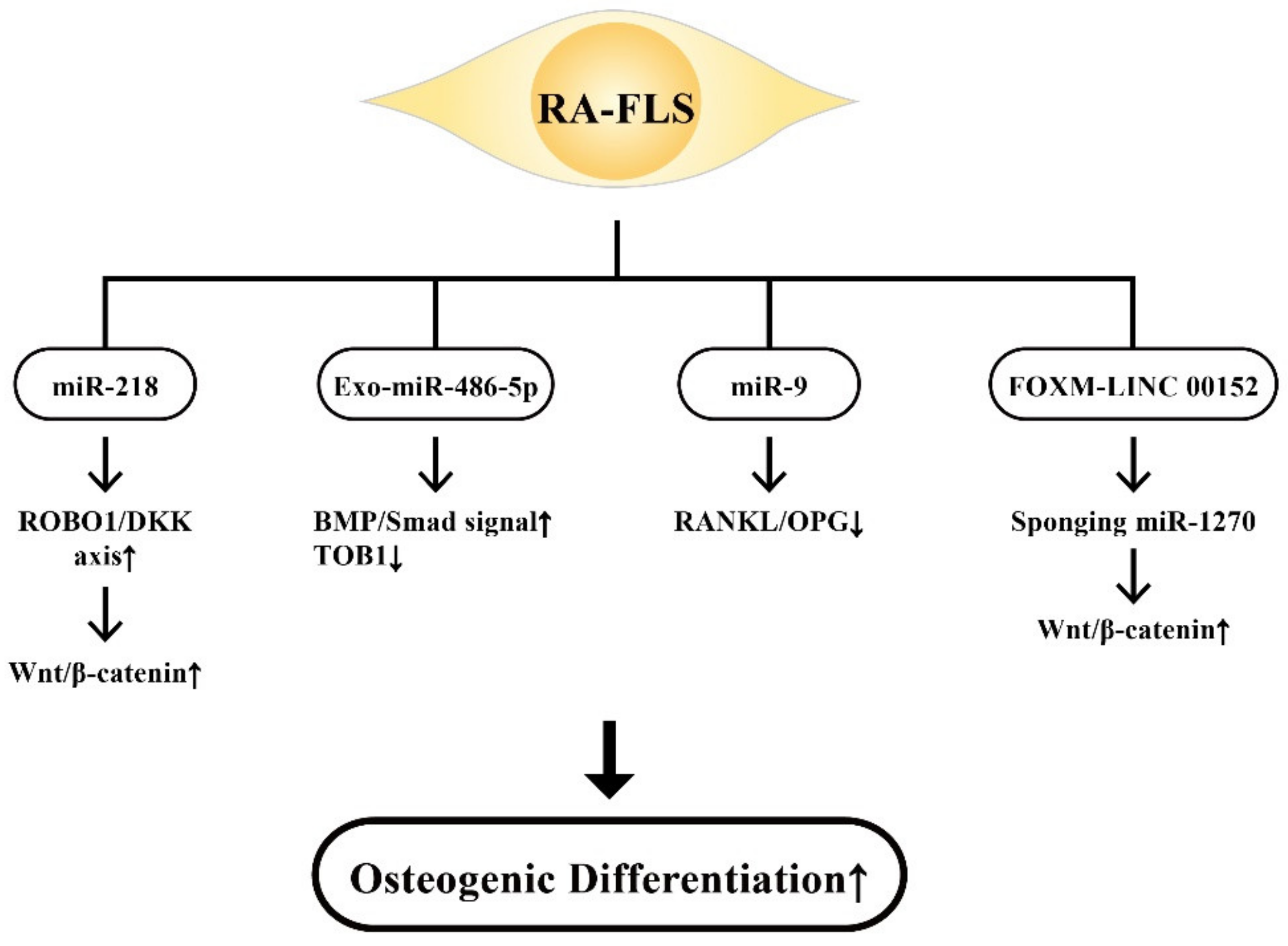
9. The Effects of ncRNAs Derived from RA-FLSs on Bone Metabolism
9.1. The Involvement of RA-FLS-Derived ncRNAs in Bone Destruction
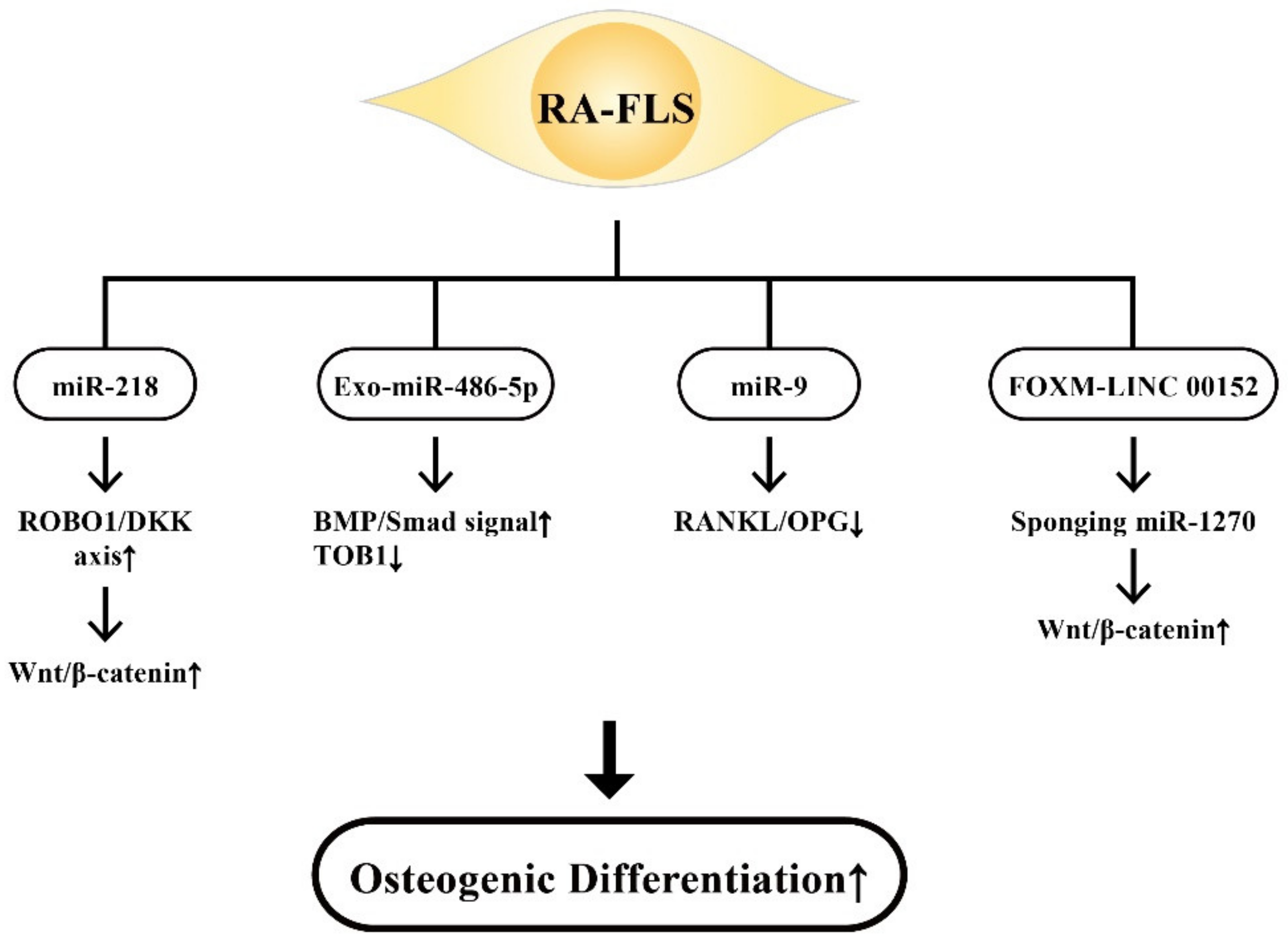
9.2. The Involvement of RA-FLS-Derived ncRNAs on Osteogenic Differentiation
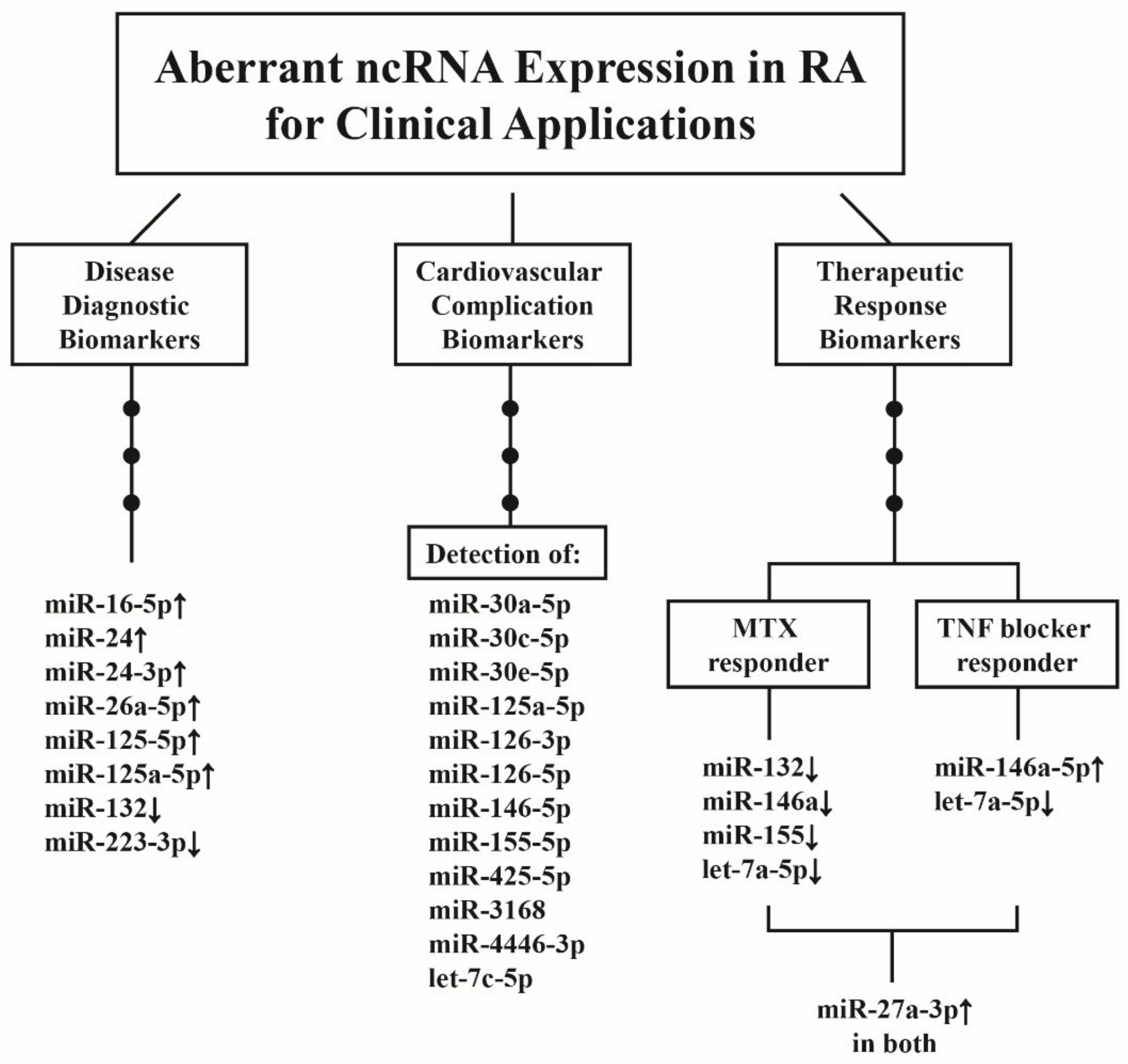
10. Clinical Applications of Aberrant ncRNA Expressions in RA Patients
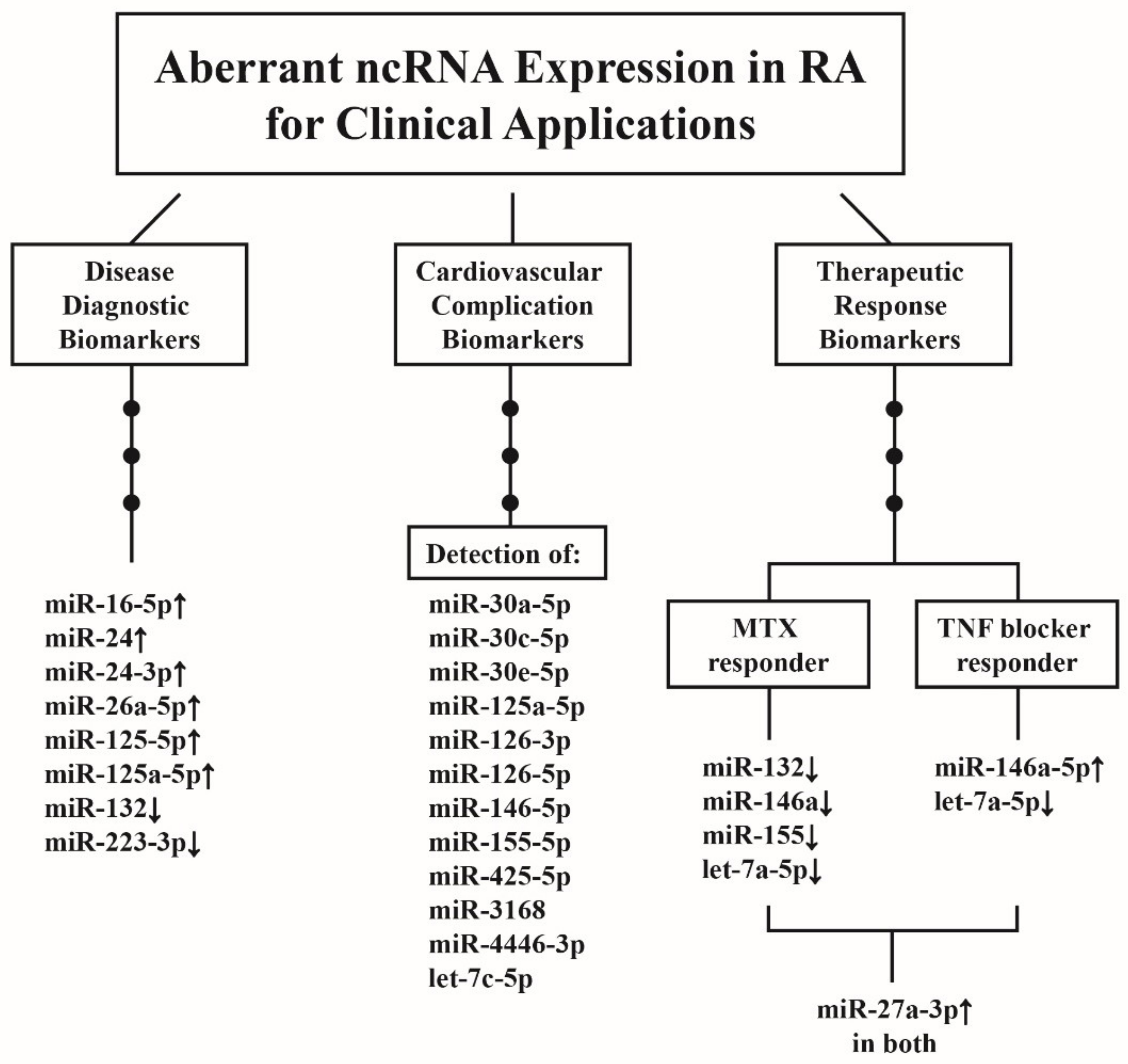
10.1. ncRNAs as Disease Biomarkers for RA Diagnosis
10.2. ncRNAs as Predictor for CV Complications in Patients with RA
10.3. ncRNAs as Biomarkers for Therapeutic Response in Patients with RA
10.4. Therapeutic Potential of the Engineered-Exosomes Containing ncRNAs for RA Treatment
11. Conclusions and Prospects
- (1)
- The study of the effects of sex hormones (estrogen and androgen) on the epigenetic regulation of cytokines during the development of RA.
- (2)
- Research and development of either agonists, antagonists for the ncRNA that can potentially be biomarkers or therapeutic agents for RA in the future, or both.
- (3)
- Investigations into the ncRNAs essential for chondrocyte regeneration to facilitate cartilage repair in rheumatoid joints.
- (4)
- Searches for crucial ncRNAs capable of actively stimulating osteogenic differentiation to prevent disability in RA.
- (5)
- Exploring the ncRNAs relevant to FLS activation to ameliorate rheumatoid inflammation as early as possible.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACPA | anti-citrullinated protein antibodies |
| Ago | argonaute |
| AKT | RAC-alpha serine/threonine-protein kinase or protein kinase B |
| Bax/Bcl-2 | Bcl-associated protein X gene/B cell lymphoma-2 regulatory protein |
| BM-MSC | bone marrow-derived mesenchymal stem cell |
| BMP | bone morphogenetic protein |
| CD | cluster of differentiation |
| C/EBP-β | CCAAT/enhancer-binding protein beta |
| CIA | collagen-induced arthritis |
| ceRNA | competing endogenous RNA |
| circRNA | circular RNA |
| CRP | C-reactive protein |
| CSF | colony stimulating factor |
| CTGF | connective tissue growth factor |
| CV | cardiovascular |
| CXC | cysteine- X-amino acid-cysteine chemokine |
| DAS28 | disease activity score 28 of rheumatoid arthritis |
| DC | dendritic cell |
| DKK | Dickkopf Wnt signaling pathway inhibitor |
| DRs | derived RNAs |
| EBNA | Epstein–Barr virus nuclear antigen |
| ESR | erythrocyte sedimentation rate |
| Exo | exosome |
| FcγR | IgG-Fc receptor |
| FLS | fibroblast-like synoviocyte |
| FOX | forkhead box protein |
| FSTL1 | follistatin-like protein 1 |
| Grp78 | glucose-regulating protein 78 |
| HAND2-AS-1 | Heart- and neural crest derivatives-expressed protein 2- antisense RNA 1 |
| HDAC | histone deacetylase |
| HLA | human leukocyte antigen |
| HOTAIR | HOX (homeobox gene) transcript antisense RNA |
| IC | immune complex |
| IFN | interferon |
| IL | interleukin |
| IRAF-6 | TNF receptor-associated factor 6 |
| IRAK-1 | IL-1 receptor-associated kinase 1 |
| IRF | interferon regulatory factor |
| ITGAV | Integrin alpha chain V gene |
| lncRNA | long non-coding RNA |
| LPS | lipopolysaccharide |
| MAPK-ERK | mitogen-activated protein kinase/extracellular signal-regulated kinases |
| miR | microRNA |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stem cell |
| mTOR | mammalian target of rapamycin |
| MyD88 | myeloid differentiation factor 88 |
| ncRNA | non-coding RNA |
| NEAT | Nuclear enriched abundant transcript |
| NET | neutrophil extracellular trap |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer |
| OB | osteoblast |
| OC | osteoclast |
| OPG | osteoprotegerin |
| PAD | peptidylarginine deiminase |
| PBMC | peripheral blood mononuclear cell |
| PVT1 | plasmacytoma variant translocation 1 |
| MNC | mononuclear cell |
| PDK4 | pyruvate dehydrogenase kinase 4 |
| PGRN | progranulin |
| PMN | polymorphonuclear neutrophil |
| PTEN | phosphatase and tensin homolog |
| RA | rheumatoid arthritis |
| RA-FLS | rheumatoid arthritis-fibroblast-like synoviocyte |
| RANK | receptor activator of NF-κB |
| RANKL | receptor activator of NK-κB ligand |
| RF | rheumatoid factor |
| RISC | RNA induced silencing complex |
| ROBO | roundabout guidance receptor |
| SATB2 | special AT-rich sequence binding protein 2 |
| SF | synovial fluid |
| Smad | small mothers against decapentaplegia protein |
| snoRNA | small nucleolar RNA |
| Sp7 | specific protein 7 |
| sRNA | endogenous small RNA |
| STAT | signal transducer and activator of transcription |
| T-bet | T box expressed in T cells |
| TBS | trabecular bone score |
| tDR | transfer-derived RNA |
| TGF-β | transforming growth factor beta |
| Th17 | helper T cell-17 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| Treg | regulatory T cell |
| TRIF | Toll/IL-1 receptor (TIR) domain-containing adaptor inducing interferon- β factor |
| VEGF | vascular endothelium-derived growth factor |
| Wnt | wingless and Int-1 protein in Drosophila |
| yDR | Y RNA, |
| YY1 | yingyang 1 |
References
- Mateen, S.; Zafar, A.; Moin, S.; Khan, A.Q.; Zubair, S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 2016, 455, 161–171. [Google Scholar] [CrossRef]
- Angelotti, F.; Parma, A.; Cafaro, G.; Capecchi, R.; Alunno, A.; Puxeddu, I. One year in review 2017: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2017, 35, 368–378. [Google Scholar] [PubMed]
- Zamanpoor, M. The genetic pathogenesis, diagnosis and therapeutic insight of rheumatoid arthritis. Clin. Genet. 2019, 95, 547–557. [Google Scholar] [CrossRef]
- Gourley, M.; Miller, F.W. Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat. Clin. Pract. Rheumatol. 2007, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Su, B. Small but influential: The role of microRNAs on gene regulatory network and 3′UTR evolution. J. Genet. Genomics 2009, 36, 1–6. [Google Scholar] [CrossRef]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, S.; Tomari, Y. Argonaute-mediated translational repression (and activation). Fly 2009, 3, 204–206. [Google Scholar] [CrossRef]
- Ateş, A.; Kinikli, G.; Turgay, M.; Akay, G.; Tokgöz, G. Effects of rheumatoid factor isotypes on disease activity and severity in patients with rheumatoid arthritis: A comparative study. Clin. Rheumatol. 2007, 26, 538–545. [Google Scholar] [CrossRef]
- Soltys, A.J.; Hay, F.C.; Bond, A.; Axford, J.S.; Jones, M.G.; Randen, I.; Thompson, K.M.; Natvig, J.B. The binding of synovial tissue-derived human monoclonal immunoglobulin M rheumatoid factor to immunoglobulin G preparations of differing galactose content. Scand. J. Immunol. 1994, 40, 135–143. [Google Scholar] [CrossRef]
- Young, A.; Sumar, N.; Bodman, K.; Goyal, S.; Sinclair, H.; Roitt, I.; Isenberg, D. Agalactosyl IgG: An aid to differential diagnosis in early synovitis. Arthritis Rheum. 1991, 34, 1425–1429. [Google Scholar] [CrossRef]
- Van Zeben, D.; Rook, G.A.; Hazes, J.M.; Zwinderman, A.H.; Zhang, Y.; Ghelani, S.; Rademacher, T.W.; Breedveld, F.C. Early agalactosylation of IgG is associated with a more progressive disease course in patients with rheumatoid arthritis: Results of a follow-up study. Br. J. Rheumatol. 1994, 33, 36–43. [Google Scholar] [CrossRef]
- Lu, M.-C.; Hsieh, S.-C.; Lai, N.-S.; Li, K.-J.; Wu, C.-H.; Yu, C.-L. Comparison of anti-agalactosyl IgG antibodies, rheumatoid factors, and anti-cyclic citrullinated peptide antibodies in the differential diagnosis of rheumatoid arthritis and its mimics. Clin. Exp. Rheumatol. 2007, 25, 716–721. [Google Scholar] [PubMed]
- Masson-Bessière, C.; Sebbag, M.; Durieux, J.J.; Nogueira, L.; Vincent, C.; Girbal-Neuhauser, E.; Durroux, R.; Cantagrel, A.; Serre, G. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin. Exp. Immunol. 2000, 119, 544–552. [Google Scholar] [CrossRef]
- Masson- Bessière, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nagueira, L.; Vincent, C.; Senshu, T.; Serre, G. The major synovial targets of the rheumatoid arthritis-specific anti-filaggrin autoantibodies are deiminated forms of the α- and β-chains of fibrin. J. Immunol. 2001, 166, 4177–4184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.M.T.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A.C. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Vincent, C.; Nogueira, L.; Clavel, C.; Sebbag, M.; Serre, G. Autoantibodies to citrullinated proteins: APCA. Autoimmunity 2005, 38, 17–24. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-C.; Lai, N.-S.; Yu, H.-C.; Huang, H.-B.; Hsieh, S.-C.; Yu, C.-L. Anti-citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocytes/macrophages and stimulate tumor necrosis factor α production. Arthritis Rheum. 2010, 62, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via surface-expressed citrullinated Grp78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.S.; Yu, H.C.; Yu, C.L.; Koo, M.; Huang, H.B.; Lu, M.C. Anti-citrullinated protein antibodies suppress let-7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015, 220, 1351–1358. [Google Scholar] [CrossRef]
- Lu, M.H.; Yu, C.L.; Yu, H.C.; Huang, H.B.; Koo, M.; Lai, N.S. Anti-citrullinated protein antibodies promote apoptosis of mature human Saos-2 osteoblasts via cell-surface binding to citrullinated heat shock protein 60. Immunobiology 2016, 221, 76–83. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Pratesi, F.; Dioni, I.; Tommasi, C.; Alcaro, M.C.; Paolini, I.; Barbetti, F.; Boscaro, F.; Panza, F.; Puxeddu, I.; Rovero, P.; et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann. Rheum. Dis. 2014, 73, 1414–1422. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Ge, C.; Tong, D.; Liang, B.; Lönnoblom, E.; Schneider, N.; Hagert, C.; Viljanen, J.; Ayoglu, B.; Stawikowska, R.; Nilsson, P.; et al. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight 2017, 2, e93688. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-C.; Lu, M.-C. The roles of anti-citrullinated protein antibodies in the immunopathogenesis of rheumatoid arthritis. Tzu Chi Med. J. 2019, 31, 5–10. [Google Scholar]
- Wu, C.Y.; Yang, H.Y.; Lai, J.H. Anti-citrullinated protein antibodies in patients with rheumatoid arthritis: Biological effects and mechanisms of immunopathogenesis. Int. J. Mol. Sci. 2020, 21, 4015. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silmam, A.J.; Funovits, J.; Felson, D.T.; Binghan, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; de Jong, B.A.; van den Hoogen, F.H.; van de Putte, L.B.; van Venrooij, W.J. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis specific autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantapää-Dahlqvist, S.; de Jong, B.A.W.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- Sokolove, J.; Bloomberg, R.; Deane, K.D.; Lahey, L.J.; Derber, L.A.; Chandra, P.E.; Edison, J.D.; Gilliland, W.R.; Tibshirani, R.J.; Morris, J.M.; et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE 2012, 7, e35296. [Google Scholar] [CrossRef]
- van der Helm-van Mil, A.H.M.; Verpoort, K.N.; Breedveld, F.C.; Toes, R.E.M.; Huizinga, T.W.J. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R949–R958. [Google Scholar] [CrossRef] [Green Version]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.K.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very recent onset rheumatoid arthritis: Clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology 2007, 46, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darrah, E.; Giles, J.T.; Ols, M.L.; Bull, H.G.; Andrade, F.; Rosen, A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci. Transl. Med. 2013, 5, 186ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darrah, E.; Giles, J.T.; Davis, R.L.; Naik, P.; Wang, H.; Konig, M.F.; Cappelli, L.C.; Bingham, C.O., 3rd; Danoff, S.K.; Andrade, F. Autoantibodies to peptidylarginine deimidase 2 are associated with less severe disease in rheumatoid arthritis. Front. Immunol. 2018, 9, 2696. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, R.; Paimela, L.; Julkunen, H.; Tienari, P.J.; Leirisalo-Repo, M.; Palosuo, T.; Vaarala, O. Peptidylarginine deiminase, the arginine to citrulline converting enzyme, is frequently recognized by sera of patients with rheumatoid arthritis, systemic lupus erythematosus, and primary Sjögren’s syndrome. Scand. J. Rheumatol. 2003, 32, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Badillo-Soto, M.A.; Rodriguez-Rodriguez, M.; Pérez-Pérez, M.E.; Daza-Benitez, L.; Bollain-y-Goytia, J.J.; Carrillo-Jiménez, M.A.; Avalos-Diaz, E.; Herrera-Esparza, R. Potential protein targets of the peptidylarginine deiminase 2 and peptidylarginine deiminase 4 enzymes in rheumatoid synovial tissue and its possible meaning. Eur. J. Rheumatol. 2016, 3, 44–49. [Google Scholar] [CrossRef]
- Bawadekar, M.; Shim, D.; Johnson, C.J.; Warner, T.F.; Rebernick, R.; Damgaard, D.; Nielsen, C.H.; Pruijn, G.J.M.; Nett, J.E.; Shelef, M.A. Peptidylarginine deiminases 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J. Autoimmun. 2017, 80, 39–47. [Google Scholar] [CrossRef]
- Jonsson, M.K.; Kantyka, T.; Falkowski, K.; Aliko, A.; Aga, A.B.; Lillegraven, S.; Sexton, J.; Fevang, B.T.; Mydel, P.; Haavardsholm, E.A. Peptidylarginine deiminase 4 (PAD4) activity in early rheumatoid arthritis. Scand. J. Rheumatol. 2020, 49, 87–95. [Google Scholar] [CrossRef]
- Kolarz, B.; Ciesla, M.; Dryglewska, M.; Majdan, M. Peptidyl arginine deiminase type 4 gene promoter hypo-methylation in rheumatoid arthritis. J. Clin. Med. 2020, 9, 2049. [Google Scholar] [CrossRef]
- Teo, C.Y.; Shave, S.; Chor, A.L.T.; Salleh, A.B.; Rahman, M.B.B.A.; Walkinshaw, M.D.; Tejo, B.A. Discovery of a new class of inhibitors for the protein arginine deiminase 4 (PAD4) by structure-based virtual screening. BMC Bioinform. 2012, 13 (Suppl. S17), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleng, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4000 samples from North America and Sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koushik, S.; Joshi, N.; Nagaraju, S.; Mahmood, S.; Mudeenahally, K.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. PAD4: Pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin. Therapeut. Targets 2017, 21, 433–447. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Guderud, K.; Mæhlen, M.T.; Nordang, G.B.N.; Viken, M.K.; Andreassen, B.K.; Molberg, Ø.; Flåm, S.T.; Lie, B.A. Lack of association among peptidyl arginine deiminase type 4 autoantibodies, PADI4 polymorphisms, and clinical characteristics in rheumatoid arthritis. J. Rheumatol. 2018, 45, 1211–1219. [Google Scholar] [CrossRef]
- Martinez-Prat, L.; Palterer, B.; Vitiello, G.; Parronchi, P.; Robinson, W.H.; Mahler, M. Autoantibodies to protein-arginine deiminase (PAD) 4 in rheumatoid arthritis: Immunological and clinical significance, and potential for precision medicine. Expert Rev. Clin. Immunol. 2019, 15, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Auger, I.; Balandraud, N.; Massy, E.; Hemon, M.F.; Peen, E.; Arnoux, F.; Mariot, C.; Martin, M.; Lafforgue, P.; Busnel, J.-M.; et al. Peptidylarginine deiminase autoimmunity and the development of anti-citrullinated protein antibody in rheumatoid arthritis: The hapten-carrier model. Arthritis Rheumatol. 2020, 72, 903–911. [Google Scholar] [CrossRef]
- Darrah, E.; Davis, R.; Curran, A.M.; Naik, P.; Chen, R.; Na, C.H.; Giles, J.T.; Andrade, F. Citrulline not a major determinant in the recognition of peptidylarginine deiminase 2 and 4 by autoantibodies in rheumatoid arthritis. Arthritis Rheumatol. 2020, 72, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Great therapeutic potential of peptidylarginine deiminase 4 (PAD4) inhibitors: Treatment of rheumatoid arthritis, epigenetic tools, regulation of pluripotency in stem cells, and more. ACS Med. Chem. Lett. 2017, 8, 19–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawez, A.; Al-Haidari, A.; Mahi, R.; Rahman, M.; Thorlacius, H. MiR-155 regulates PAD4-dependent formation of neutrophil extracellular traps. Front. Immunol. 2019, 10, 2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS ONE 2013, 8, e53823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Ilik, I.A.; Qu, K.; Georqiev, P.; Chu, C.; Akhtar, A.; Chang, H.Y. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat. Biotechnol. 2014, 32, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Bayoumi, A.S.; Sayed, A.; Broskova, Z.; Teoh, J.-P.; Wilson, J.; Su, H.; Tang, Y.-L.; Kim, I.-M. Crosstalk between long noncoding RNAs and microRNAs in health and disease. Int. J. Mol. Sci. 2016, 17, 356. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Mullen, G.E.D.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003, 24, 528–533. [Google Scholar] [CrossRef]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. How Toll-like receptors signal: What we know and what we don’t know. Curr. Opin. Immunol. 2006, 18, 3–9. [Google Scholar] [CrossRef]
- Lindsay, M.A. microRNAs and the immune response. Trends Immunol. 2008, 29, 343–351. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Friggeri, A.; Yang, Y.; Park, Y.-J.; Tsuruta, Y.; Abraham, E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc. Natl. Acad. Sci. USA 2009, 106, 15819–15824. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Song, Y.; Liu, Y.; Chen, Q.; Han, Q.; Chen, W.; Pan, T.; Zhang, Y.; Cao, X.; Wang, Q. MicroRNA-92a negatively regulates Toll-like receptor (TLR)-triggered inflammatory response in macrophages by targeting MKK4 kinase. J. Biol. Chem. 2013, 288, 7956–7967. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Cui, H.; Xie, N.; Tan, Z.; Yang, S.; Icyuz, M.; Thannickal, V.J.; Abraham, E.; Liu, G. miR-125a-5p regulates differential activation of macrophages and inflammation. J. Biol. Chem. 2013, 288, 35428–35436. [Google Scholar] [CrossRef] [Green Version]
- Soldano, S.; Trombetta, A.C.; Contini, P.; Tomatis, V.; Ruaro, B.; Brizzolara, R.; Montagna, P.; Sulli, A.; Paolino, S.; Pizzorni, C.; et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1842–1845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, M.; Ericsson, A.C. Function of macrophages in disease: Current understanding on molecular mechanisms. Front. Immunol. 2021, 12, 620510. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.-C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Padilla, M.; Mata-Haro, V. Regulation of TLR signaling pathways by microRNAs: Implications in inflammatory diseases. Centr. Eur. J. Immunol. 2018, 43, 482–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique signatures of long non-coding RNA expression in response to virus infection and altered innate immune signaling. mBio 2010, 1, e00206. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Gong, A.Y.; Wang, Y.; Ma, S.; Chen, X.; Chen, J.; Su, C.J.; Shibata, A.; Strauss-Soukup, J.K.; Drescher, K.M.; et al. LincRNA-Cox 2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J. Immunol. 2016, 196, 2799–2808. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Ming, Z.; Gong, A.-Y.; Wang, Y.; Chen, X.; Hu, G.; Zhou, R.; Shibata, A.; Swanson, P.C.; Chen, X.-M. A long noncoding RNA, lincRNA-Tnfaip3, act as a coregulator of NF-κB to modulate inflammatory gene transcription in mouse macrophages. FASEB J. 2017, 31, 1215–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chao, T.-C.; Patil, V.S.; Qin, Y.; Tiwari, S.K.; Chiou, J.; Dobin, A.; Tsai, C.-M.; Li, Z.; Dang, J.; et al. The long noncoding RNA ROCK1 regulates inflammatory gene expression. EMBO J. 2019, 38, e100041. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gu, L.-N.; Zhang, J.; Pan, J.; Zhang, J.M.; Zhao, D.Y.; Liu, F. LncRNA-AK149641 regulates the secretion of tumor necrosis factor-α in P815 mast cells by targeting the nuclear factor-kappa B signaling pathway. Sci. Rep. 2020, 10, 16655. [Google Scholar] [CrossRef] [PubMed]
- Mathy, N.W.; Chen, X.-M. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J. Biol. Chem. 2017, 292, 12375–12382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Liu, H.; Chen, F. Functioning of long noncoding RNAs expressed in macrophage in the development of atherosclerosis. Front. Pharmacol. 2020, 11, 567582. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J. Immunol. 2010, 184, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, Y.; Guo, Q.; Zou, L.; Zhang, J.; Fang, Y.; Zhang, J.; Zhang, J.; Fu, X.; Liu, H.; et al. Altered microRNA expression profile with miR-146a upregulation in CD4+T cells from Patients with rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R81. [Google Scholar] [CrossRef] [Green Version]
- Lai, N.-S.; Yu, H.-C.; Tung, C.-H.; Huang, K.-Y.; Huang, H.-B.; Lu, M.-C. The role of aberrant expression of T cell miRNAs affected by TNF-α in the immunopathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 261. [Google Scholar] [CrossRef] [Green Version]
- Kikodze, N.; Pantsulaia, I.; Chikovani, T. The role of T regulatory and TH17 cells in the pathogenesis of rheumatoid arthritis (review). Georgian Med. News 2016, 261, 62–68. [Google Scholar]
- Dong, L.; Wang, X.; Tan, J.; Li, H.; Qian, W.; Chen, J.; Chen, Q.; Wang, J.; Xu, W.; Tao, C.; et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J. Cell. Mol. Med. 2014, 18, 2213–2224. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Liu, W.; Xue, B.; Zhang, L.; Liu, X.-Y.; Liu, B.; Wang, Y.; Cai, Y.; Duan, R. Upregulated expression of microRNA-16 correlates with Th17/Treg cell imbalance in patients with rheumatoid arthritis. DNA Cell Biol. 2016, 35, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Xiang, H.; Yang, J.; Hong, L.; Zhang, L.; Liu, Y.; Feng, X.; Cai, C. Dendritic cells from rheumatoid arthritis patient peripheral blood induce Th17 cell differentiation via miR-363/integrin av/TGF-β axis. Scand. J. Immunol. 2017, 85, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Kmiołek, T.; Rzeszotarska, E.; Wajda, A.; Walczuk, E.; Kuca-Warnawin, E.; Romanowska-Próchnicka, K.; Stypinska, B.; Majewski, D.; Jagodzinski, P.P.; Pawlik, A.; et al. The interplay between transcriptional factors and microRNAs as an important factor for Th17/Treg balance in RA patients. Int. J. Mol. Sci. 2020, 21, 7169. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Yu, C.L.; Chen, H.C.; Yu, H.C.; Huang, H.B.; Lai, N.S. Increased miR-223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin-like growth factor-1-mediated interleukin-10 production. Clin. Exp. Immunol. 2014, 177, 641–651. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Zhu, B.; Duan, T.; Xu, Q.; Wang, R.; Lu, L.; Jiao, Z. Plasma microRNA expression profiles in Chinese patients with rheumatoid arthritis. Oncotarget 2015, 6, 42557. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Solus, J.F.; Sheng, Q.; Ye, F.; Song, H.; Wu, Q.; Guo, Y.; Oeser, A.M.; Allen, R.M.; Vickers, K.C.; et al. The endogenous plasma small RNAome of rheumatoid arthritis. ACR Open Rheumatol. 2020, 2, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, Z.; Salmaninejad, A.; Hosseini, N.; Shahbakhsh, Y.; Fadaee, E.; Shahrzad, M.K.; Fadaei, S. MicroRNA and exosome: Key players in rheumatoid arthritis. J. Cell Biochem. 2019, 120, 10930–10944. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M.; Zhao, Y.; Wu, X.-D.; Wang, M.-J.; Yu, H.; Lu, J.-J.; Hu, Y.-J.; Huang, Q.-C.; Huang, R.-Y.; Lu, C.-J. Novel findings from determination of common expressed plasma exosomal microRNAs in patients with psoriatic arthritis, psoriasis vulgaris, rheumatoid arthritis, and gouty arthritis. Discov. Med. 2019, 28, 47–68. [Google Scholar]
- Liu, C.; Pan, A.; Chen, X.; Tu, J.; Xia, X.; Sun, L. MiR-5571-3p and miR-135b-5p, derived from analyses of microRNA profile sequencing, correlate with increased disease risk and activity of rheumatoid arthritis. Clin. Rheumatol. 2019, 38, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hou, C. miR-138 activates NF-κB signaling and PGRN to promote rheumatoid arthritis via regulating HDAC4. Biochem. Biophys. Res. Commun. 2019, 519, 166–171. [Google Scholar] [CrossRef]
- Evangelatos, G.; Fragoulis, G.E.; Koulouri, V.; Lambrou, G.I. MicroRNAs in rheumatoid arthritis: From pathogenesis to clinical impact. Autoimmun. Rev. 2019, 18, 102391. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yan, S.; Yang, J.; Lu, H.; Xu, D.; Wang, Z. Non-coding RNAs in rheumatoid arthritis: From bench to bedside. Front. Immunol. 2019, 10, 3129. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.B.; Medvedev, A.E. Long noncoding RNAs as regulators of Toll-like receptor signaling and innate immunity. J. Leukoc. Biol. 2016, 99, 839–850. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Han, J.; Kim, Y.; Lee, M.; Jin, E.J. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin. Exp. Med. 2015, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Messemaker, T.C.; Frank-Bertoncelj, M.; Marques, R.B.; Adriaans, A.; Bakker, A.M.; Daha, N.; Gay, S.; Huizinga, T.W.; Toes, R.E.M.; Mikkers, H.M.M.; et al. A novel long non-coding RNA in the rheumatoid arthritis risk locus TRAF1-C5 influences C5 mRNA levels. Genes Immun. 2016, 17, 85–92. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, S.; Yu, L.; Qu, B.; Xu, L.; Liu, L.; Sun, H.; Li, C.; Shi, Y.; Liu, H. Long noncoding RNA profiling revealed differentially expressed lncRNAs associated with disease activity in PBMCs from patients with rheumatoid arthritis. PLoS ONE 2017, 12, e0186795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Xu, C.; Li, X.; Zeng, L.; Ye, J.; Guo, Y.; Huang, Z.; Li, J. Comprehensive analysis of long non-coding RNA and mRNA expression profiles in rheumatoid arthritis. Exp. Ther. Med. 2017, 14, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. Long non-coding RNAs target pathogenetically relevant genes and pathways in rheumatoid arthritis. Cells 2019, 8, 816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayama, T.; Marr, A.K.; Kino, T. Differential expression of glucocorticoid receptor noncoding RNA repressor Gas5 in autoimmune and inflammatory diseases. Horm. Metab. Res. 2016, 48, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-C.; Yu, H.-C.; Yu, C.-L.; Huang, H.-B.; Koo, M.; Tung, C.H.; Lai, N.-S. Increased expression of long noncoding RNAs LOC100652951 and LOC100506036 in T cells from patients with rheumatoid arthritis facilitates the inflammatory responses. Immunol. Res. 2016, 64, 576–583. [Google Scholar] [CrossRef]
- Moharamoghli, M.; Hassan-Zadeh, V.; Dolatshahi, E.; Alizadeh, Z.; Farazmand, A. The expression of GASS, THRIL, and RMRP lncRNAs is increased in T cells of patients with rheumatoid arthritis. Clin. Rheumatol. 2019, 38, 3073–3080. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Ren, S.; Liu, Y.; Zhou, H.; Tang, X.; Yang, J.; Tian, J.; Xu, P.; Xu, H.; Wang, S. Elevated expression of the long noncoding RNA IFNG-AS1 in the peripheral blood from patients with rheumatoid arthritis. J. Immunol. Res. 2020, 2020, 6401978. [Google Scholar] [CrossRef] [PubMed]
- Shui, X.; Chen, S.; Lin, J.; Kong, J.; Zhou, C.; Wu, J. Knockdown of lncRNA NEAT1 inhibits Th17/CD4+T cell differentiation through reducing the STAT3 protein level. J. Cell Physiol. 2019, 234, 22477–22484. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, Y.; Yang, L.; Hou, X.; Wang, J.; Gu, W.; Wang, X.; Liu, L.; Zhang, J.; Lu, H. Long noncoding RNAs expression profile and functional networks in rheumatoid arthritis. Oncotarget 2017, 8, 95280–95292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.J.; Wei, Q.F.; Wang, S.J.; Zhang, H.J.; Zhang, X.Y.; Geng, Q.; Cui, Y.H.; Wang, X.H. LncRNA HOTAIR alleviates rheumatoid arthritis by targeting miR-138 and inactivating NF-κB pathway. Int. Immunopharmacol. 2017, 50, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lui, Y.; Meng, F.; Xia, Z.; Wu, X.; Fang, Y.; Zhang, C.; Zhang, Y.; Liu, D. LncRNA MEG3 inhibits rheumatoid arthritis through miR-141 and inactivation of AKT/mTOR signalling pathway. J. Cell. Mol. Med. 2019, 23, 7116–7120. [Google Scholar] [CrossRef] [Green Version]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, R.; Rasool, M. Fibroblast-like synoviocytes-dependent effector molecules as a critical mediator for rheumatoid arthritis: Current status and future directions. Int. Rev. Immunol. 2017, 36, 20–30. [Google Scholar] [CrossRef]
- Mousavi, M.J.; Karami, J.; Aslani, S.; Tahmasebi, M.N.; Vaziri, A.S.; Jamshidi, A.; Farhadi, E.; Mahmoudi, M. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from friend to foe. Auto Immun. Highlights 2021, 12, 3. [Google Scholar] [CrossRef]
- Ospelt, C.; Neidhart, M.; Gay, R.E.; Gay, S. Synovial activation in rheumatoid arthritis. Front. Biosci. 2004, 9, 2323–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.; Wang, K.; Huang, M.; Qiu, Q.; Xiao, Y.; Shi, M. Halofuginone inhibits TNF-α-induced the migration and proliferation of fibroblast-like synoviocyte from rheumatoid arthritis patients. Int. Immunopharmacol. 2017, 43, 187–194. [Google Scholar] [CrossRef]
- Chen, Y.; Xian, P.-F.; Yang, L.; Wang, S.-X. MicroRNA-21 promotes proliferation of fibroblast-like synoviocytes through mediation of NF-κB nuclear translocation in a rat-model of collagen-induced rheumatoid arthritis. Biomed. Res. Int. 2016, 9279078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Xing, S.; Liu, M.; Deng, W.; Wang, Y.; Huang, Z.; Huang, Y.; Huang, X.; Wu, C.; Guo, X.; et al. MiR-26a-5p enhances cells proliferation, invasion, and apoptosis resistance of fibroblast-like synoviocytes in rheumatoid arthritis by regulating PTEN/PI3K/AKT pathway. Biosci. Rep. 2019, 39, BSR20182192. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Lu, Y.; Zong, M.; Tan, Q.; Fan, L. Hypoxia-induced miR-91-C/EBPβ signaling regulates cell proliferation and apoptosis of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.-L.; Shi, G.-R.; Xie, J.; Du, X.-Z.; Yang, H. MicroRNA-27a inhibits cell migration and invasion of fibroblast-like synoviocytes by targeting follistatin-like protein 1 in rheumatoid arthritis. Mol. Cells 2016, 39, 611–618. [Google Scholar] [CrossRef]
- Li, S.; Jin, Z.; Lu, X. MicroRNA-192 suppresses cell proliferation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes by downregulating caveolin 1. Mol. Cell. Biochem. 2017, 432, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-J.; Li, X.-W.; Lu, J.-L.; Long, Z.-X.; Liang, J.-Q.; Wei, S.-B.; Lu, C.-X.; Lu, W.-Z. MiR-20a regulates fibroblast-like synoviocyte proliferation and apoptosis in rheumatoid arthritis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3886–3893. [Google Scholar]
- Liu, J.; Fei, D.; Xing, J.; Du, J. MicroRNA-29a inhibits proliferation and induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes by repressing STAT3. Biomed. Pharmacother. 2017, 96, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Wangyang, Y.; Yi, L.; Wang, T.; Feng, Y.; Liu, G.; Li, D.; Zheng, X. MiR-199a-3p inhibits proliferation and induces apoptosis in rheumatoid arthritis. fibroblast-like synoviocyte via suppressing retinoblastoma 1. Biosci. Rep. 2018, 38, BSR20180982. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Lv, F.; Zhang, H.; Wang, X.; Geng, Q.; Zhang, X.; Li, T.; Wang, S.; Wang, Y.; Cui, Y. MicroRNA-101-3p inhibits fibroblast-like synoviocyte proliferation and inflammation in rheumatoid arthritis by targeting PTGS2. Biosci. Rep. 2020, 40, BSR20191136. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Ge, Y.; Zhou, Y.; Wang, J.; Xie, X.; Li, S.; Tang, M.; Xu, L.C.; Tian, J. miR-124a inhibits the proliferation and inflammation in rheumatoid arthritis fibroblast-like synoviocytes via targeting PIK3/NF-κB pathway. Cell. Biochem. Funct. 2019, 37, 208–215. [Google Scholar] [CrossRef]
- Lin, K.; Su, H.-Y.; Jiang, L.-F.; Chu, T.-G.; Li, Z.; Chen, X.-L.; Gao, W.-Y. Influences of miR-320a on proliferation and apoptosis of fibroblast-like synoviocyte in rheumatoid arthritis through targeting MAPK-ERK1/2. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1907–1914. [Google Scholar]
- Wang, Y.; Jiao, T.; Fu, W.; Zhao, S.; Yang, L.; Xu, N.; Zhang, N. miR-410-3p regulates proliferation and apoptosis of fibroblast-like synovioocytes by targeting YY1 in rheumatoid arthritis. Biomed. Pharmacother. 2019, 119, 109426. [Google Scholar] [CrossRef]
- Li, D.; Zhou, Q.; Hu, G.; Wang, G. MiRNA-506 inhibits rheumatoid arthritis fibroblast-like synoviocytes proliferation and induces apoptosis by targetting TLR4. Biosci. Rep. 2019, 39, BSR20182500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, K.; Yuan, X.; Xu, N.; Zhao, S.; Hou, L.; Yang, L.; Zhang, N. miR-431-5p regulates cell proliferation and apoptosis in fibroblast-like synoviocytes in rheumatoid arthritis by targeting XIAP. Arthritis Res. Ther. 2020, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhang, H.; Chang, J.; Lu, M.; Gao, W.; Liu, W.; Li, Y.; Yin, L.; Wang, X.; et al. Identification of a novel microRNA-141-3p/forkhead box C1/β-catenin axis associated with rheumatoid arthritis synovial fibroblast function in vivo and in vitro. Theranostics 2020, 10, 5412–5434. [Google Scholar] [CrossRef]
- Ye, Y.; Gao, X.; Yang, N. LncRNA ZFAS1 promotes cell migration and invasion of fibroblast-like synoviocytes by suppression of miR-27a in rheumatoid arthritis. Hum. Cell 2018, 31, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mo, B.Y.; Guo, X.H.; Yang, M.R.; Liu, F.; Bi, X.; Liu, Y.; Fang, L.K.; Luo, X.Q.; Wang, J.; Bellanti, J.A.; et al. Long non-coding RNA GAPLINC promotes tumor-like biologic behaviors of fibroblast-like synoviocytes as microRNA sponging in rheumatoid arthritis patients. Front. Immunol. 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Guo, X.H.; Mo, B.Y.; Wang, M.L.; Luo, X.Q.; Chen, Y.X.; Liu, F.; Olsen, N.; Pan, Y.F.; Zheng, S.G. LncRNA PICSAR promotes cell proliferation, migration, and invasion of fibroblast-like synoviocytes by sponging miR-4701-5p in rheumatoid arthritis. EBioMedicine 2019, 50, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kong, X.; Hu, H.; Shi, S. Knockdown of long non-coding RNA PVT1 induces apoptosis of fibroblast-like synoviocytes through modulating miR-543-dependent SCUBE2 in rheumatoid arthritis. J. Orthop. Surg. Res. 2020, 15, 142. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yi, S.; Liu, Y. Long non-coding RNA PVT1 can regulate the proliferation and inflammatory responses of rheumatoid arthritis fibroblast-like synoviocytes by targeting microRNA-145-5p. Hum. Cell 2020, 33, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, L.; Yuan, X.; Xu, N.; Zhao, S.; Yang, L.; Zhang, N. LncRNA NEAT1 targets fibroblast-like synoviocyte in rheumatoid arthritis via the miR-410-3p/yy1 axis. Front. Immunol. 2020, 11, 1975. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wang, R.; Zhou, W.; Cai, X.; Ye, Z. LncRNA NEAT1 regulates the proliferation and production of the inflammatory cytokines in rheumatoid arthritis fibroblast-like synoviocytes by targeting miR-204-5p. Hum. Cell 2021, 34, 372–382. [Google Scholar] [CrossRef]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, H.; Xia, Y.; Yang, F.; Lu, Y. Therapeutic potential of mesenchymal cell-derive miRNA-150-5p-expressing exosomes in rheumatoid arthritis mediated by the modulation of MMP14 and VEGF. J. Immunol. 2018, 201, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhu, L.; In, I.I.; Chen, Y.; Jia, N.; Zhu, W. Bone marrow-derived mesenchymal stem cells-secreted exosomal microRNA-192-5p delays inflammatory response in rheumatoid arthritis. Int. Immunopharmacol. 2020, 78, 105985. [Google Scholar] [CrossRef]
- Meng, Q.; Qiu, B. Exosomal microRNA-320a derived from mesenchymal stem cells regulates rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing CXCL9 expression. Front. Physiol. 2020, 11, 441. [Google Scholar] [CrossRef]
- Su, Y.; Liu, Y.; Ma, C.; Guan, C.; Ma, X.; Meng, S. Mesenchymal stem cell-originated exosomal lncRNA HAND2-AS1 impairs rheumatoid arthritis fibroblast-like synoviocyte activation through miR-143-3p/TNFAIP3/NF-κB pathway. J. Orthop. Surg. Res. 2021, 16, 116. [Google Scholar] [CrossRef]
- Takahashi, N.; Akatsu, T.; Udagawa, N.; Sasaki, T.; Yamaguchi, A.; Moseley, J.M.; Martin, T.J.; Suda, T. Osteoblastic cells are involved in osteoclast formation. Endocrinology 1988, 123, 2600–2602. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocrinol. Rev 1999, 20, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Kobayshi, Y.; Uehara, S.; Udagawa, N.; Takahashi, N. Regulation of bone metabolism by Wnt signals. J. Biochem. 2016, 159, 387–392. [Google Scholar] [CrossRef]
- Ruaro, B.; Casabella, A.; Paolino, S.; Pizzorni, C.; Ghio, M.; Seriolo, C.; Molfetto, L.; Odetti, P.; Smith, V.; Cutolo, M. Dikkopf-1 (DKK-1) serum levels in systemic sclerosis and rheumatoid arthritis patients: Correlation with the trabecular bone score (TBS). Clin. Rheumatol. 2018, 37, 3057–3062. [Google Scholar] [CrossRef]
- Wei, J.; Shi, Y.; Zheng, L.; Zhou, B.; Inose, H.; Wang, J.; Guo, X.E.; Grosschedl, R.; Karsenty, G. miR-34s inhibit osteoblast proliferation and differentiation in the mouse by targeting SATB2. J. Cell Biol. 2012, 197, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Tian, Q.; Ling, S.; Liu, Y.; Yang, S.; Shao, Z. miR-145 suppresses osteogenic differentiation by targeting Sp7. FEBS Lett. 2013, 587, 3027–3031. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Farina, N.H.; Matzelle, M.M.; Fanning, P.J.; Lian, J.B.; Gravallese, E.M. Synovium-derived microRNAs regulate bone pathways in rheumatoid arthritis. J. Bone Miner. Res. 2017, 32, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Wang, Z.; Wang, L.; Shi, J.; Guo, X.; Zhou, W.; Wu, X.; Liu, Y.; Zhang, W.; Yang, H.; et al. Downregulation of miR-106b attenuates inflammatory responses and joint damage in collagen-induced arthritis. Rheumatology 2017, 56, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, X.; Yang, M.; Ruan, W.; Wei, W.; Gu, D.; Wang, J.; Guo, X.; Guo, L.; Yuan, Y. miR-145-5p increases osteoclast numbers in vitro and aggravates bone erosion in collagen-induced arthritis by targeting osteoprotegerin. Med. Sci. Monit. 2018, 24, 5292–5300. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.-R.; Fan, X.-L.; Lin, P.; Yang, H.; Chen, X.-Z.; Xu, X.-D. miR-206 inhibits osteogenic differentiation of bone marrow mesenchymal stem cells by targetting glutaminase. Biosci. Rep. 2019, 39, BSR20181108. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Fang, Y.; Rao, Y.; Tan, W.; Zhou, W.; Wu, X.; Zhang, C.; Zhang, Y.; Liu, Y.; Sunagawa, M.; et al. Synovial fibroblast-derived exosomal microRNA-106b suppresses chondrocyte proliferation and migration in rheumatoid arthritis via down-regulation of PDK4. J. Mol. Med. 2020, 98, 409–423. [Google Scholar] [CrossRef]
- Adami, G.; Fassio, A.; Rossini, M.; Caimmi, C.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D. Osteoporosis in rheumatic diseases. Int. J. Mol. Sci. 2019, 20, 5867. [Google Scholar] [CrossRef] [Green Version]
- Marinova-Mutafchieva, L.; Williams, R.O.; Funa, K.; Maini, R.N.; Zvaifler, N.J. Inflammation is preceded by tumor necrosis factor-dependent infiltration of mesenchymal cells in experimental arthritis. Arthritis Rheum. 2002, 46, 507–513. [Google Scholar] [CrossRef]
- Choi, H.S.; Ryu, C.J.; Choi, H.M.; Park, J.S.; Lee, J.H.; Kim, K.I.; Yang, H.-I.; Yoo, M.C.; Kim, K.S. Effects of the pro-inflammatory milieu on the dedifferentiation of cultured fibroblast-like synoviocytes. Mol. Med. Rep. 2012, 5, 1023–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Lim, S.-K. Role of miRNAs in bone and their potential as therapeutic targets. Curr. Opin. Pharmacol. 2014, 16, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Fukui, S.; Takatani, A.; Shimizu, T.; Umeda, M.; Nishino, A.; Igawa, T.; Koga, T.; Kawashiri, S.; Ichinose, K.; et al. Osteogenic differentiation of fibroblast-like synovial cells in rheumatoid arthritis is induced by microRNA-218 through a ROBO/Slit pathway. Arthritis Res. Ther. 2018, 20, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liu, M.; Luo, X.; Peng, L.; Zhao, Z.; He, C.; He, Y. Exosomal miRNA-486-5p derived from rheumatoid arthritis fibroblast-like synoviocytes induces osteoblast differentiation through the Tob1./BMP/Smad pathway. Biomater. Sci. 2020, 8, 3430–3442. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yasuda, S.; Kono, M.; Kudo, Y.; Shimamura, S.; Kono, M.; Fujieda, Y.; Kato, M.; Oku, K.; Shimizu, T.; et al. MciroRNA-9 ameliorates destructive arthritis through down-regulation of NF-κB1-RANKL pathway in fibroblast-like synoviocytes. Clin. Immunol. 2020, 212, 108348. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, P.; Chen, M.; Chen, D.; Cheng, Y.; He, L. FOXM1/LINC00152 feedback loop regulates proliferation and apoptosis in rheumatoid arthritis fibroblast-like synoviocytes via Wnt/β-catenin signaling pathway. Biosci. Rep. 2020, 40, BSR20191900. [Google Scholar] [CrossRef]
- Xia, X.; Tang, X.; Wang, S. Roles of circRNAs in autoimmune diseases. Front. Immunol. 2019, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Yu, X.; Huang, J.; Dai, Y. Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Mol. Med. Rep. 2017, 16, 8029–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Zeng, L.; Zeng, L.; Rao, J.; Zhang, L.; Guo, Y.; Huang, Z.; Li, J. Expression and clinical significance of circular RNAs hsa-circ-0000175 and hsa-circ-0008410 in peripheral blood mononuclear cells from patients with rheumatoid arthritis. Int. J. Mol. Med. 2020, 45, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liu, J.; Fan, C.; Wang, J.; Li, W. lncRNAS56464.1 as a ceRNA promotes the proliferation of fibroblast-like synoviocytes in experimental arthritis via the Wnt signaling pathway and sponges miR-152-3p. Int. J. Mol. Med. 2021, 47, 17. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Yoshitomi, H.; Tanida, S.; Ishikawa, M.; Nishitani, K.; Ito, H.; Nakamura, T. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 2010, 12, R86. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Furu, M.; Yoshitomi, H.; Ishikawa, M.; Shibuya, H.; Hashimoto, M.; Imura, Y.; Fujii, T.; Ito, H.; Mimori, T.; et al. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS ONE 2013, 8, e69118. [Google Scholar] [CrossRef] [Green Version]
- Dunaeva, M.; Blom, J.; Thurlings, R.; Pruijn, G.J.M. Circulating serum miR-223-3p, and miR-16-5p as possible biomarkers of early rheumatoid arthritis. Clin. Exp. Immunol. 2018, 193, 376–385. [Google Scholar] [CrossRef] [Green Version]
- López-Pedrera, C.; Pérez-Sánchez, C.; Ramos-Casals, M.; Santos-Gonzalez, M.; Rodriguez-Ariza, A.; Cuadrado, M.J. Cardiovascular risk in systemic autoimmune diseases: Epigenetic mechanisms of immune regulatory functions. Clin. Dev. Immunol. 2012, 2012, 974648. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Solus, J.F.; Vickers, K.C.; Oeser, A.M.; Raggi, P.; Stein, C.M. Utility of select plasma microRNAs for disease and cardiovascular risk assessment in patients with rheumatoid arthritis. J. Rheumatol. 2015, 42, 1746–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormseth, M.J.; Solus, J.F.; Sheng, Q.; Chen, S.-C.; Ye, F.; Wu, Q.; Oeser, A.M.; Allen, R.; Raggi, P.; Vickers, K.C.; et al. Plasma miRNAs improve the prediction of coronary atherosclerosis in patients with rheumatoid arthritis. Clin. Rheumatol. 2021, 40, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Patro, P.S.; Aggarwal, A. MicroRNA-132, miR-146a, and miR-155 as potential biomarkers of methotrexate response in patients with rheumatoid arthritis. Clin. Rheumatol. 2019, 38, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, Y.; Qu, H.; Fang, J.; Ye, M.; Yin, W. Correlation of microRNA expression profile with clinical response to tumor necrosis factor inhibitor in treating rheumatoid arthritis patients: A prospective cohort study. J. Clin. Lab. Anal. 2019, 33, e22953. [Google Scholar] [CrossRef] [Green Version]
- Sode, J.; Krintel, S.B.; Carlsen, A.L.; Hetland, M.L.; Johansen, J.S.; Hørslev-Petersen, K.; Stengaard-Pedersen, K.; Ellingsen, T.; Burton, M.; Junker, P.; et al. Plasma microRNA profiles in patients with early rheumatoid arthritis responding to adalimumab plus methotrexate vs methotrexate alone: A placebo-controlled clinical trial. J. Rheumatol. 2018, 45, 53–61. [Google Scholar] [CrossRef]
- Jin, S.; Chen, H.; Li, Y.; Zhong, H.; Sun, W.; Wang, J.; Zhang, T.; Ma, J.; Yan, S.; Zhang, J.; et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann. Rheum. Dis. 2018, 77, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-G.; Zhang, Y.; Ju, W.-F.; Li, C.-Y.; Mu, Y.-C. MiR-21 relieves rheumatoid arthritis in rats via targeting Wnt signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23 (Suppl. S3), 96–103. [Google Scholar]
- Yen, R.W.Y.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar]
- Li, Z.; Li, Y.; Li, Q.; Zhang, Z.; Jiang, L.; Li, X. Role of miR-9-5p in preventing peripheral neuropathy in patients with rheumatoid arthritis by targeting REST/miR-132 pathway. In Vitro Cell. Develop. Biol. Anim. 2019, 55, 52–61. [Google Scholar] [CrossRef]
- Najm, A.; Masson, F.-M.; Preuss, P.; Georges, S.; Ory, B.; Quillard, T.; Sood, S.; Goodyear, C.S.; Veale, D.J.; Fearon, U.; et al. MicroRNA-17-5p reduces inflammation and bone erosions in mice with collagen-induced arthritis and directly targets the JAK/STAT pathway in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2020, 72, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ma, J.; Zhao, H.; Xiao, C.; Zhong, H.; Ling, H.; Xie, Z.; Tian, Q.; Chen, H.; Zhang, T.; et al. Resolvin D1 suppresses pannus formation via decreasing connective tissue growth factor caused by upregulation of miRNA-146a-5p in rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-Y.; Hsieh, S.-C.; Liu, C.-W.; Lu, C.-H.; Liao, H.-T.; Chen, M.-H.; Li, K.-J.; Wu, C.-H.; Shen, C.-Y.; Kuo, Y.-M.; et al. The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications. Int. J. Mol. Sci. 2021, 22, 5689. https://doi.org/10.3390/ijms22115689
Tsai C-Y, Hsieh S-C, Liu C-W, Lu C-H, Liao H-T, Chen M-H, Li K-J, Wu C-H, Shen C-Y, Kuo Y-M, et al. The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications. International Journal of Molecular Sciences. 2021; 22(11):5689. https://doi.org/10.3390/ijms22115689
Chicago/Turabian StyleTsai, Chang-Youh, Song-Chou Hsieh, Chih-Wei Liu, Cheng-Hsun Lu, Hsien-Tzung Liao, Ming-Han Chen, Ko-Jen Li, Cheng-Han Wu, Cheih-Yu Shen, Yu-Min Kuo, and et al. 2021. "The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications" International Journal of Molecular Sciences 22, no. 11: 5689. https://doi.org/10.3390/ijms22115689
APA StyleTsai, C.-Y., Hsieh, S.-C., Liu, C.-W., Lu, C.-H., Liao, H.-T., Chen, M.-H., Li, K.-J., Wu, C.-H., Shen, C.-Y., Kuo, Y.-M., & Yu, C.-L. (2021). The Expression of Non-Coding RNAs and Their Target Molecules in Rheumatoid Arthritis: A Molecular Basis for Rheumatoid Pathogenesis and Its Potential Clinical Applications. International Journal of Molecular Sciences, 22(11), 5689. https://doi.org/10.3390/ijms22115689