5.3. Synthetic Procedures
5.3.1. (anti-11-Oxo[4.3.3]propellan-8-yl) N-(2-methoxy-5-methylphenyl)carbamate (anti-7) and (syn-11-Oxo[4.3.3]propellan-8-yl) N-(2-methoxy-5-methylphenyl)carbamate (syn-7)
According to General Procedure A, stereoisomeric hydroxyketones anti-6 and syn-6 (1.3 g, 6.69 mmol), 2-methoxy-5-methylphenyl isocyanate (1.4 g, 8.71 mmol) and the catalyst Bu2Sn(OAc)2 (0.36 mL, 1.34 mmol) were reacted in THF (25 mL) and worked-up. The residue was purified by fc (5 cm, cyclohexane:ethyl acetate = 1:0–7:3, 50 mL).
anti-7 (Rf = 0.52): Pale yellow solid, mp 143–144 °C, yield 0.86 g (36%), C21H27NO4 (357.2). MS (EI): m/z (%) = 357 [M+], 181 [M-C12H17O]+, 137 [M-C13H17O3]+, 122 [M-C13H18NO3]+. Exact mass (APCI): m/z = 358.1979 (calcd. 358.2013 for C21H28NO4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3292 (ν N-H), 2931 (ν C-H aliphatic), 1732 (ν C=Oketone), 1697 (ν C=Ocarbamate), 1597 (δ N-H). 1H NMR (CDCl3): δ (ppm) = 1.32–1.45 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.75 (dd, J = 14.9/4.1Hz, 2H, 7-Hanti, 9-Hanti), 2.22 (s, 3H, CH3) 2.24 (d, J = 19 Hz, 2H, 10-Hsyn, 12-Hsyn,), 2.35 (d, J = 19 Hz, 2H, 10-Hanti, 12-Hanti), 2.36 (dd, J = 14.8/8.8 Hz, 2H, 7-Hsyn, 9-Hsyn), 3.77 (s, 3H, OCH3), 5.32 (tt, J = 8.4/4.4 Hz, 1H, 8-H), 6.67 (d, J = 8.3 Hz, 1H, 3-HN-Ar), 6.71 (dd, J = 1.6/8.3 Hz, 1H, 4-HN-Ar), 7.06 (s, broad, 1H, NH carbamate), 7.83 (s, broad, 1H, 6-HN-Ar). 13C NMR (CDCl3): δ (ppm) = 21.2 (1C, CH3), 21.4 (2C, C-3, C-4), 32.1 (2C, C-2, C-5), 43.9 (2C, C-7, C-9), 47.7, (2C, C1, C-6), 49.9 (2C, C-10, C-12), 55.9 (1C, OCH3), 75.6 (1C, C-8), 110.0 (1C, C-3N-Ar), 118.9 (1C, C- N-Ar), 123.2 (1C, C-4N-Ar), 127.4 (1C, C-1N-Ar), 130.7 (1C, C-5N-Ar), 145.7 (1C, C-2N-Ar), 153.3 (1C, NH(CO)O), 219.2 (1C, C=Oketone). Purity (HPLC): 95.9% (tR = 21.40 min).
syn-7 (Rf = 0.48): Pale yellow solid, mp 115–118 °C, yield 0.74 g (31%), C21H27NO4 (357.2). MS (EI): m/z (%) = 357 [M+], 181 [M-C12H17O]+, 137 [M-C13H17O3]+, 122 [M-C13H18NO3]+. Exact mass (APCI): m/z = 358.1991 (calcd. 358.2013 for C21H28NO4 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 2931 (ν C-H aliphatic), 1737 (ν C=Oketone), 1720 (ν C=Ocarbamate), 1597 (δ N-H). 1H NMR (CDCl3): δ (ppm) = 1.33–1.43 (m, 4H, 2-Heq, 3-Heq, 4-Heq, 5-Heq), 1.47–1.51 (m, 2H, 2-Hax, 4-Hax), 1.62–1.68 (m, 2H, 3-Hax, 5-Hax), 1.97 (dd, J = 15.2/3.6 Hz, 2H, 7-Hsyn, 9-Hsyn), 2.10 (d, J = 19.2 Hz, 2H, 10-Hanti, 12-Hanti,), 2.15 (dd, J = 15.2/8.4 Hz, 2H, 7-Hanti, 9-Hanti), 2.21 (d, J = 19.2 Hz, 2H, 10-Hsyn, 12-Hsyn,), 2.23 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 5.27 (tt, J = 8.7/3.7 Hz, 1H, 8-H), 6.68 (d, J = 8.4 Hz, 1H, 3-HN-Ar), 6.72 (dd, J = 2/8.4 Hz, 1H, 4-HN-Ar), 7.11 (s, broad, 1H, NH), 7.85 (s, broad, 1H, 6-HN-Ar). 13C NMR (CDCl3): δ (ppm) = 21.2 (1C, CH3), 21.7 (2C, C-3, C-4), 31.9 (2C, C-2, C-5), 43.9 (2C, C-7, C-9), 47.6 (2C, C-1, C-6), 49.7 (2C, C-10, C-12), 56.0 (1C, OCH3), 75.6 (1C, C-8), 110.1 (1C, C-3N-Ar), 119.0 (1C, C-6N-Ar), 123.1 (1C, C-4N-Ar), 127.4 (1C, C-1N-Ar), 130.8 (1C, C-5N-Ar), 145.7 (1C, C-2N-Ar), 153.4 (1C, NH(CO)O), 218.7 (1C, C=Oketone). Purity (HPLC): 95.5% (tR = 21.79 min).
5.3.2. [(8-anti-11-anti and 8-anti-11-syn)-11-(3,4-Dimethoxbenzylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4b)
NaBH(OAc)3 (0.47 g, 2.24 mmol) was added to a solution of ketone anti-7 (0.20 g, 0.56 mmol), 3,4-dimethoxybenzylamine (0.11 g, 0.67 mmol) and acetic acid (32 μL, 0.56 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 48 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate = 7:3 to 5:5, 10 mL, Rf = 0.25, cyclohexane:ethyl acetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4b and anti,syn-4b as brown oil, yield 0.24 g (85%). C30H40N2O5 (508.6). Exact mass (APCI): m/z = 509.2982 (calcd.509.3010 for C30H41N2O5 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3329 (ν N-H), 2927 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.29–1.49 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.61 (dd, J = 13.4/6.6 Hz, 2 × 0.5H, 7-H, 9-H), 1.67–1.75 (m, 4 × 0.5H, 7-H, 9-H, 10-H, 12-H), 1.92 (dd, J = 14.4/4.8 Hz, 2 × 0.5H, 10-H, 12-H), 1.98–2.10 (m, 2H, 7-H, 9-H), 2.14–2.26 (m, 2H, 10-H, 12-H), 2.28 (s, 3H, CH3), 3.37–3.51 (m, 2 × 0.5H, 11-H), 3.70 (s, 2H, NCH2Ar), 3.82 (s, 3 × 0.5H, p-OCH3), 3.84 (s, 3 × 0.5H, p-OCH3), 3.86 (s, 3 × 0.5H, OCH3Arylcarbamate), 3.88 (s, 3 × 0.5H, m-OCH3), 3.89 (s, 3 × 0.5H, m-OCH3), 5.19–5.31 (m, 2 × 0.5H, 8-H), 6.73 (dd, J = 8.3/3.5 Hz, 1H, 6-HBn), 6.77 (m, 1H, 5-HBn), 6.81 (dd, J = 8.1/2.1 Hz, 1H, 4-HAr), 6.85 (d, J = 8.4 Hz, 1H, 3-HAr), 6.90 (t, J = 2.1 Hz, 1H, 2-HBn), 7.13 (s, 0.5H, NH), 7.18 (s, 0.5H, NH), 7.93 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.8, 20.9 (2C, C-3, C-4), 21.2 (1C, CH3), 32.4, 32.7 (2C, C-2, C-5), 44.5, 44.8 (2C, C-10, C-12), 45.0 (2C, C-7, C-9), 49.7 (2C, C-1, C-6), 55.2 (1C, NHCH2Ph), 55.7 (1C, C-11), 55.9, 59.1 (3C, 3 × OCH3), 76.2 (1C, C-8), 110.2 (1C, C-3Ar), 111.2 (1C, C-2Bn), 112.1 (1C, C-5Bn), 119.0 (1C, C-6Ar), 121.1 (1C, C-6Bn), 122.9 (1C, C-4Ar), 127.8 (1C, C-1Ar), 129.5 (1C, C-1Bn), 130.8 (1C, C-5Ar), 145.7 (1C, C-2Ar), 148.1, 148.8 (2C, C-3Bn, C-4Bn) 153.6 (1C, C=O). anti,anti-4b:anti,syn-4b = 1:1. Purity (HPLC): 96.0% (tR = 20.31 min).
5.3.3. [(8-anti-11-anti and 8-anti-11-syn)-11-(4-Chlorobenzylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4c)
NaBH(OAc)3 (0.47 g, 2.24 mmol) was added to a solution of ketone anti-7 (0.20 g, 0.56 mmol), 4-chlorobenzylamine (95 mg, 0.67 mmol) and acetic acid (32 μL, 0.56 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 6 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate:methanol = 5:3.5:1.5, 10 mL, Rf = 0.40) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4c and anti,syn-4c as yellow oil, yield 0.12 g (45%). C28H35ClN2O3 (482.2). Exact mass (APCI): m/z = 483.2387 (calcd.483.2409 for C28H36ClN2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3329 (ν N-H), 2927 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.30–1.53 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.59–1.76 (m, 2H, 7-H, 9-H), 1.89–2.09 (m, 4H, 10-CH2, 12-CH2), 2.14–2.27 (m, 2H, 7-H, 9-H), 2.29 (s, 3H, CH3), 3.35–3.48 (m, 2 × 0.5H, 11-H), 3.73 (s, 2H, NCH2Ph), 3.85 (s, 3H, OCH3), 5.18–5.31 (m, 2 × 0.5H, 8-H), 6.73 (dd, J = 8.3/2.1 Hz, 1H, 4-HAr), 6.77 (d, J = 8.3 Hz, 1H, 3-HAr), 7.13 (s, 0.5H, NH), 7.18 (s, 0.5H, NH), 7.25–7.36 (m, 4H, 4-chlorophenyl), 7.93 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.0, 21.1 (2C, C-3, C-4), 21.5 (1C, CH3), 32.4, 32.8 (2C, C-2, C-5), 44.7, 44.8, 45.0, 45.2 (4C, C-7, C-9, C-10, C-12), 49.7 (2C, C-1, C-6), 50.5 (1C, NCH2Ph), 55.7, 55.9 (2 × 0.5C, OCH3), 57.0 (1C, C-11), 76.0, 76.5 (2 × 0.5C, C-8), 109.9 (1C, C-3Ar), 118.9 (1C, C-6Ar), 122.9 (1C, C-4Ar), 127.6, 128.6 (2C, C-3Bn, C-5Bn), 129.6, 129.8 (2C, C-2Bn, C-6Bn), 130.7 (1C, C-5Ar), 138.7 (1C, C-1Bn) 145.6 (1C, C-2Ar), 153.6 (1C, C=O). anti,anti-4c:anti,syn-4c = 1:1. Purity (HPLC): 92.6% (tR = 20.85 min).
5.3.4. [(8-anti-11-anti and 8-anti-11-syn)-11-(3,4-Dichlorobenzylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4d)
NaBH(OAc)3 (0.47 g, 2.24 mmol) was added to a solution of ketone anti-7 (0.20 g, 0.56 mmol), 3,4-dichlorobenzylamine (0.12 g, 0.67 mmol) and acetic acid (32 μL, 0.56 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 6 d. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate:methanol = 5:3.5:1.5, 10 mL, Rf = 0.49) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4d and anti,syn-4d as yellow oil, yield 0.17 g (59%). C28H34Cl2N2O3 (517.5). Exact mass (APCI): m/z = 517.2015 (calcd.517.2019 for C28H35Cl2N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3329 (ν N-H), 2927 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.28–1.55 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.60–1.75 (m, 2 × 1.5H, 7-H, 9-H), 1.88–2.10 (m, 4H, 10-CH2, 12-CH2), 2.14–2.25 (m, 2 × 0.5H, 7-H, 9-H), 2.28 (s, 3H, CH3), 3.34–3.48 (m, 2 × 0.5H, 11-H), 3.72 (s, 2H, NCH2Ph), 3.83 (s, 3H, OCH3), 5.17–32 (m, 2 × 0.5H, 8-H), 6.73 (dd, J = 8.2/1.8 Hz, 1H, 4-HAr), 6.77 (d, J = 8.7 Hz, 1H, 3-HAr), 7.13 (s, 1H, NH), 7.20 (dd, J = 7.4/3.2 Hz, 1H, 5-H3,4-dichlorophenyl), 7.38 (dd, J = 8.2/5.3 Hz, 1H, 6-H3,4-dichlorophenyl), 7.46 (dd, J = 5.0/2.1 Hz, 1H, 2-H3,4-dichlorophenyl), 7.92 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.0 (1C, CH3), 21.1, 21.5 (2C, C-3, C-4), 32.4, 32.8 (2C, C-2, C-5), 44.6, 44.8, 45.0, 45.1 (4C, C-7, C-9, C10, C-12), 49.8, 50.5 (2C, C1, C-6), 51.7 (1C, NCH2Ph), 55.9 (1C, OCH3), 57.1 (1C, C-11), 76.0, 76.4 (2 × 0.5C, C-8), 110.0(1C, C-3Ar), 118.9 (1C, C-6Ar), 122.9 (1C, C-4Ar),125. 6, 127.8 (2C, C-23,4-dichlorophenyl, C-63,4-dichlorophenyl), 103.3, 130.4, 130.5, 130.7, 130.8 (5C, C-33,4-dichlorophenyl, C-43,4-dichlorophenyl, C-53,4-dichlorophenyl, C-1Ar, C-5Ar), 132.5 (1C, C-13,4dichlorophenyl) 146.0 (1C, C-2Ar), 153.6 (1C, C=O). anti,anti-4d:anti,syn-4d = 1:1. Purity (HPLC): 88.6% (tR = 21.17 min).
5.3.5. [(8-anti-11-anti and 8-anti-11-syn)-11-(2,4-Dimethylbenzylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4e)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 3,4-dimethylbenzylamine (45 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 20 mL, Rf = 0.21) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4e and anti,syn-4e as brown oil, yield 0.12 g (93%). C30H40N2O3 (476.7). Exact mass (APCI): m/z = 477.3159 (calcd. 477.3112 for C30H41N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3329 (ν N-H), 2927 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.24–1.70 (m, 9H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(0.5H), 9-CH2(0.5H)), 1.97–2.12 (m, 3H, 7-CH2(0.5H), 9-CH2(0.5H)), 10-CH2(1H), 12-CH2(1H)), 2.14–2.37 (m, 13H, 7-CH2(1H), 9-CH2(1H), 10-CH2(1H), 12-CH2(1H), CH3), 3.45–3.51 (m, 0.5H, 11-H), 3.51–3.58 (m, 0.5H, 11-H), 3.76 (s, 2H, NCH2Ph), 3.82 (s, 3 × 0.5H, OCH3), 3.85 (s, 3 × 0.5H, OCH3), 5.20 (tt, J = 8.4/4.0 Hz 0.5H, 8-H), 5.28 (tt, J = 8.4/5.0 Hz, 0.5H, 8-H), 6.71–6.80 (m, 2H, 3-HAr, 4-HAr), 6.95–7.03 (m, 2H, 5-H2,4-diMePhenyl, 6-H2,4-diMePhenyl), 7.11 (d, J = 3.8 Hz, 1H, 3-H2,4-diMePhenyl), 7.36 (s, 1H, NH), 7.92 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 19.0 (1C, CH3), 20.8, 21.1, 21.4 (4C, C-3, C-4, 2 × CH3), 32.5, 32.8 (2C, C-2, C-5), 41.8, 44.6, 45.0 (4C, C-7, C-9, C-10, C-12), 49.7, 50.3 (2C, C-1, C-6), 55.7 (0.5C, C-11), 55.9 (1C, OCH3), 57.2 (0.5C, C-11), 75.8 (0.5C, C-8), 76.4 (0.5C, C-8), 110.0 (1C, C-3Ar), 119.1 (1C, C-6Ar), 122.8 (1C, C-4Ar), 127.0 (1C, C-52,4-diMePhenyl), 127.5, 127.7 (2 × 0.5C, C-1Ar), 129.0 (1C, C-32,4-diMePhenyl), 130.6, 130.7 (2 × 0.5C, C-5Ar), 131.5 (2C, C-12,4-diMePhenyl, C-62,4-diMePhenyl), 136.5 (1C, C-22,4-diMePhenyl), 137.8 (1C, C-42,4-diMePhenyl), 145.6 (1C, C-2Ar), 153.7 (1C, C=O). anti,anti-4e:anti,syn-4e = 1:1. Purity (HPLC): 91.3% (tR = 21.61 min).
5.3.6. [(8-anti-11-anti and 8-anti-11-syn)-11-[(3,5-Bis(trifluoromethyl)benzylamino]-[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4f)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 3,5-bis(trifluoromethyl)benzylamine (82 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate = 7:3–1:1, 20 mL, Rf = 0.14, cyclohexane:ethyl acetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4f and anti,syn-4f as brown oil, yield 95 mg (59%). C30H34F6N2O3 (584.6). Exact mass (APCI): m/z = 585.2596 (calcd. 585.2546 for C30H34F6N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3433 (ν N-H), 2931 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.31–1.52 (m, 10H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(1H), 9-CH2(1H)), 1.70 (dd, J = 14.0/5.4 Hz, 2 × 0.5H, 7-H, 9-H), 1.93 (dd, J = 14.4/4.9 Hz, 2 × 0.5H, /-H, 9-H), 2.02–2.13 (m, 2H, 10-H, 12-H), 2.16–2.27 (m, 2H, 10-H, 12-H), 2.28 (m, 3H, CH3), 3.38–3.52 (m, 2 × 0.5H, 11-H), 3.83 (s, 3H, OCH3), 3.88 (s, 2H, NCH2Ph), 5.19–5.33 (m, 2 × 0.5H, 8-H), 6.73 (d, J = 8.3 Hz, 1H, 3-HAr), 6.77 (dd, J = 8.8/3.8 Hz, 1H, 4-HAr), 7.13 (s, 1H, NH), 7.77 (s, 1H, 4-H3,5-diCF3Ph), 7.85 (d, J = 4.9 Hz, 2H, 2-H3,5-diCF3Ph, 6-H3,5-diCF3Ph), 7.92 (s, 1H, 6-HAr). A signal for the NH protons is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.0 (1C, CH3), 21.1, 21.4 (2C, C-3, C-4), 32.4, 32.8 (2C, C-2, C-5), 44.9 (4C, C-7, C-9, C-10, C-12), 49.8, 50.4 (2C, C-1, C-6), 57.5, 56.3 (2 × 0.5C, C-11), 75.9, 76.4 (2 × 0.5C, C-8), 110.0 (1C, C-3Ar), 118.9 (1C, C-6Ar), 121.3 (d, J = 7.53 Hz, 1C, C-43,5-diCF3Ph), 122.9 (1C, C-4Ar), 126.2 (q, J = 262.3 Hz, 2C, CF3), 128.6 (2C, C-23,5-diCF3Ph, C-63,5-diCF3Ph), 130.7 (2C, C-1Ar, C-5Ar), 130.9 (2C, C-33,5-diCF3Ph, C-53,5-diCF3Ph), 141.4 (1C, C-13,5-diCF3Ph), 145.6 (1C, C-2Ar), 153.6 (1C, C=O). anti,anti-4f:anti,syn-4f = 1:1. Purity (HPLC): 92.0% (tR = 22.40 min).
5.3.7. [(8-anti-11-anti and 8-anti-11-syn)-11-(4-Nitrobenzylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4g)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 4-nitrobenzylamine hydrochloride (70 mg, 0.34 mmol) and NEt3 (58 μL, 0.42 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 24 h. Then acetic acid (16 μL, 0.28 mmol) was added and the mixture was stirred for additional 24 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.29) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4g and anti,syn-4g as yellow oil, yield 90 mg (65%). C28H35N3O5 (493.6). MS (ESI): m/z = 494 [M+H]+. Exact mass (APCI): m/z = 494.2680 (494.2649 calcd. for C28H36N3O5 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 2931 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H), 1519 (ν N-O), 1342 (ν N-O). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.36–1.55 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.68 (m, 4H, 7-H, 9-H, 10-H, 12-H), 1.90–2.11 (m, 2H, 10-H, 12-H), 2.17–2.27 (m, 2H, 7-H, 9-H), 2.28 (s, 3H, CH3), 3.38–3.43 (m, 0.4H, 11-H), 3.46 (tt, J = 7.4, 5.8 Hz, 0.6H), 3.82 (s, 3 × 0.4H, OCH3), 3.83 (s, 3 × 0.6H, OCH3), 3.87 (s, 2H, NCH2Ph), 5.22 (tt, J = 8.2, 5.3 Hz, 0.6H, 8-H), 5.28 (tt, J = 8.9/4.8 Hz, 0.4H, 8-H), 6.74 (dd, J = 8.3/1.3 Hz, 1H, 3-HAr), 6.77 (dd, J = 8.4/2.1 Hz, 1H, 4-HAr), 7.10 (s, 1H, NH), 7.51–7.56 (m, 2H, 2-H4-NO2Ph, 6-H4-NO2Ph), 7.91 (s, 1H, 6-HAr), 8.15–8.19 (m, 2H, 3-H4-NO2Ph, 5-H4-NO2Ph). A signal for the NH proton is not seen in the spectrum. Signals for the OH and NH protons are not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 21.0, 21.1, 21.5 (3C, C-3, C-4, CH3), 32.4, 32.8 (2C, C-2, C-5), 44.9, 45.0 (4C, C-7, C-9, C-10, C-12), 49.8, 50.5 (2C, C-1, C-6), 52.1 (1C, NCH2Ph), 55.9 (1C, OCH3), 56.1 (0.6C, C-11), 57.3 (0.4C, C-11), 76.0 (0.6C, C-8), 76.5 (0.4C, C-8), 110.0 (1C, C-3Ar), 118.9 (1C, C-6Ar), 122.9 (1C, C-4Ar), 123.8 (2C, C-34-NO2Ph, C-54-NO2Ph), 127.5 (1C, C-1Ar), 129.0 (2C, C-24-NO2Ph, C-64-NO2Ph), 130.7 (1C, C-5Ar), 145.6 (1C, C-2Ar), 147.2 (1C, C-14-NO2Ph), 148.3 (1C, C-44-NO2Ph), 153.5 (1C, C=O). anti,anti-4g:anti,syn-4g = 6:4. Purity (HPLC): 98.0% (tR = 20.04 min).
5.3.8. [(8-anti-11-anti and 8-anti-11-syn)-11-[(4-Dimethylamino)benzylamino]-[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4h)
NaBH(OAc)3 (0.12 g, 0.57 mmol) was added to a solution of 4v (0.10 g, 0.28 mmol) and 4-(dimethylamino)benzaldehyde (50 mg, 0.34 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 20 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.30) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4h and anti,syn-4h as yellow oil, yield 70 mg (50%). C30H41N3O3 (491.7). Exact mass (APCI): m/z = 492.3228 (calcd. 492.3221 for C30H42N3O3 [M+2H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (ν N-H), 2927 (ν C-H aliphatic), 1724 (ν C=O), 1612 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.28–1.60 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.61–1.88 (m, 3H, 7-H, 9-H, 10-H(0.5H), 12-H(0.5H)), 1.91–2.25 (m, 5H, 7-H, 9-H, 10-H(1.5H), 12-H(1.5H)), 2.28 (s, 3H, CH3), 2.89 (s, 3H, NCH3), 2.91 (s, 3H, NCH3), 3.39–3.54 (m, 1H, 2 × 0.5 H, 11-H), 3.71 (s, 2H, NCH2Ph), 3.82 (s, 3 × 0.5H, OCH3), 3.84 (s, 3 × 0.5H, OCH3), 5.17–5.31 (m, 2 × 0.5H, 8-H), 6.66–6.71 (m, 2H, 3-H4-diMePh, 5-H4-diMePh), 6.73–6.79 (m, 2H, 3-HAr, 4-HAr), 7.12 (s, 0.5H, NH), 7.22–7.26 (m, 2H, 2-H4-diMePh, 6-H4-diMePh), 7.29 (s, 0.5H, NH), 7.92 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.9, 21.1, 21.4 (3C, C-3, C-4, CH3), 32.5, 32.8 (2C, C-2, C-5), 40.7, 40.8 (2C, N(CH3)2), 44.8, 45.0 (4C, C-7, C-9, C-10, C-12), 49.6, 50.3 (2C, C-1, C-6), 55.9 (0.5C, C-11), 56.3 (1C, OCH3), 60.5 (0.5C, C-11), 75.9 (0.5C, C-8), 76.4 (0.5, C-8), 110.0 (1C, C-3Ar), 112.7 (2C, C-34-diMePh, C-54-diMePh), 119.1 (1C, C-6Ar), 122.8 (1C, C-4Ar), 127.7 (1C, C-1Ar), 129.9 (2C, C-24-diMePh, C-64-diMePh), 130.1 (1C, C-14-diMePh), 130.7 (1C, C-5Ar), 145.8 (1C, C-2Ar), 150.2 (1C, C-44-diMePh), 153.7 (1C, C=O). anti,anti-4h:anti,syn-4h = 1:1. Purity (HPLC): 98.4% (tR = 17.89 min).
5.3.9. [(8-anti-11-anti and 8-anti-11-syn)-11-[(Furan-2-yl-mehtyl)amino][4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4i)
NaBH(OAc)3 (0.36 g, 1.67 mmol) was added to a solution of ketone anti-7 (0.15 g, 0.42 mmol), furfurylamine (0.15 g, 0.55 mmol) and acetic acid (24 μL, 0.42 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 48 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.59) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4i and anti,syn-4i as dark yellow oil, yield 0.15 mg (79%). C26H34N2O4 (438.6). MS (ESI): m/z = 439 [M+H]+. Exact mass (APCI): m/z = 439.2605 (calcd. 439.2591 for C26H35N2O4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 2931 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.29–1.54 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.61 (dd, J = 13.3/6.8 Hz, 2 × 0.5H, 10-H, 12-H), 1.67–1.73 (m, 2H, 7-H, 9-H), 1.90 (dd, J = 14.4/4.9 Hz, 2 × 0.5H, 10-H, 12-H), 1.96–2.07 (m, 2H, 7-H, 9-H), 2.13–2.26 (m, 2H, 7-H, 9-H), 2.28 (m, 3H, CH3), 3.35–3.49 (m, 2 × 0.5H, 11-H), 3.78 (s, 2H, NCH2Furyl), 3.84 (s, 3H, OCH3), 5.16–5.32 (m, 2 × 0.5H, 8-H), 6.18–6.24 (m, 1H, 3-HFuran), 6.29–6.33 (m, 1H, 4-HFuran), 6.71–6.79 (m, 2H, 3-HAr, 4-HAr), 7.13 (s, 0.5H, NH), 7.20 (s, 0.5H, NH), 7.32–7.38 (m, 1H, 5-HFuran), 7.92 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. Signals for the OH and NH protons are not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.9, 21.1 (2C, C-3, C-4), 21.5 (1C, CH3), 32.4, 32.8 (2C, C-2, C-5), 44.5, 44.9, 45.1, 45.2 (5C, C-7, C-9, C-10, C-12, NCH2Furan), 49.7, 50.4 (2C, C-1, C-6), 55.5 (0.5C, C-11), 55.9 (1C, OCH3), 56.8 (0.5C, C-11), 76.0 (0.5C, C-8), 76.4 (0.5C, C-8), 107.2, 107.6 (1C, C-3Furan), 109.9 (1C, C-3Ar), 110.4, 110.6 (1C, C-4Furan), 118.9 (1C, C-6Ar), 122.9(1C, C-4Ar), 127.5 (1C, C-1Ar), 130.6 (1C, C-5Ar), 142.0, 142.3 (1C, C-5Furan), 145.6 (1C, C-2Ar), 146.0 (1C, C-2Furan), 153.6 (1C, C=O). anti,anti-4i:anti,syn-4i = 6:4. Purity (HPLC): 91.4% (tR = 19.33 min).
5.3.10. [(8-syn-11-syn and 8-syn-11-anti)-11-[(Furan-2-yl-mehtyl)amino][4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-syn-4i)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone syn-7 (0.10 g, 0.28 mmol), furfurylamine (29 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 5 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1 to 1:1, 10 mL, Rf = 0.18) to obtain a mixture of diastereoisomeric aminocarbamates syn,syn-4i and anti,syn-4i as yellow oil, yield 84 mg (66%). C26H34N2O4 (438.6). MS (ESI): m/z = 439 [M+H]+. Exact mass (APCI): m/z = 439.2631 (calcd. 439.2591 for C26H35N2O4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 2931 (ν C-H aliphatic), 1720 (ν C=O), 1597 (δ N-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.33–1.58 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.70 (dd, J = 13.5/7.3 Hz, 2 × 0.5H, 7-H, 9-H), 1.80–1.90 (m, 3H), 1.97–2.07 (m, 3H), 2.29 (s, 3H, CH3), 2.29–2.35 (m, 2 × 0.5H, 7-H, 9-H), 3.29–3.35 (m, 0.5H, 11-H), 3.35–3.41 (m, 0.5H, 11-H), 3.80–3.85 (m, 5H, OCH3, NCH2Furyl), 5.20 (tt, J = 8.3/4.7 Hz, 0.5H, 8-H), 5.31 (tt, J = 8.4/4.2 Hz, 0.5H, 8-H), 6.30 (dd, J = 6.2/3.5 Hz, 1H, 3-HFuran), 6.33 (d, J = 2.8/2.0 Hz, 1H, 4-HFuran), 6.73 (dd, J = 8.2/3.2 Hz, 1H, 3-HAr), 6.77 (dd, J = 7.1/5.0 Hz, 1H, 4-HAr), 7.12 (s, 1H, NH), 7.38 (ddd, J = 4.0/1.8/0.9 Hz, 1H, 5-HFuran), 7.92 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 21.1, 21.2, 21.3 (3C, C-3, C-4, CH3), 32.1, 33.1 (2C, C-2, C-5), 44.3, 44.6, 45.0, 45.0 (5C, C-7, C-9, C-10, C-12, NCH2Furyl), 49.5, 50.0 (2C, C-1, C-6), 55.7 (0.5C, C-11), 55.9 (1C, OCH3), 56.4 (0.5C, C-11), 76.0 (0.5C, C-8), 76.1 (0.5C, C-8), 108.5 (1C, C-3Furan), 110.0 (1C, C-3Ar), 110.6 (1C, C-4Furan), 119.0 (1C, C-2Furan), 122.8 (C-4Ar), 127.5 (C-1Ar), 130.7 (1C, C-5Ar), 142.4 (1C, C-5Furan), 145.6 (1C, C-2Ar), 153.5 (1C, C=O). syn,syn-4i:syn,anti-4i = 1:1. Purity (HPLC): 97.6% (tR = 19.35 min).
5.3.11. [(8-anti-11-anti and 8-anti-11-syn)-11-[2-(Indol-3-yl)ethylamino][4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4k)
NaBH(OAc)3 (62 mg, 0.29 mmol) was added to a solution of ketone anti-7 (35 mg, 0.10 mmol), tryptamine (20 mg, 0.12 mmol) and acetic acid (6 μL, 0.10 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 96 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.18,) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4k and anti,syn-4k as brown oil, yield 20 mg (41%). C31H39N3O3 (501.7). Exact mass (APCI): m/z = 502.3190 (calcd. 502.3064 for C31H40N3O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (ν N-H), 2927 (ν C-H aliphatic), 1716 (ν C=O), 1597 (δ N-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25–1.49 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.56 (dd, J = 14.5/5.0 Hz, 2 × 0.5H, 7-H, 9-H), 1.98–2.18 (m, 7H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 12-CH2), 2.27 (s, 3H, CH3), 3.17–3.25 (m, 2H, NCH2CH2Indole), 3.39–3.46 (m, 2H, NCH2CH2Indole), 3.59–3.66 (m, 0.6H, 11-H), 3.70 (tt, J = 9.4/7.3 Hz, 0.4H, 11-H), 3.77 (s, 3 × 0.4H, OCH3), 3.78 (s, 3 × 0.6H, OCH3), 5.15 (tt, J = 7.9/5.0 Hz, 0.6H, 8-H), 5.23 (tt, J = 8.1/4.9 Hz, 0.4H, 8-H), 6.71 (d, J = 8.2 Hz, 1H, 3-HAr), 6.74–6.78 (m, 1H, 4-HAr), 7.04–7.07 (m, 2H, 5-HIndole, 6-HIndole), 7.12–7.17 (m, 1H, 2-HIndole), 7.33 (ddd, J = 8.1/2.1/1.0 Hz, 1H, 7-HIndole), 7.59 (s, 1H, NHcarbamate) 7.63 (ddd, J = 8.2/2.5/1.0 Hz, 1H, 4-HIndole), 7.86–7.89 (s, 1H, 6-HAr), 8.41 (s, 1H, NHIndole). A signal for the NH proton is not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.4 (1C, CH3), 20.7, 21.1 (2C, C-3, C-4), 22.6 (1C, NCH2CH2Indole), 32.3, 32.8 (2C, C-2, C-5), 40.9, 41.6 (2C, C-10, C-12), 44.4, 45.1 (2C, C-7, C-9), 47.8 (1C, NCH2CH2Indole), 49.4, 49.8 (2C, C-1, C-6), 55.9 (1C, OCH3), 56.7 (0.4C, C-11), 58.0 (0.6C, C-11), 75.5 (0.4C, C-8), 76.2 (0.6C, C-8), 110.0 (1C, C-3Ar), 110.5, 111.6 (2 × 0.5C, C-7Indole), 118.7 (1C, C-4Indole), 119.7 (1C, C-6Ar), 119.8 (1C, C-5Indole), 122.4 (1C, C-2Indole), 123.1 (2C, C-4Ar, C-6Indole), 126.9 (1C, C-3aIndole), 127.7 (1C, C-1Ar), 130.6 (1C, C-5Ar), 136.5 (1C, C-7aIndole), 146.3 (1C, C-2Ar), 153.8 (1C, C=O). anti,anti-4k:anti,syn-4k = 6:4. Purity (HPLC): 66.3%, light sensitive (tR = 20.8 min).
5.3.12. [(8-syn-11-syn and 8-syn-11-anti)-11-[(2-(Indol-3-yl)ethylamino][4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-syn-4k)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone syn-7 (0.10 g, 0.28 mmol), tryptamine (52 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 5 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.15,) to obtain a mixture of diastereoisomeric aminocarbamates syn,syn-4k and syn,anti-4k as brown oil, yield 60 mg (41%). C31H39N3O3 (501.7). Exact mass (APCI): m/z = 502.3078 (calcd. 502.3064 for C31H40N3O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3421 (ν N-H), 2931 (ν C-H aliphatic), 1716 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.26–1.58 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.70–1.97 (m, 6H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 12-CH2), 2.28 (s, 3H, CH3), 2.30–2.43 (m, 2H, 7-H, 9-H), 3.11–3.21 (m, 2H, NCH2CH2Indolyl), 3.31–3.39 (m, 2H, NCH2CH2Indole), 3.57–3.66 (m, 1H, 11-H), 3.83 (m, 3H, OCH3), 5.02–5.10 (m, 0.4H, 8-H), 5.22 (tt, J = 8.1/4.0 Hz, 0.6H, 8-H), 6.73 (d, J = 8.3 Hz, 1H, 3-HAr), 6.78 (dd, J = 8.3/2.1 Hz, 1H, 4-HAr), 7.02–7.17 (m, 4H, 2-HIndole,5-HIndole, 6-HIndole, NHCarbamate), 7.35 (d, J =, 1H, 7-HIndole), 7.61 (d, J = 7.9 Hz, 1H, 4-HIndole), 7.89 (s, 1H, 6-HAr), 8.74 (s, 1H, NHIndole). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.7, 21.1 (3C, C-3, C-4, CH3), 22.9 (1C, NCH2CH2Indole), 31.9, 32.9 (2C, C-2, C-5), 41.5, 41.6 (2C, C-10, C-12), 44.8, 45.0 (2C, C-7, C-9), 47.8 (1C, NCH2CH2Indole), 49.1, 49.8 (2C, C-1, C-6), 55.9 (1C, OCH3), 56.7 (0.4C, C-11), 57.6 (0.6C, C-11), 75.6 (0.4C, C-8), 76.1 (0.6C, C-8), 110.0 (1C, C-3Ar), 110.8, 111.6 (2 × 0.5C, C-7Indole), 118.7 (1C, C-4Indole), 119.1 (1C, C-6Ar), 119.6 (1C, C-5Indole), 122.3 (1C, C-2Indole), 123.1 1 (2C, C-4Ar, C-6Indole), 127.0 (1C, C-3aIndole), 127.5 (1C, C-1Ar), 130.7 (1C, C-5Ar), 136.5 (1C, C-7aIndole), 145.7 (1C, C-2Ar), 153.4 (1C, C=O). syn,syn-4k:syn,anti-4k = 4:6. Purity (HPLC): 97.8% (tR = 20.97 min).
5.3.13. [(8-anti-11-anti and 8-anti-11-syn)-11-(Phenylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4l)
NaBH(OAc)3 (0.30 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), aniline (31.3 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 48 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate = 8:2, 5 mL, Rf = 0.82, cyclohexane:ethyl acetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4l and anti,syn-4l as pale yellow oil, yield 84 mg (69%). C27H34N2O3 (434.6). Exact mass (APCI): m/z = 435.2599 (calcd.435.2642 for C27H35N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3394 (ν N-H), 2931 (ν C-H aliphatic), 1720 (ν C=O), 1600 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.32–1.54 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.66 (dd, J = 13.6/6.0 Hz, 2 × 0.5H, 10-H, 12-H), 1.72–1.82 (m, 2H, 7-H, 9-H), 1.90 (dd, J = 14.4/4.9 Hz, 2 × 0.5H, 10-H, 12-H), 2.21–2.35 (m, 7H, CH3, 7-H, 9-H, 10-H, 12-H), 3.85 (s, 3H, OCH3), 3.99–4.07 (m, 0.5H, 11-H), 4.07–4.12 (m, 0.5H, 11-H), 5.29 (tt, J = 8.5/4.8 Hz, 1H, 8-H), 6.61 (dd, J = 8.3/3.2 Hz, 2H, 2-HPh, 6-HPh), 6.70 (td, J = 7.2/1.3 Hz, 1H, 4HPh), 6.75 (d, J = 8.3 Hz, 1H, 3-HAr), 6.78 (d, J = 8.2 Hz, 1H, 4-HAr), 7.13–7.22 (m, 3H, 3-HPh, 5-Ph, NH), 7.94 (s, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.9 (2 × 0.5C, C-3, C-4), 21.1 (1C, CH3), 21.4 (2 × 0.5C, C-3, C-4), 32.3, 32.7 (2C, C-2, C-5), 44.6, 44.9 (4C, C7, C-9, C-10, C-12), 49.8, 50.4 (2C, C-1, C-6), 55.9 (1C, OCH3), 60.5 (1C, C-11), 75.9, 76.3 (2 × 0.5C, C-8), 110.0 (1C, C-3Ar), 119.0 (1C, C-4Ph), 119.1 (1C, C-6Ar), 123.0 (1C, C-4Ar), 129.5 (4C, C-2Ph, C-3Ph, C-5Ph, C-6Ph), 130.7 (2C, C-1Ar, C-5Ar), 145.6 (1C, C-2Ar), 145.8 (1C, C-1Ph), 153.6 (1C, C=O). anti,anti-4l:anti,syn-4l = 1:1. Purity (HPLC): 98.2% (tR = 20.55 min).
5.3.14. [(8-anti-11-anti and 8-anti-11-syn)-11-(4-Methoxyphenylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4m)
NaBH(OAc)3 (0.47 g, 2.24 mmol) was added to a solution of ketone anti-7 (0.20 g, 0.56 mmol), 4-methoxyphenylamine (87 mg, 0.67 mmol) and acetic acid (32 μL, 0.56 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:Et2O = 8:2 to 1:1, 10 mL, Rf = 0.56, cyclohexane:ethylacetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4m and anti,syn-4m as brown oil, yield 0.19 g (74%). C28H36N2O4 (464.6). Exact mass (APCI): m/z = 465.2721 (calcd.465.2748 for C28H35Cl2N2O4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3390 (ν N-H), 2931 (ν C-H aliphatic), 1724 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.30–1.53 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.64 (dd, J = 13.6/6.1 Hz, 2 × 0.5H, 7-H, 9-H), 1.70–1.80 (m, 2H, 10-H, 12-H), 1.90 (dd, J = 14.4/4.8 Hz, 2 × 0.5H, 7-H, 9-H), 2.17–2.32 (m, 7H, CH3, 7-H, 9-H, 10-H, 12-H), 3.74 (s, 3H, OCH3 4-OMephenyl), 3.85 (s, 3H, OCH3 Ar), 3.94–4.08 (m, 2 × 0.5H, 11-H), 5.23–5.33 (m, 2 × 0.5H, 8-H), 6.56–6.62 (m, 2H, 3-HAr, 4-HAr), 6.73–6.80 (m, 4H, 2-H4-OMephenyl, 3-H4-OMephenyl,, 5-H4-OMephenyl, 6-H4-OMephenyl), 7.18 (s, 1H, NH), 7.93 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.2, 21.2, 21.6 (3C, C-3, C-4, CH3), 32.3, 32.7 (2C, C-2, C-5), 44.7, 44.8, 45.5, 45.9 (4C, C-7, C-9, C-10, C-12), 50.0, 50.6 (2C, C-1, C-6), 52.9, 53.0 (2 × 0.5C, C-11), 55.9, 56.0 (2C, 2 × OCH3), 76.0, 76.3 (2 × 0.5C, C-8), 110.0 (1C, C-3Ar), 115.0 (4C, C-24-OMephenyl, C-34-OMephenyl, C-54-OMephenyl, C-64-OMephenyl), 118.9 (1C, C-6Ar), 122.9 (1C, C-4Ar), 130.7, 130.7 (2C, C-1Ar, C-5Ar), 145.6 (1C, C-2Ar), 146.0 (1C, C-14-OMephenyl), 152.3 (1C, C-44-OMephenyl), 153.6 (1C, C=O). anti,anti-4m:anti,syn-4m = 1:1. Purity (HPLC): 96.4% (tR = 20.53 min).
5.3.15. [(8-anti-11-anti and 8-anti-11-syn)-11-(3-Chloro-4-methoxyphenylamino)-[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4n)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 3-chloro-4-methoxyaniline (53 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:Et2O = 7:3 to 1:1, 10 mL, Rf = 0.51, cyclohexane:ethylacetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4n and anti,syn-4n as brown oil, yield 0.13 g (63%). C28H35ClN2O4 (499.0). Exact mass (APCI): m/z = 499.2403 (calcd.499.2358 for C28H36ClN2O4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3390 (ν N-H), 2931 (ν C-H aliphatic), 1720 (ν C=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.33–1.50 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.62–1.92 (m, 4H, 7-H, 9-H, 10-H, 12-H), 2.19–2.32 (m, 7H, 7-H, 9-H, 10-H, 12-H, CH3), 3.82 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.90–4.04 (m, 2 × 0.5H, 11-H), 5.23–5.33 (m,2 × 0.5H, 8-H), 6.49–6.58 (m, 1H, 2-H4-Cl-5-MeOPhen), 6.67–6.83 (m, 4H, 5-H4-Cl-5-MeOPhen, 6-H4-Cl-5-MeOPhen, 3-HAr, 4-HAr), 7.18 (s, 1H, NHcarbamate), 7.93 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.8 (1C, CH3), 21.1 (2C, C-3, C-4), 32.3, 32.7 (2C, C-2, C-5), 44.5, 44.9 (4C, C-7, C-9, C-10, C-12), 49.8, 50.3 (2C, C-1, C-6), 55.9 (2C, 2 × OCH3), 56.9 (2 × 0.5C, C-11), 75.7, 76.3 (2 × 0.5C, C-8), 110.0 (1C, C-3Ar), 113.8 (1C, C-64-Cl-5-MeOPhen), 114.9 (1C, C-24-Cl-5-MeOPhen), 118.9 (1C, C-6Ar), 119.2 (1C, C-54-Cl-5-MeOPhen), 123.0 (1C, C-4Ar), 123.6 (1C, C-34-Cl-5-MeOPhen), 127.5 (1C, C-1Ar), 130.7 (1C, C-5Ar), 138.2 (1C, C-14-Cl-5-MeOPhen), 145.7, 147.7, (2C, C-2Ar, C-44-Cl-5-MeOPhen) 153.6 (1C, C=O). anti,anti-4n:anti,syn-4n = 1:1. Purity (HPLC): 95.2% (tR = 21.27 min).
5.3.16. [(8-anti-11-anti and 8-anti-11-syn)-11-(4-Aminophenylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4o)
NaBH(OAc)3 (0.47 g, 2.24 mmol) was added to a solution of ketone anti-7 (0.20 g, 0.56 mmol), p-phenylenediamine (97 mg, 0.90 mmol) and acetic acid (32 μL, 0.56 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, ethyl acetate:methanol = 9.5:0.5–8:2, 10 mL, Rf = 0.11, cyclohexane:ethylacetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4o and anti,syn-4o as violet oil, yield 0.19 g (76%). C27H35N2O3 (449.6). Exact mass (APCI): m/z = 450.2720 (calcd. 450.2751 for C27H36N3O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3417 (ν N-H2), 3290 (ν N-H), 2931 (ν C-H aliphatic), 1716 (ν C=O), 1604 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.37–1.51 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.63–92 (m, 4H, 7-H, 9-H, 10-H, 12-H), 2.17–2.28 (m, 4H, 7-H, 9-H, 10-H, 12-H), 2.29 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 3.92–4.05 (m, 2 × 0.5H, 11-H), 5.22–5.32 (m, 2 × 0.5H, 8-H), 6.53–6.66 (m, 4H, 2-H4-aminophenyl, 3-H4-aminophenyl, 5-H4-aminophenyl, 6-H4-aminophenyl), 6.74 (dd, J = 8.3/1.4 Hz, 1H, 4-HAr), 6.78 (d, J = 8.5 Hz, 1H, 3-HAr), 7.14 (s, 0.5H, NH), 7.21 (s, 0.5H, NH), 7.28 (s, 1H, NHcarbamate), 7.93 (s, 1H, 6-HAr). A signal for the NH protons is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.1, 21.4 (3C, C-3, C-4, CH3), 32.0, 32.7 (2C, C-2, C-5), 45.0, 45.4 (4C, C-7, C-9, C-10, C-12), 50.0 (2C, C-1, C-6), 55.9 (2C, C-11, OCH3), 77.4 (2 × 0.5C, C-8), 110.1(1C, C-3Ar), 119.0 (1C, C-6Ar), 123.1 (5C, C-24-aminophenyl, C-34-aminophenyl, C-54-aminophenyl, C-64-aminophenyl, C-4Ar), 130.6, 131.3 (3C, C-14-aminophenyl, C-44-aminophenyl,C-5Ar), 145.7 (1C, C-2Ar), 151.8 (1C, C=O). anti,anti-4o:anti,syn-4o = 1:1. Purity (HPLC): 97.8% (tR = 19.33 min).
5.3.17. [(8-anti-11-anti and 8-anti-11-syn)-11-(Phenethylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4p)
NaBH(OAc)3 (0.67 g, 3.20 mmol) was added to a solution of ketone anti-7 (0.23 g, 0.64 mmol), 2-phenylethanamine (0.1 g, 0.85 mmol) and acetic acid (38 μL, 0.64 mmol) in 1,2-dichloroethane (10 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 6 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, methanol:ethyl acetate = 9:1, 5 mL, Rf = 0.27, cyclohexane:ethyl acetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4p and anti,syn-4p as pale yellow oil, yield 0.23 g (76%). C29H38N2O3 (462.6). Exact mass (APCI): m/z = 463.2952 (calcd.463.2955 for C29H39N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3379 (νN-H), 2931 (ν C-H aliphatic), 1732 (νC=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.27–1.45 (m, 6H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H)), 1.51 (m, 2 × 0.5H, 2-H, 5-H), 1.60 (m, 2 × 0.5H, 2-H, 5-H), 1.67 (dd, J = 14.4/5.1 Hz, 2 × 0.5H, 7-H, 9-H), 1.98–2.08 (m, 4H, 10-CH2, 12-CH2), 2.13–2.24 (m, 3H, 7-CH2(1.5H), 9-CH2(1.5H)), 2.29 (s, 3H, CH3), 3.00–3.21 (m, 4H, NCH2CH2Ph), 3.50–3.60 (m, 0.5H, 11-H), 3.64 (q, J = 9.6/9.0 Hz, 0.5H, 11-H), 3.81 (s, 3 × 0.5H, OCH3), 3.83 (s, 3 × 0.5H, OCH3), 5.20 (tt, J = 7.7/5.1 Hz, 0.5H, 8-H), 5.28 (tt, J = 8.4/4.1 Hz, 0.5H, 8-H), 6.73 (dd, J = 8.2/2.1 Hz, 1H, 3-HAr), 6.77 (dd, J = 8.2/2.1 Hz, 1H, 4-HAr), 7.13 (s, 1H, NH), 7.19–7.42 (m, 5H, Ph), 7.91 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.6 (1C, CH3), 21.0, 21.1 (2C, C-3, C-4), 32.5, 32.9 (2C, C-2, C5), 34.4 (1C, NCH2CH2Ph), 44.6 (2C, C-10, C-12), 45.2 (2C, C-7, C-9), 49.5 (1C, NCH2CH2Ph), 50.0 (2C, C-1, C-6), 55.8, 55.9 (1C, OCH3), 56.6, 57.9 (2 × 0.5 C, C-11), 76.2 (1C, C-8), 109.9 (1C, C-3Ar), 119.3 (1C, C-6Ar), 122.9 (1C, C-4Ar), 126.8 (1C, C-4Ph), 127.3 (2C, C-3Ph, C-5Ph), 127.6 (2C, C-2Ph, C-6Ph), 128.9 (1C, C-1Ar), 130.6 (1C, C-5Ar), 137.9 (1C, C-1Ph), 145.6, 145.9 (1C, C-2Ar), 153.4, 153.7 (1C, C=O). anti,anti-4p:anti,syn-4p = 1:1. Purity (HPLC): 96.8% (tR = 21.20 min).
5.3.18. [(8-anti-11-anti and 8-anti-11-syn)-11-(3-Phenylpropylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4q)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.13 g, 0.36 mmol), 3-phenypropan-1-amine (64 mg, 0.47 mmol) and acetic acid (30 μL, 0.36 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:Et2O = 8:2–1:1, 10 mL, Rf = 0.56, cyclohexane:ethylacetate = 7:3) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4q and anti,syn-4q as pale yellow oil, yield 0.12 g (63%). C30H40N2O3 (476.7). Exact mass (APCI): m/z = 477.3134 (calcd. 477.3112 for C30H41N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3267 (νN-H), 2931 (ν C-H aliphatic), 1720 (νC=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.13–1.65 (m, 9H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(0.5H), 9-CH2(0.5H)), 1.92–2.28 (m, 12H, 7-CH2(1.5H), 9-CH2(1.5H), 10-CH2, 12-CH2, CH3, NCH2CH2CH2Ph), 2.58–2.66 (m, 2H, NCH2CH2CH2Ph), 2.81–2.88 (m, 2H, NCH2CH2CH2Ph), 3.52–3.68 (m, 2 × 0.5H, 11-H), 3.79 (s, 3 × 0.5H, OCH3), 3.84 (s, 3 × 0.5 H, OCH3), 5.20 (m, 2 × 0.5H, 8-H), 6.70–6.80 (m, 2H, 3-HAr, 4-HAr), 7.13–7.18 (m, 4H, 2-HPh, 3-HPh, 5-HPh, 6-HPh), 7.22–7.26 (m, 1H, 4-HPh), 7.35 (s, 1H, NHcarbamate), 7.62 (s, 1H. NH), 7.92 (d, J = 2.0 Hz, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.4 (1C, CH3), 20.8, 21.1 (2C, C-3, C-4), 29.4 (NCH2CH2CH2Ph), 32.4 (NCH2CH2CH2Ph), 32.7, 33.1 (2C, C-2, C-5), 40.9, 41.6, 44.3, 45.1 (4C, C-7, C-9, C-10, C-12), 47.1 (NCH2CH2CH2Ph), 49.3, 49.7 (2C, C-1, C-6), 55.9, 56.0 (2 × 0.5C, C-11), 56.7, 58.0 (2 × 0.5C, OCH3), 76.0 (1C, C-8), 110.2 (1C, C-3Ar), 119.5 (1C, C-6Ar), 123.0 (1C, C-4Ar), 126.5 (1C, C-4), 128.5 (4C, C-2Ph, C-3Ph, C-5Ph, C-6Ph), 128.7(1C, C-1Ar), 130.7 (1C, C-5Ar), 140.0 (1C, C-1Ph), 146.1 (1C, C-2Ar), 153.7 (1C; C=O). anti,anti-4q:anti,syn-4q = 1:1. Purity (HPLC): 97.9% (tR = 21.61 min).
5.3.19. [(8-anti-11-anti and 8-anti-11-syn)-11-[(3-Aminopropyl)amino][4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4r)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), propane-1,3-diamine (62 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 96 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1–1:1, 10 mL, Rf = 0.18) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4r and anti,syn-4r as colorless solid, mp 74–76 °C, yield 80 mg (66%). C24H37N3O3 (415.6). Exact mass (APCI): m/z = 417.2856 (calcd. 417.2991 for C24H39N3O3 [M+2H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (ν N-H), 2927 (ν C-H aliphatic), 1720 (ν C=O), 1597 (δ N-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.26–1.51 (m, 10H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, NCH2CH2CH2NH2), 1.63–2.25 (m, 10H, 7-CH2, 9-CH2, 10-CH2, 12-CH2, NCH2CH2CH2NH2), 2.24 (s, 3 × 0.5H, CH3), 2.28 (s, 3 × 0.5H, CH3), 2.82–2.88 (m, 2H, NCH2CH2CH2NH2), 3.39–3.53 (m, 2 × 0.5H, 11-H), 3.79 (s, 3 × 0.5H, OCH3), 3.86 (s, 3 × 0.5H, OCH3), 5.17–5.30 (m, 2 × 0.5H, 8-H), 6.69–6.78 (m, 2H, 3-HAr, 4-HAr), 7.14 (s, 1H, NH), 7.89 (s, 1H, 6-HAr). Signals for the NH protons are not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.7 (1C, CH3), 21.1, 21.4 (2C, C-3, C-4), 32.0, 32.3, 32.3, 32.8 (3C, C-2, C-5, NCH2CH2CH2NH2), 44.5, 44.9 (4C, C-7, C-9, C-10, C-12), 47.6 (1C, NCH2CH2CH2NH2), 49.6 (1C, NCH2CH2CH2NH2), 50.2 (2C, C-1, C-6), 55.9 (1C, OCH3), 58.3 (0.5C, C-11), 59.1 (0.5C, C-11), 75.6 (0.5C, C-8), 76.0 (0.5C, C-8), 110.1 (1C, C-3Ar), 119.3 (1C, C-6Ar), 123.0 (C-4Ar), 127.6 (1C, C-1Ar), 130.6 (1C, C-5Ar), 145.9 (1C, C-2Ar), 153.6 (1C, C=O). anti,anti-4r:anti,syn-4r = 1:1. Purity (HPLC): 92.3% (tR = 20.97 min).
5.3.20. [(8-anti-11-anti and 8-anti-11-syn)-11-(2-Hydroxyethyl-1-amino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4s)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 2-aminoethanol (20 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 7 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, ethyl acetate:methanol = 8:2–1:1, 10 mL, Rf = 0.11, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4s and anti,syn-4s as yellow oil, yield 35 mg (33%). C23H34N2O4 (402.5). Exact mass (APCI): m/z = 403.2626 (calcd. 403.2591 for C23H35N2O4 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (ν N-H), 2927 (ν C-H aliphatic), 1724 (νC=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.28–1.53 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.59–1.77 (m, 2H, 7-H, 9-H), 1.87–2.27 (m, 6H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 12-CH2), 2.28 (m, 3H, CH3), 2.76–2.84 (m, 2H, NCH2CH2OH), 3.37–3.53 (m, 2 × 0.5H, 11-H), 3.66–3.71 (m, 2H, NCH2CH2OH), 3.84 (s, 3H, OCH3), 5.18–5.31 (m, 2 × 0.5H, 8-H), 6.73 (dd, J = 8.3/2.1 Hz, 1H, 4-HAr), 6.77 (d, J = 8.3 Hz, 1H, 3-HAr), 7.14 (s, 1H, NH), 7.92 (s, 1H, 6-HAr). Signals for the OH and NH protons are not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.9 (1C, CH3), 21.1, 21.4 (2C, C-3, C-4), 32.4, 32.9 (2C, C-2, C-5), 44.5, 44.8, 45.0 (4C, C-7, C-9, C-10, C-12), 49.6 (2C, C-1, C-6), 50.2, 50.4 (2 × 0.5C, NCH2CH2OH), 55.9 (1C, OCH3), 56.5 (0.5C, C-11), 57.7 (0.5C, C-11), 60.8 (1C, NCH2CH2OH), 75.9 (0.5C, C-8), 76.4 (0.5C, C-8), 109.9(1C, C-3Ar), 118.9 (1C, C-6Ar), 122.9 (1C, C-4Ar), 127.5 (1C, C-1Ar), 130.6 (1C, C-5Ar), 145.6 (1C, C-2Ar), 153.4 (1C, C=O). anti,anti-4s:anti,syn-4s = 1:1. Purity (HPLC): 92.5% (tR = 17.37 min).
5.3.21. [(8-anti-11-anti and 8-anti-11-syn)-11-(5-Hydroxypentylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4t)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), 5-aminopentan-1-ol (35 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 11 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, ethyl acetate:methanol = 8:2–1:1, 20 mL, Rf = 0.23, ethyl acetate:methanol = 1:1) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4t and anti,syn-4t as yellow oil, yield 25 mg (21%). C26H40N2O4 (444.6). Exact mass (APCI): m/z = 445.3066 (calcd. 445.3061 for C26H40N2O4 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 3330 (νO-H), 2931 (ν C-H aliphatic), 1724 (νC=O), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.25–1.48 (m, 10H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, NCH2CH2CH2CH2CH2O), 1.51–1.71 (m, 6H, 7-H, 9-H, 10-H, 12-H, NCH2CH2CH2CH2CH2O), 1.95–2.25 (m, 6H, 7-H, 9-H, 10-H, 12-H, NCH2CH2CH2CH2CH2O), 2.27 (s, 3 × 0.5H, CH3), 2.29 (s, 3 × 0.5H, CH3), 2.93 (m, 2H, NCH2CH2CH2CH2CH2O), 3.65–3.80 (m, 3H, 11-H, CH2O), 3.81 (s, 3 × 0.5H, OCH3), 3.84 (m, 3 × 0.5H, OCH3), 5.17–5.31 (m, 2 × 0.5H, 8-H), 6.70–6.80 (m, 2H, 3-HAr, 4-HAr), 7.16 (s, 1H, NH), 7.88 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. Signals for the OH and NH protons are not seen in the spectrum. 13C NMR (100 MHz, CDCl3): δ(ppm) = 20.6 (2C, C-3, C-4), 21.1 (1C, CH3), 23.3 (1C, NCH2CH2CH2CH2CH2O), 25.8 (1C, NCH2CH2CH2CH2CH2O), 31.3 (1C, NCH2CH2CH2CH2CH2O), 32.7 (2C, C-2, C-5), 41.0 (2C, C-10, C-12), 45.1 (2C, C-7, C-9), 47.2 (1C, NCH2CH2CH2CH2CH2O), 49.6 (2C, C-1, C-6), 55.9 (1C, OCH3), 56.8 (0.5C, C-11), 58.2 (0.5C, C-11), 61.9 (1C, NCH2CH2CH2CH2CH2O), 75.5 (0.5C, C-8), 76.2 (0.5C, C-8), 110.2 (1C, C-3Ar), 119.5 (1C, C-6Ar), 123.1 (1C, C-4Ar), 127.5 (1C, C-1Ar), 130.6 (1C, C-5Ar), 146.0 (1C, C-2Ar), 153.5 (1C, C=O). anti,anti-4t:anti,syn-4t = 1:1. Purity (HPLC): 94.4% (tR = 22.40 min).
5.3.22. [(8-anti-11-anti and 8-anti-11-syn)-11-(Isobutylamino)[4.3.3]propellan-8-yl] N-(2-methoxy-5-methylphenyl)carbamate (8-anti-4u)
NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of ketone anti-7 (0.10 g, 0.28 mmol), isobutylamine (25 mg, 0.34 mmol) and acetic acid (16 μL, 0.28 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 6 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 20 mL, Rf = 0.23, ethyl acetate:methanol = 1:1) to obtain a mixture of diastereoisomeric aminocarbamates anti,anti-4u and anti,syn-4u as yellow oil, yield 0.11 g (92%). C25H38N2O3 (414.6). Exact mass (APCI): m/z = 415.2969 (calcd. 415.2955 for C25H39N2O3 [M+H]+). FT-IR (ATR, film): (ν (cm−1) = 3429 ν N-H), 2927 (νC-H aliphatic), 1724 (ν C=O), 1597 (δN-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.00 (d, J = 6.6 Hz, 3H, NCH2CH(CH3)2), 1.01 (d, J = 6.6 Hz, 3H, NCH2CH(CH3)2), 1.30–1.43 (m, 6H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H)), 1.49–1.62 (m, 2H, 2-H, 5-H), 1.70–1.76 (m, 1H, NCH2CH(CH3)2), 1.95–2.23 (m, 8H 7-CH2, 9-CH2, 10-CH2, 12-CH2), 2.28 (s, 3 × 0.5H, CH3), 2.29 (s, 3 × 0.5H, CH3), 2.55 (d, J = 6.9 Hz, 1H, NCH2CH(CH3)2), 2.58 (d, J = 6.9 Hz, 1H, NCH2CH(CH3)2), 3.48–3.54 (m, 0.5H, 11-H), 3.55–3.62 (m, 0.5H, 11-H), 3.82 (s, 1.5H, OCH3), 3.84 (s, 1.5H, OCH3), 5.21 (tt, J = 8.0/5.5 Hz, 0.5H, 8-H), 5.26 (tt, J = 8.4/4.2 Hz, 0.5H, 8-H), 6.71–6.79 (m, 2H, 3-HAr, 4-HAr), 7.14 (s, 1H, NH), 7.91 (s, 1H, 6-HAr). A signal for the NH proton is not seen in the spectrum. Signals for the OH and NH protons are not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 21.0, 21.1, 21.1, 21.2 (5C, C-3, C-4, CH3, NCH2CH(CH3)2), 32.6, 33.0 (2C, C-2, C-5), 44.7, 45.3 (5C, C-7, C-9, C-10, C-12, NCH2CH(CH3)2), 49.3, 50.0 (2C, C-1, C-6), 55.9 (2C, OCH3, NCH2CH(CH3)2), 56.8 (0.5C, C-11), 58.2 (0.5C, C-11), 75.7 (0.5C, C-8), 76.3 (0.5C, C-8), 110.1, 119.1, 122.9, 127.6, 130.7, 145.8, 153.6. anti,anti-4u:anti,syn-4u = 1:1. Purity (HPLC): 95.8% (tR = 20.07 min).
5.3.23. [(8-anti-11-anti and 8-anti-11-syn)-11-(Benzylamino)[4.3.3]propellan-8-ol (11-anti-13)
NaBH(OAc)3 (0.33 g, 1.55 mmol) was added to a solution of hydroxyketone anti-11 (0.10 g, 0.52 mmol), benzylamine (72 mg, 0.68 mmol) and acetic acid (30 μL, 0.52 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 8 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.28) to obtain a mixture of diastereoisomeric aminoalcohols anti,anti-13 and anti,syn-13 as colorless solid, mp 121–123 °C, yield 86 mg (61%). C19H27NO (285.4). MS (ESI): m/z = 286 [M+H]+. Exact mass (APCI): m/z = 286.2145 (calcd.286.2165 for C19H28NO [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3313 (δ O-H), 2931 (ν C-H aliphatic). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.24–1.40 (m, 6H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H)), 1.40–1.45 (m, 2H, 7-H, 9-H, 10-H, 12-H), 1.50–1.62 (m, 2H, 2-CH2(1H), 5-CH2(1H)), 1.96 (dd, J = 13.8/7.1 Hz, 2 × 0.3H, 10-H, 12-H), 2.03–2.16 (m, 4H, 7-H, 9-H, 10-H, 12-H), 2.30–2.36 (m, 2 × 0.3H, 7-H, 9-H), 3.45–3.51 (m, 0.3H, 11-H), 3.60–3.66 (m, 0.7H, 11-H), 3.93 (s, 2 × 0.3H, NCH2Ph), 3.95 (s, 2 × 0.7H, NCH2Ph), 4.32 (tt, J = 7.1/5.3 Hz, 0.7H, 8-H), 4.40 (tt, J = 8.0/4.2 Hz, 0.3H, 8-H), 7.31–7.40 (m, 3H, 3-HPh, 4-HPh, 5-HPh), 7.59 (dd, J = 6.7/1.3 Hz, 2H, 2-HPh, 6-HPh). Signals for the NH and OH protons are not observed in the spectrum 13C NMR (150 MHz, CDCl3): δ (ppm) = 19.5 (2C, C-3, C-4), 32.8 (2C, C-2, C-5), 41.1 (2C, C-10, C-12), 48.9 (2C, C-7, C-9), 49.9 (2C, C-1, C-6), 50.6 (1C, NCH2Ph), 54.4 (0.7C, C-11), 56.0 (0.3C, C-11), 71.6 (0.7C, C-8), 72.6 (0.3C, C-8), 129.0 (1C, C-4Ph), 129.3 (2C, C-3Ph, C-5Ph), 130.5(2C, C-2Ph, C-6Ph). anti,anti-13:anti,syn-13 = 7:3. Purity (HPLC): 93.3% (tR = 13.61, 15.15 min).
5.3.24. [(8-syn-11-syn and 8-syn-11-anti)-11-(Benzylamino)[4.3.3]propellan-8-ol (11-syn-13)
NaBH(OAc)3 (0.33 g, 1.55 mmol) was added to a solution of hydroxyketone syn-11 (0.10 g, 0.52 mmol), benzylamine (72 mg, 0.68 mmol) and acetic acid (30 μL, 0.52 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 8 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 5.5:3.5:1, 10 mL, Rf = 0.28) to obtain a mixture of diastereoisomeric aminoalcohols syn,syn-13 and syn,anti-13 as colorless oil, yield 90 mg (64%). C19H27NO (285.4). MS (ESI): m/z = 286 [M+H]+. Exact mass (APCI): m/z = 286.2215 (calcd.286.2165 for C19H28NO [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3290 (ν O-H), 2927 (ν C-H aliphatic). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.2–61.39 (m, 2H, 3-CH2(1H), 4-CH2(1H)), 1.44–1.61 (m, 6H, 2-CH2, 3-CH2(1H), 4-CH2(1H), 5-CH2), 1.62–1.88 (m, 6H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 12-CH2), 1.93 (dd, J = 13.3/8.3 Hz, 2 × 0.5H, 7-H, 9-H), 2.15 (dd, J = 13.8/7.5 Hz, 2 × 0.5H, 7-H, 9-H), 3.28 (q, J = 8.2 Hz, 0.5H, 11-H), 3.35 (q, J = 8.4 Hz, 0.5H, 11-H), 3.82 (s, 2H, NCH2Ph), 4.33 (tt, J = 7.5/5.5 Hz, 0.5H, 8-H), 4.54 (tt, J = 7.5/5.4 Hz, 0.5H, 8-H), 7.27–7.32 (m, 1H, 4-HPh), 7.32–7.36 (m, 2H, 3-HPh, 5-HPh), 7.39–7.43 (m, 2H, 2-HPh, 6-HPh). Signals for the NH and OH protons are not observed in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.2, 20.6 (2C, C-3, C-4), 32.1, 33.4 (2C, C-2, C-5), 43.6, 47.9 (4C, C-7, C9, C-10, C-12), 49.3 (2C, C-1, C-6), 51.0 (1C, NCH2Ph), 55.3 (0.5C, C-11), 55.6 (0.5C, C-11), 71.3 (0.5C, C-8), 72.1 (0.5C, C-8), 128.3 (1C, C-4Ph), 128.7, 128.8 (2C, C-3Ph, C-5Ph), 129.3, 129.5 (2C, C-2Ph, C-6Ph), 134.4 (1C, C-1Ph). syn,syn-13:syn,anti-13 = 1:1. Purity (HPLC): 93.1% (tR = 12.42, 13.90 min).
5.3.25. (11′-anti)-Spiro-([1,3]dioxolane-2,8′-(N-benzyl[4.3.3]propellan))-11′-amine (anti-14) and (11′-syn)-spiro-([1,3-dioxolane-2,8′-(N-benzyl[4.3.3]propellan))-11′-amine (syn-14)
Under N2, NaBH(OAc)3 (1.35 g, 6.37 mmol) was added to a solution of monoketal 12 (0.5 g, 2.12 mmol), benzylamine (0.45 g, 4.24 mmol) and acetic acid (0.12 mL, 2.12 mmol) in 1,2-dichloroethane (15 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 24 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, cyclohexane:Et2O: NEt3 = 5.5:3.5:1, 20 mL).
syn-14 (Rf = 0.43) Pale yellow oil, yield 0.19 g (28%). C21H29NO2 (327.5). MS (ESI): m/z = 328 [M+H]+. Exact mass (APCI): m/z = 328.2295 (328.2271 calcd. for C21H30NO2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 2927 (ν C-H aliphatic). 1H NMR (400 MHz, Toluene-d8): δ (ppm) = 1.60–1.65 (m, 4H, 3′-CH2, 4′-CH2,), 1.67–1.83 (m, 6H, 2′-CH2, 5′-CH2, 10′-CH2(1H), 12′-CH2(1H)), 2.11 (d, J = 14.0 Hz, 2H, 7′-H, 9′-H), 2.16–2.24 (m, 4H; 7′-H, 9′-H, 10′-H, 12′-H), 3.49 (tt, J = 8.5/5.6 Hz, 1H, 8′-H), 3.67–3.69 (m, 4H, OCH2CH2O), 3.76 (s, 2H, NCH2Ph), 7.23 (m, 1H, 4-HPh), 7.38 (dd, 2H, J = 8.7/6.4 Hz, 2H, 3-HPh, 5-HPh), 7.49 (m, 2H, 2-HPh, 6-HPh). A signal for the NH proton is not observed in the spectrum. 13C NMR (100 MHz, Toluene-d8): δ (ppm) = 21.9 (2C, C-3′, C-4′), 32.4 (2C, C-2′, C-5′), 44.9 (2C, C-10′, C-12′), 49.2 (2C, C-7′, C-9′), 49.6 (2C, C-1′, C-6′), 53.4 (1C, NCH2Ph), 56.7 (C-11′), 63.9 (1C, C-8′), 64.0 (2C, OCH2CH2O), 105,1 (1C, C-4Ph), 126.9 (2C, C-3Ph, C-5Ph), 128.4 (2C, C-2Ph, C-6Ph), 141.1 (1C, C-1Ph). Purity (HPLC): 83.8% (tR = 15.67 min).
anti-14 (Rf = 0.38) Pale yellow oil, yield 0.21 g (30%). C21H29NO2 (327.5). MS (ESI): m/z = 328 [M+H]+. Exact mass (APCI): m/z = 328.2270 (328.2271 calcd. for C21H30NO2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 2927 (ν C-H aliphatic). 1H NMR (400 MHz, Toluene-d8): ν (ppm) = 1.39–1.52 (m, 4H, 3′-CH2, 4′-CH2,), 1.55–1.61 (m, 2H, 2′-H, 5′-H), 1.67–1.73 (m, 2H, 2′-H, 5′-H), 1.87 (dd, J = 13.2/6.3 Hz. 2H, 10′-Hanti, 12′-Hanti), 2.05 (dd, J = 13.2/8.5 Hz, 2H, 10′-Hsyn, 12′-Hsyn), 2.26 (d, J = 14.1 Hz, 2H, 7′-H, 9′-H), 2.40 (d, J = 14.1 Hz, 2H, 7′-H, 9′-H), 3.44 (tt, J = 8.3/6.3 Hz, 1H, 11′-H), 3.70 (s, 4H, OCH2CH2O), 3.78 (s, 2H, NCH2Ph), 7.26 (m, 1H, 4-HPh), 7–38 (m, 2H, 3-HPh, 5-HPh), 7.49 (d, 2H, J = 7.6 Hz, 2-HPh, 6-HPh). A signal for the NH proton is not observed in the spectrum. 13C NMR (100 MHz, Toluene-d8): δ (ppm) = 21.9 (2C, C-3′, C-4′), 31.9 (2C, C-2′, C-5′), 45.1 (2C, C-10′, C-12′), 49.4 (2C, C-7′, C-9′), 49.8 (2C, C-1′, C-6′), 53.3 (1C, NCH2Ph), 56.9 (1C, C-11′), 63.9 (1C, C-8′), 64.0 (2C, OCH2CH2O), 117.5 (1C, C-4Ph), 126.9 (2C, C-3Ph, C-5Ph), 128.4 (2C, C-2Ph, C-6Ph), 141.6 (1C, C-1Ph). Purity (HPLC): 98.2% (tR = 16.24 min).
5.3.26. syn- and anti-N-[2-(Indol-3-yl)ethyl]-[4.3.3]propellan-8-amine (15)
Under N2, NaBH(OAc)3 (0.3 g, 1.42 mmol) was added to a solution of monoketone 10 (0.1 g, 0.56 mmol), tryptamine (0.14 g, 8.41 mmol) and acetic acid (0.32 μL, 0.56 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 5 days. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 8:1:1 5 mL, Rf = 0.35) to obtain a mixture of diastereoisomeric amines syn-15 and anti-15 as a brown solid, mp 109–111 °C, yield 60 mg (33%). C22H30N2 (322.5). MS (ESI): m/z = 323 [M+H]+. Exact mass (APCI): m/z = 323.2506 (calcd. 323.2482 for C22H31N2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (νN-H), 2927 (ν C-H aliphatic). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.40–1.90 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.54–1.64 (m, 5H, 10-CH2, 11-CH2(1H), 12-CH2), 1.68–1.71 (m, 3H, 7-CH2(1H), 9-CH2(1H), 11-CH2(1H)), 1.83–1.86 (m, 2 × 0.5H, 7-H, 9-H), 1.89–1.93 (2 × 0.5H, 7-H, 9-H), 2.77 (s, broad, 1H, NH), 3.06–3.10 (m, 2H, NCH2CH2), 3.16–3.19 (m, 2H, NCH2CH2), 3.45 (m, 2 × 0.5H, 8-H), 7.06–7.10 (m, 2H, 5-Hindole, 6-Hindole), 7.15 (s, 1H, 2-Hindole), 7.35 (d, J = 8.1 Hz, 1H, 7-Hindole), 7.56 (d, J = 7.2 Hz, 1H, 4-Hindole), 8.95 (s, broad, 1H, NHindole). 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.5, 20.9 (2C, C-3, C-4), 21.4, 22.3 (2 × 0.5 C, C-11), 23.2 (1C, NCH2CH2), 31.9, 32.6 (2C, C-2, C-5), 37.6, 39.1 (2C, C-10, C-12), 41.9, 42.2 (2C, C-7, C-9), 47.5 (1C, NCH2CH2), 49.9, 50.5 (2C, C-1, C-6), 56.5, 57.5 (2 × 0.5C, C-8), 111.4 (1C, C-3indole), 111.5 (1C, C-7indole), 118.7 (1C, C-4indole), 119.7 (1C, C-5indole), 122.3 (1C, C-2indole), 122.9 (1C, C-6indole), 127.1 (1C, C-3aindole), 136.56 (1C, C-7aindole). syn-15:anti-15 = 1:1. Purity (HPLC): 96.8% (tR = 19.28 min).
5.3.27. syn- and anti-N-(4-(Dimethylaminobenzyl)-[4.3.3]propellan-8-amine (17)
Under N2, NaBH(OAc)3 (89 mg, 0.42 mmol) was added to a solution of propellanamine syn-16/anti-16 (50 mg, 0.28 mmol) and 4-(dimethylamino)benzaldehyde (44 mg, 0.29 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 72 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 8:1:1 20 mL, Rf = 0.32) to obtain a mixture of diastereoisomeric amines syn-17 and anti-17 as a yellow solid, mp 89–91 °C, yield 86 mg (98%). C21H32N2 (312.5). MS (ESI): m/z = 313 [M+H]+. Exact mass (APCI): m/z = 313.2673 (calcd. 313.2638 for C21H33N2 [M+H]+). 1H NMR (600 MHz, CDCl3): δ(ppm) = 1.27–1.50 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.54–1.78 (m, 7H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 11-CH2(1H), 12-CH2), 1.79–1.81 (m, 1H, 11-CH2(1H), 1.86 (dd, J = 13.1/8.0 Hz, 2 × 0.5H, 7-H, 9-H), 1.92 (dd, J = 13.1/8.2, 2 × 0.5H, 7-H, 9-H), 2.04 (s, 1H, NH), 2.91 (s, 6H, N(CH3)2), 3.31–3.38 (m, 2 × 0.5H, 8-H), 3.70 (s, 2H, NCH2Ar), 6.69 (d, J = 8.6 Hz, 2H, 3-HAr, 5-HAr), 7.25 (d, J = 8.6 Hz, 2H, 2-HAr, 6-HAr). 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.7, 20.9 (2C, C-3, C-4), 21.2 (1C, C-11), 31.9, 32.6 (2C, C-2, C-5), 37.6, 38.9 (2C, C-10, C-12), 40.6 (2C, N(CH3)2), 43.6, 44.1 (2C, C-7, C-9), 49.9, 50.4 (2C, C-1, C-6), 51.1 (1C, NCH2Ar), 55.0, 55.9 (2 × 0.5C, C-8), 112.5 (2C, C-3Ar, C-5Ar), 129.9 (3C, C-1Ar, C-2Ar, C-6Ar), 150.1 (1C, C-4Ar). syn-17:anti-17 = 1:1. Purity (HPLC): 96.1% (tR = 15.12 min).
5.3.28. syn- and anti-N-Cyclohexylmethyl[4.3.3]propellan-8-amine (18)
Under N2, NaBH(OAc)3 (71 mg, 0.34 mmol) was added to a solution of propellanamine syn-16/anti-16 (30 mg, 0.17 mmol) and cyclohexanecarbaldehyde (15 μL, 0.19 mmol) in 1,2-dichloroethane (5 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 12 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate:methanol = 8:1:1 20 mL, Rf = 0.53) to obtain a mixture of diastereoisomeric amines syn-18 and anti-18 as a colorless solid, mp 108–111 °C, yield 42 mg (91%). C19H33N (275.5). MS (ESI): m/z = 276 [M+H]+. Exact mass (APCI): m/z = 276.2710 (calcd. 276.2686 for C19H34N [M+H]+). 1H NMR (600 MHz, CDCl3): δ (ppm) = 0.89–0.98 (m, 2H, 2-CH2Cy(1H), 6-CH2Cy(1H), 1.16 (ttd, J = 12.5/3.3/1.2 Hz, 1H, 4-CH2Cy(1H)), 1.21–1.42 (m, 8H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H), 3-CH2Cy(1H), 5-CH2Cy(1H)), 1.45–1.87 (m, 16H, 2-CH2(1H), 5-CH2(1H), 7-CH2(1H), 9-CH2(1H), 10-CH2, 11-CH2, 12-CH2, 1-HCy, 2-CH2Cy(1H), 3-CH2Cy(1H), 4-CH2Cy(1H), 5-CH2Cy(1H), 6-CH2Cy(1H)), 1.89 (dd, J = 13.1/8.0 Hz, 2 × 0.5H, 7-H, 9-H), 1.96 (dd, J = 13.1/8.3 Hz, 2 × 0.5H, 7-H, 9-H), 2.57 (dd, J = 6.8/1.5 Hz, 2H, NCH2Cy), 3.37–3.45 (m, 2 × 0.5 H, 8-H). A signal for the NH proton is not seen in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.5, 20.9 (2C, C-3, C-4), 21.5, 22.5 (2 × 0.5C, C-11), 25.8 (2C, C-3Cy, C-5Cy), 26.4 (1C, C-4Cy), 31.5 (2C, C-2Cy, C-6Cy), 32.1, 32.8 (2C, C-2, C-5), 36.0, 36.1 (2 × 0.5C, C-1Cy), 37.8, 39.5 (2C, C-10, C-12), 42.9, 43.4 (2C, C-7, C-9), 49.8, 50.4 (2C, C-1, C-6), 54.2 (1C, NCH2Cy), 56.9, 57.8 (2 × 0.5C, C-8). ). syn-18:anti-18 = 1:1. Purity (LC-MS): 97.4% (tR = 6.957 min).
5.3.29. 11-syn-11-Benzylamino[4.3.3]propellan-11-one (syn-20) and 11-anti-11-Benzylamino[4.3.3]propellan-8-one (anti-20)
Under N2, NaBH(OAc)3 (4.40 g, 20.8 mmol) was added to a solution of diketone 19 (2.0 g, 10.4 mmol), benzylamine (1.34 g, 12.5 mmol) and acetic acid (0.60 mL, 10.40 mmol) in 1,2-dichloroethane (30 mL, dried over molecular sieves 4 Å). The mixture was stirred at rt for 96 h. Then NaOH (1 M) was added (pH 8–10), the mixture was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine (1×), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (5 cm, cyclohexane:ethyl acetate: NEt3 = 6.95:2.95:0.1 to 2.95:6.95:0.1, 20 mL).
syn-20 (Rf = 0.29, cyclohexane: ethyl acetate: NEt3 = 69.5:29.5:1) Pale yellow solid, mp 77–79 °C, yield 0.46 g (16%). C19H25NO (283.4). Exact mass (APCI): m/z = 284.1973 (calcd. 284.2009 for C19H26NO [M+H]+). FT-IR (ATR, film): ν (cm−1) = 2927 (ν C-H aliphatic), 1724 (ν C=O). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.31–1.39 (m, 4H, 3-CH2, 4-CH2), 1.44–1.50 (m, 2H, 2-CH2(1H), 5-CH2(1H)), 1.61–1.66 (m, 2H, 2-CH2(1H), 5-CH2(1H)), 1.71 (dd, J = 13.7/5.6 Hz, 2H, 10-Hsyn, 12-Hsyn), 1.89 (dd, J = 13.7/8.6 Hz, 2H, 10-Hanti, 12-Hanti), 2.06 (d, J = 19.1 Hz, 2H, 7-Hanti, 9-Hanti), 2.18 (d, J = 19.1 Hz, 2H, 7-Hsyn, 9-Hsyn), 3.37 (tt, J = 8.6/5.6 Hz, 1H, 11-H), 3.67 (s, 2H, NCH2Ph), 7.16–7.20 (m, 1H, 4-HPh), 7.23–7.28 (m, 4H, 2-HPh, 3-HPh, 5-HPh, 6-HPh). A signal for the NH proton is not observed in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) =21.9 (2C, C-3, C-4), 32.7 (2C, C-2, C-5), 44.5 (2C, C-10, C-12), 47.8 (2C, C-1, C-6), 50.0 (2C, C-7, C-9), 53.1 (1C, NCH2Ph), 56.1 (1C, C-11), 127.2 (1C, C-4Ph), 128.4 (2C, C-3Ph, C-5Ph), 128.7 (2C, C-2Ph, C-6Ph), 140.5 (1C, C-1Ph), 219.7 (1C, C=O). Purity (HPLC): 74.7% (tR = 13.83 min).
anti-20 (Rf = 0.19, cyclohexane:ethyl acetate: NEt3 = 6.95:2.95:0.1) Pale yellow oil, yield 0.24 g (9%). C19H25NO (283.4). Exact mass (APCI): m/z = 284.2069 (calcd. 284.2009 for C19H26NO [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 2927 (ν C-H aliphatic), 1724 (δ C=O). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.31–1.50 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.54 (dd, J = 13.6/5.9 Hz, 2H, 10-Hanti, 12-Hanti), 2.16 (dd, J = 13.6/8.5 Hz, 2H, 10-Hsyn, 12-Hsyn), 2.27 (d, J = 19.5 Hz, 2H, 7-Hsyn, 9-Hsyn), 2.44 (d, J = 19.5 Hz, 2H, 7-Hanti, 9-Hanti), 3.51 (tt, J = 8.5/5.9 Hz, 1H, 11-H), 3.70 (s, 2H, NCH2Ph), 7.22–7.33 (m 5H, Ph). A signal for the NH proton is not observed in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.6 (2C, C-3, C-4), 32.2 (2C, C-2, C-5), 44.7 (2C, C-10, C-12), 47.9 (2C, C-1, C-6), 50.6 (2C, C-7, C-9), 52.9 (1C, NCH2Ph), 56.3 (1C, C-11), 127.1 (1C, C-4Ph), 128.3 (2C, C-3Ph, C-5Ph), 128.5 (2C, C-2Ph, C-6Ph), 140.5 (1C, C-1Ph), 219.9 (1C, C=O). Purity (HPLC): 73.8% (tR = 14.05 min).
5.3.30. 1 -Benzyl-3-(2-methoxy-5-methylphenyl)-1-(syn-11-oxo-[4.3.3]propellan-8-yl)urea (syn-21)
According to the General Procedure A, amine syn-20 (0.20 g, 0.70 mmol), 2-methoxy-5-methylphenyl isocyanate (0.14 g, 0.84 mmol) and Bu2Sn(OAc)2 (26 mg, 0.07 mmol) were dissolved in THF (15 mL) and the mixture was stirred at rt for 18 h. The crude product was purified by fc (3 cm, cyclohexane:ethyl acetate = 7:3, 20 mL, Rf = 0.44). Colorless solid, mp 168–170 °C, yield 0.22 g (70%). C28H34N2O3 (446.3). Exact mass (APCI): m/z = 447.2666 (calcd. 447.2642 for C28H35N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3390 (ν N-H), 2927 (ν C-H aliphatic), 1732 (ν C=O ketone), 1658 (νC=O urea), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.38–1.51 (m, 6H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H)), 1.57–1.66 (m, 2H, 2-CH2(1H), 5-CH2(1H)), 1.92 (dd, J = 13.8/9.1 Hz, 2H, 7-H, 9-H), 2.03 (dd, J = 13.8/9.3 Hz, 2H, 7-H, 9-H), 2.25 (s, 3H, CH3), 2.28 (d, J = 18.4 Hz, 2H, 10-H, 12-H), 2.33 (d, J = 18.4 Hz, 2H, 10-H, 12-H), 3.46 (s, 3H, OCH3), 4.60 (s, 2H, NCH2Ph), 5.33 (p, J = 9.2 Hz, 1H, 8-H), 6.59 (d, J = 8.2 Hz, 1H, 3-HAr), 6.68 (dd, J = 8.2/2.1 Hz, 1H, 4-HAr), 6.95 (s, 1H, NH), 7.31–7.44 (m, 5H, Ph), 7.97 (d, J = 2.1 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.1 (1C, CH3), 21.8 (2C, C-3, C-4), 33.4 (2C, C-2, C-5), 40.8 (2C, C-7, C-9), 46.4 (2C, C-1, C-6), 47.5 (1C, NCH2Ph), 50.0 (2C, C-10, C-12), 53.8 (1C, C-8), 55.8 (1C, OCH3), 109.9 (1C, C-3Ar), 119.7 (1C, C-6Ar), 122.2 (1C, C-4Ar), 126.5 (1C, C-4Ph), 127.9 (4C, C-2Ph, C-3Ph, C-5Ph, C-6Ph), 128.8 (1C, C-1Ar), 130.7 (1C, C-5Ar), 137.7 (1C, C-1Ph), 145.7 (1C, C-2Ar), 156.1 (1C,C=O urea), 218.6 (1C,C=O ketone). Purity (HPLC): 95.0% (tR = 22.67 min).
5.3.31. 1 -Benzyl-3-(2-methoxy-5-methylphenyl)-1-(anti-11-oxo-[4.3.3]propellan-8-yl)urea (anti-21)
According to the General Procedure A, amine anti-20 (0.22 g, 0.78 mmol), 2-methoxy-5-methylphenyl isocyanate (0.15 g, 0.94 mmol) and Bu2Sn(OAc)2 (27 mg, 0.08 mmol) were dissolved in THF (20 mL) and the mixture was stirred at rt for 18 h. The crude product was purified by fc (3 cm, cyclohexane:ethyl acetate = 7:3, 20 mL, Rf = 0.40). Pale yellow solid, mp 133–136 °C, yield 0.28 g (82%). C28H34N2O3 (446.3). Exact mass (APCI): m/z = 447.2639 (calcd. 447.2642 for C28H35N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3425 (ν N-H), 2935 (ν C-H aliphatic), 1735 (νC=O ketone), 1654 (ν C=O urea), 1597 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.35–1.52 (m, 6H, 2-CH2(1H), 3-CH2, 4-CH2, 5-CH2(1H)), 1.58–1.66 (m, 2H, 2-CH2(1H), 5-CH2(1H)), 1.81 (dd, J = 13.5/9.6 Hz, 2H, 7-Hanti, 9-Hanti), 2.15 (dd, J = 13.5/9.1 Hz, 2H, 7-Hsyn, 9-Hsyn), 2.26 (s, 3H, CH3), 2.32 (s, 4H, 10-CH2, 12-CH2), 3.45 (s, 3H, OCH3), 4.51 (s, 2H, NCH2Ph), 5.33 (q, J = 9.4 Hz, 1H, 8-H), 6.59 (d, J = 8.2 Hz, 1H, 3-HAr), 6.68 (dd, J = 8.2/1.6 Hz, 1H, 4-HAr), 6.94 (s, 1H, NH), 7.29–7.43 (m, 5H, Ph), 7.97 (d, J = 2.1 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.2 (1C, CH3), 21.5 (2C, C-3, C-4), 32.7 (2C, C-2, C-5), 41.1 (2C, C-7, C-9), 46.7 (2C, C-1, C-6), 48.2 (1C, NCH2Ph), 51.1 (2C, C-10, C-12), 55.2 (1C, C-8), 55.9 (1C, OCH3), 110.0 (1C, C-3Ar), 119.7 (1C, C-6Ar), 122.3 (1C, C-4Ar), 126.5 (1C, C-4Ph), 128.0 9 (4C, C-2Ph, C-3Ph, C-5Ph, C-6Ph), 128.9 (1C, C-1Ar), 129.3, 130.8 (1C, C-5Ar), 137.4 (1C, C-1Ph), 145.8 (1C, C-2Ar), 156.1 (1C,C=O urea), 219.1 (1C,C=O ketone). Purity (HPLC): 80.8% (tR = 22.66 min).
5.3.32. 1 -Benzyl-1-{(8-syn,11-anti)-11-hydroxy[4.3.3]propellan-8-yl}-3-(2-methoxy-5-methylphenyl)urea (syn,anti-22) and 1-benzyl-1-{(8-syn,11-syn)-11-hydroxy[4.3.3]propellan-8-yl}-3-(2-methoxy-5-methylphenyl)urea (syn,syn-22)
NaBH4 (30 mg, 0.79 mmol) was added to a solution of the ketone syn-21 (0.35 g, 0.78 mmol) in a mixture of THF and methanol (9:1, 15 mL). The mixture was stirred at rt for 30 min, then water (1 mL) was added and stirred for additional 10 min. After evaporation of the organic solvent under vacuum, ethyl acetate (10 mL) was added. The mixture was washed with NaOH (1 M, 5 mL) and brine (5 mL), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (4 cm, petroleum ether:ethyl acetate = 8:2 to 6.5:3.5, 20 mL).
syn,anti-22 (Rf = 0.38, cyclohexane:ethyl acetate = 7:3): Colorless solid, mp 177–179 °C, yield 0.15 g (43%). C28H36N2O3 (448.6). Exact mass (APCI): m/z = 449.2870 (calcd. 449.2799 for C28H37N2O3 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3383 (ν O-H), 2927 (ν C-H aliphatic), 1639 (ν C=Ourea), 1535 (δN-H). 1H NMR (400 MHz, CDCl3): 1.34–1.44 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.67–1.73 (m, 4H, 7-Hanti, 9-Hanti, 10-Hanti, 12-Hanti), 1.88 (dd, J = 12.9/7.4 Hz, 2H, 7-Hsyn, 9-Hsyn), 2.02 (dd, J = 13.6/7.0 Hz, 2H, 10-Hsyn, 12-Hsyn), 2.26 (s, 3H, CH3), 3.52 (s, 3H, OCH3), 4.34 (tt, J = 7.0/5,9 Hz, 1H, 11-H), 4.56 (s, 2H, NCH2Ph), 5.17 (tt, J = 10.9/7.4 Hz, 1H, 8-H), 6.61 (d, J = 8.2 Hz, 1H, 3-HAr), 6.68 (ddd, J = 8.2/2.1/0.8 Hz, 1H, 4-HAr), 7.06 (s, broad, 1H, NH), 7.26–7.40 (m, 5H, Ph), 8.04 (d, J = 2.1 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 18.7 (1C, CH3), 21.2 (2C, C-3, C-4), 32.9 (2C, C-2, C-5), 41.2 (2C, C-7, C-9), 46.8 (1C, NCH2Ph), 47.3 (2C, C-1, C-6), 50.2 (2C, C-10, C-12), 52.7(1C, C-8), 55.8 (1C, OCH3), 71.8 (1C, C-11), 109.9 (1C, C-3Ar), 119.6 (1C, C-6Ar), 122.0 (1C, C-4Ar), 126.6 (1C, C-4Ph), 127.5 (2C, C-3Ph, C-5Ph), 128.9 (2C, C-2Ph, C-6Ph), 129.0(1C, C-1Ar), 130.7 (1C, C-5Ar), 138.5 (1C, C-1Ph), 145.7 (1C, C-2Ar), 156.1 (1C, C=O). Purity (HPLC): 94.9% (tR = 22.69 min).
syn,syn-22 (Rf = 0.36, cyclohexane:ethyl acetate = 7:3): Pale yellow solid, mp 148–151 °C, yield 0.10 g (28%). C28H36N2O3 (448.6). Exact mass (APCI): m/z = 449.2770 (calcd. 449.2799 for C28H37N2O3 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3437 (ν O-H), 2931 (ν C-H aliphatic), 1643 (ν C=Ourea), 1535 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.39–1.51 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.62–1.82 (m, 6H, 7-CH2, 9-CH2, 10-Hsyn, 12-Hsyn), 2.03 (dd, J = 13.9/7.0 Hz, 2H, 10-Hanti, 12-Hanti), 2.25 (s, 3H, CH3), 3.48 (s, 3H, OCH3), 4.51 (s, 2H, NCH2Ph), 4.54 (tt, J = 7.1/6.2, Hz, 1H, 11-H), 5.01 (tt, J = 10.6/7.9 Hz, 1H, 8-H), 6.59 (d, J = 8.2 Hz, 1H, 3-HAr), 6.68 (dd, J = 8.2, 2.1 Hz, 1H, 4-HAr), 7.05 (s, broad, 1H, NH), 7.27–7.42 (m, 5H, Ph), 7.99 (d, J = 2.1 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.1 (1C, CH3), 21.1 (2C; C-3, C-4), 34.0 (2C, C-2, C-5), 42.3 (2C, C-7, C-9), 48.0 (2C, C-1, C-6), 46.9 (1C, NCH2Ph) 49.4 (2C, C-10, C-12), 53.6 (1C, C-8), 55.8 (1C, OCH3), 72.6 (1C, C-11), 109.9 (1C, C-3Ar), 119.7 (1C, C-6Ar), 122.2 (1C, C-4Ar), 126.7 (1C, C-4Ph), 127.8 (2C, C-3Ph, C-5Ph), 128.8 (2C, C-2Ph, C-6Ph), 129.0 (1C, C-1Ar), 130.7 (1C, C-5Ar), 137.8 (1C, C-1Ph), 145.8 (1C, C-2Ar), 156.0 (1C, C=O). Purity (HPLC): 97.7% (tR = 22.15 min).
5.3.33. 1 -Benzyl-1-{(8-anti,11-anti)-11-hydroxy[4.3.3]propellan-8-yl}-3-(2-methoxy-5-methylphenyl)urea (anti,anti-22) and 1-benzyl-1-{(8-anti,11-syn)-11-hydroxy[4.3.3]propellan-8-yl}-3-(2-methoxy-5-methylphenyl)urea (anti,syn-22)
NaBH4 (9 mg, 0.24 mmol) was added to a solution of the ketone anti-21 (0.15 g, 0.23 mmol) in a mixture of THF and methanol (9:1, 15 mL). The mixture was stirred at rt for 30 min, then water (1 mL) was added and stirred for additional 10 min. After evaporation of the organic solvent under vacuum, ethyl acetate (10 mL) was added. The mixture was washed with NaOH (1 M, 5 mL) and brine (5 mL), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (4 cm, petroleum ether:ethyl acetate = 8:2 to 6.5:3.5, 20 mL).
anti,anti-22 (Rf = 0.23, cyclohexane:ethyl acetate = 7:3): Colorless solid, mp 126–128 °C, yield 50 mg (33%). C28H36N2O3 (448.6). Exact mass (APCI): m/z = 449.2793 (calcd. 449.2799 for C28H37N2O3 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3394 (νO-H), 2924 (νC-H aliphatic), 1635 (ν C=Ourea), 1535 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.24–1.54 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.67 (dd, J = 14.0/4.6 Hz, 2H, 10-Hanti, 12-Hanti), 1.95 (m, 4H, 7-CH2, 9-CH2), 2.07 (dd, J = 14.0/7.6 Hz, 2H, 10-Hsyn, 12-Hsyn), 2.25 (s, 3H, CH3), 3.45 (s, 3H, OCH3), 4.46 (tt, J = 7.6/4.6 Hz, 1H, 11-H), 4.57 (s, 2H, NCH2Ph), 5.25 (p, J = 9.5 Hz, 1H, 8-H), 6.58 (d, J = 8.2 Hz, 1H, 3-HAr), 6.66 (dd, J = 7.9/1.3 Hz, 1H, 4-HAr), 6.96 (s, 1H, NH), 7.26–7.40 (m, 5H, Ph), 8.01 (d, J = 2.1 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 20.8 (1C, CH3), 21.1 (2C, C-3, C-4), 33.1 (2C, C-2, C-5), 43.1 (2C, C-7, C-9), 46.9 (1C, NCH2Ph), 48.4 (2C, C-10, C-12), 49.3 (2C, C-1, C-6), 55.0 (1C, C-8), 55.8 (1C, OCH3), 73.3 (1C, C-11), 109.9 (1C, C-3Ar), 119.6 (1C, C-6Ar), 122.0 (1C, C-4Ar), 126.6 (1C, C-4Ph), 127.6 (2C, C-3Ph, C-5Ph), 129.0 (2C, C-2Ph, C-6Ph), 129.1 (1C, C-1Ar), 130.7 (1C, C-5Ar), 138.1 (1C, C-1Ph), 145.7 (1C, C-2Ar), 156.1 (1C, C=O). Purity (HPLC): 95.5% (tR = 21.94 min).
anti,syn-22 (Rf = 0.26, cyclohexane:ethyl acetate = 7:3): Pale yellow solid, mp 149–151 °C, yield 58 mg (38%). C28H36N2O3 (448.6). Exact mass (APCI): m/z = 449.2782 (calcd. 449.2799 for C28H37N2O3 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3379 (ν O-H), 2927 (ν C-H aliphatic), 1643 (ν C=Ourea), 1531 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.46–1.56 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.59 (dd, J = 12.9/10.9 Hz, 2H, 7-Hanti, 9-Hanti), 1.69 (dd, J = 13.5/6.4 Hz, 2H, 10-Hsyn, 12-Hsyn), 1.89 (d, J = 13.0/8.0 Hz, 2H, 7-Hsyn, 9-Hsyn), 1.94 (dd, J = 13.5/7.2 Hz, 2H, 10-Hanti, 12-Hanti), 2.25 (m, 3H, CH3), 3.47 (s, 3H, OCH3), 4.39 (p, J = 6.9 Hz, 1H, 11-H), 4.50 (s, 2H, NCH2Ph), 5.08 (tt, J = 10.7/7.9 Hz, 1H, 8-H), 6.59 (d, J = 8.2 Hz, 1H, 3-HAr), 6.67 (ddd, J = 8.2/2.1/0.8 Hz, 1H, 4-HAr), 6.97 (s, broad, 1H, NH), 7.27–7.41 (m, 5H, Ph), 8.00 (d, J = 2.0 Hz, 1H, 6-HAr). 13C NMR (100 MHz, CDCl3): δ (ppm) = 19.6 (1C, CH3), 21.1 (2C, C-3, C-4), 32.3 (2C, C-2, C-5), 43.7 (2C, C-7, C-9), 46.7 (1C, NCH2Ph), 47.6 (2C, C-10, C-12), 47.9 (2C, C-1, C-6), 53.9 (1C, C-8), 55.8 (1C, OCH3), 71.5 (1C, C-11), 109.9 (1C, C-3Ar), 119.6 (1C, C-6Ar), 122.0 (1C, C-4Ar), 126.5 (1C, C-4Ph), 127.7 (2C, C-3Ph, C-5Ph), 128.9 (2C, C-2Ph, C-6Ph), 129.0 (1C, C-1Ar), 130.7(1C, C-5Ar), 138.1(1C, C-1Ph), 145.7 (1C, C-2Ar), 156.1 (1C,C=O). Purity (HPLC): 93.5% (tR = 21.57 min).
5.3.34. 1 -Phenyl-3-(syn- and anti-[4.3.3]propellan-8-yl)urea (23a)
According to General Procedure A, propellanamine 16 (90 mg, 0.50 mmol), phenyl isocyanate (72 mg, 0.6 mmol) and Bu2Sn(OAc)2 (35 mg, 0.1 mmol) were dissolved in THF (5 mL) and the mixture was stirred at rt for 30 h. The crude product was purified by fc (1 cm, cyclohexane:ethyl acetate = 8:2, 5 mL, Rf = 0.36) to obtain a mixture of diastereoisomeric urea syn-23a and anti-23a as a brown solid, mp 134–138 °C, yield 30 mg (22%). C19H26N2O (298.4). MS (ESI): m/z = 299 [M+H]+. Exact mass (APCI): m/z = 299.2131 (calcd. 299.2118 for C19H27N2O [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3429 (ν N-H), 2927 (ν C-H aliphatic), 1647 (ν C=O), 1597 (δN-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.30–1.35 (m, 4H, 2-CH2, 5-CH2), 1.37–1.44 (m, 5H, 3-CH2, 4-CH2, 7-CH2(0.5H), 9-CH2(0.5H)), 1.48 (dd, J = 13.3/7.3 Hz, 2 × 0.5H, 7-H, 9-H), 1.51–1.54 (m, 2H, 10-CH2(1H), 12-CH2(1H)), 1.57–1.66 (m, 3H, 10-CH2(1H), 11-CH2(1H), 12-CH2(1H)), 1.68–1.75 (m, 1H, 11-CH2(1H)), 2.03 (dd, J = 13.4/8.3 Hz, 2 × 0.5H, 7-H, 9-H), 2.11 (dd, J = 13.4/8.7 Hz, 2 × 0.5H, 7-H, 9-H), 4.25–4.31 (m, 2 × 0.5H, 8-H), 7.07–7.16 (m, 1H, 4-Hphenyl), 7.26–7.28 (m, 2H, 3Hphenyl, 5-Hphenyl), 7.29–7.33 (M, 2H, 2-Hphenyl, 6-Hphenyl). Signals for the NH protons are not observed in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.8, 21.0 (2C, C-3, C-4), 21.2, 21.9 (2 × 0.5C, C-11), 31.7, 32.5 (2C, C-2, C-5), 37.6, 38.5 (2C, C-10, C-12), 46.0, 45.5 (2C, C-7, C-9), 49.4 (2C, C-1, C-6), 50.1, 50.7 (2 × 0.5C, C-8), 120.8, 121,7 (2C, C-3phenyl, C-5phenyl), 129.2 (1C, C-4phenyl), 129.6, 129.9 (2C, C-2phenyl, C-6phenyl), 137.3 (1C, C-1phenyl), 156.3 (1C, C=O). syn-23a:anti-23a = 1:1. Purity (HPLC): 97.4% (tR = 21.03 min).
5.3.35. 1 -Cyclohexyl-3-(syn- and anti-[4.3.3]propellan-8-yl)urea (23b)
According to General Procedure A, propellanamine 16 (90 mg, 0.50 mmol), cyclohexyl isocyanate (75 mg, 0.6 mmol) and Bu2Sn(OAc)2 (35 mg, 0.1 mmol) were dissolved in THF (5 mL) and the mixture was stirred at rt for 24 h. The crude product was purified by fc (1 cm, cyclohexane:ethyl acetate = 7:3, 5 mL, Rf = 0.40) to obtain a mixture of diastereoisomeric urea syn-23b and anti-23b as a colorless solid, mp 187–193 °C, yield 73 mg (47%). C19H32N2O (304.5). MS (ESI): m/z = 305 [M+H]+. Exact mass (APCI): m/z = 305.2587 (calcd. 305.2587 for C19H27N2O [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3302 (ν N-H), 2924 (ν C-H aliphatic), 1616 (ν C=O), 1558 (δ N-H). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.10–1.18 (m, 2H, 2-CH2Cy(1H), 6-CH2Cy(1H)), 1.31–1.44 (m, 11H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(0.5H), 9-CH2(0.5H), 4-CH2Cy), 1.48 (d, J = 13.5/7.0 Hz, 2 × 0.5 H, 7-H, 9-H), 1.51–1.65 (m, 7H, 10-CH2, 11-CH2(1H), 12-CH2, 3-CH2Cy(1H), 5-CH2Cy(1H)), 1.68–176 (m, 3H, 11-CH2(1H), 3-CH2Cy(1H), 5-CH2Cy(1H)), 1.92 (m, 2H, 2-CH2Cy(1H), 6-CH2Cy(1H)) 2.00 (dd, J = 13.3/8.4 Hz, 2 × 0.5H, 7-H, 9-H), 2.08 (dd, J = 13.3/8.6 Hz, 2 × 0.5H, 7-H, 9-H), 3.51 (tt, J = 9.3/4.8 Hz, 2 × 0.5H, 1-HCy), 4.09–4.15 (m, 2 × 0.5H, 8-H). Signals for the NH protons are not observed in the spectrum. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.8, 20.9 (2C, C-3, C-4), 21.6 (1C, C-11), 24.8, 25.5 (2C, C-3Cy, C-5Cy), 26.9 (1C, C-4Cy), 31.9, 32.3 (2C, C-2, C-5), 33.8 (2C, C-2Cy, C-6Cy), 37.9, 38.2 (2C, C-10, C-12), 45.5, 46.0 (2C, C-7, C-9), 49.8 (1C, C-1Cy), 50.0 (1C, C-8), 50.5 (2C, C-1, C-6), 157.3 (1C, C=O). syn-23b:anti-23b = 1:1. Purity (HPLC): 98.3% (tR = 21.62 min).
5.3.36. 1 -(2-Methoxy-5-methylphenyl)-3-(syn- and anti-[4.3.3]propellan-8-yl)urea (23c)
According to General Procedure A, propellanamine 16 (90 mg, 0.50 mmol), 2-methoxy-5-methylphenyl isocyanate (98 mg, 0.6 mmol) and Bu2Sn(OAc)2 (35 mg, 0.1 mmol) were dissolved in THF (5 mL) and the mixture was stirred at rt for 30 h. The crude product was purified by fc (1 cm, cyclohexane:ethyl acetate = 7:3, 5 mL, Rf = 0.55) to obtain a mixture of diastereoisomeric urea syn-23c and anti-23c as a beige solid, mp 215–220 °C, yield 160 mg (91%). C21H30N2O2 (342.5). MS (ESI): m/z = 343 [M+H]+. Exact mass (APCI): m/z = 343.2387 (calcd. 343.2380 for C21H31N2O2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3332 (ν N-H), 2927 (νC-H aliphatic), 1639 (ν C=O), 1546 (δ N-H). 1H NMR (600 MHz, CDCl3): δ(ppm) = 1.32–1.46 (m, 9H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(0.5H), 9-CH2(0.5H)), 1.45 (dd, J = 13.3/7.3 Hz, 2 × 0.5H, 7-H, 9-H), 1.53–1.74 (m, 6H, 10-CH2, 11-CH2, 12-CH2), 2.06 (dd, J = 13.3/8.4 Hz, 2 × 0.5H, 7-H, 9-H), 2.14 (dd, J = 13.3/8.6 Hz, 2 × 0.5H, 7-H, 9-H), 2.27 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 4.28–4.31 (m, 2 × 0.5H, 8-H), 6.72 (d, J = 8.2 Hz, 1H, 3-HAr), 6.76 (d, J = 8.0 Hz, 1H, 4-HAr), 6.80 (s, broad, 1H, NH), 7.90 (s, 1H, 6-HAr). 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.9, 21.1 (2C, C-3, C-4), 21.1 (1C, CH3), 21.2, 21.9 (2 × 0.5C, C-11), 31.8, 32.5 (2C, C-2, C-5), 37.6, 38.6 (2C, C-10, C-12), 45.6, 46.2 (2C, C-7, C-9), 49.2, 49.9 (2 × 0.5C, C-8), 50.1, 50.6 (2C, C-1, C-6), 55.9 (1C, OCH3), 110.1 (1C, C-3Ar), 120.2 (1C, C-6Ar), 122.5 (1C, C-4Ar), 128.6 (1C, C-1Ar), 130.9 (1C, C-5Ar), 146.0 (1C, C-2Ar), 155.2 (1C,C=O). syn-23c:anti-23c = 1:1. Purity (HPLC): 93.8% (tR = 22.21 min).
5.3.37. 1 -(3,4-Difluorophenyl)-3-(syn- and anti-[4.3.3]propellan-8-yl)urea (23d)
According to the General Procedure A, propellanamine 16 (90 mg, 0.50 mmol), 3,4-difluorophenyl isocyanate (93 mg, 0.6 mmol) and Bu2Sn(OAc)2 (35 mg, 0.1 mmol) were dissolved in THF (5 mL) and the mixture was stirred at rt for 48 h. The crude product was purified by fc (1 cm, cyclohexane:ethyl acetate = 7:3, 5 mL, Rf = 0.56) to obtain a mixture of diastereoisomeric urea syn-23d and anti-23d as a pale orange solid, mp 168–175 °C, yield 100 mg (63%). C19H24F2N2O (334.4). MS (ESI): m/z = 335 [M+H]+. Exact mass (APCI): m/z = 335.1932 (calcd. 335.1929 for C19H25F2N2O [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3325 (ν N-H), 2927 (ν C-H aliphatic), 1643 (ν C=O), 1558 (δ N-H). 1H NMR (600 MHz, CDCl3): δ(ppm) = 1.27–1.43 (m, 9H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(0.5H), 9-CH2(0.5)), 1.45–1.64 (m, 6H, 7-CH2(0.5H), 9-CH2(0.5H), 10-CH2, 11-CH2(1H), 12-CH2), 1.67.172 (m, 1H, 11-CH2(1H)), 2.01 (dd, J = 13.4/8.4 Hz, 2 × 0.5H, 7-H, 9-H), 2.08 (dd, J = 13.8/8.7 Hz, 2 × 0.5H, 7-H, 9-H), 4.22–4.27 (m, 2 × 0.5H, 8-H), 6.89–6.91 (m, 1H, 6-HAr), 6.99–7.06 (m, 1H, 5-HAr), 7.27–7.33 (m, 1H, 2-HAr). Signals for the NH protons are not observed. 13C NMR (150 MHz, CDCl3): δ (ppm) = 20.9, 21.0 (2C, C-3, C-4), 21.1, 21.8 (2 × 0.5C, C-11), 31.7, 32.5 (2C, C-2, C-5), 37.5, 38.3 (2C, C-10, C-12), 45.5, 46.0 (2C, C-7, C-9), 49.3, 49.97 (2 × 0.5C, C-8), 50.2, 50.7 (2C, C-1, C-6), 110.0 (d, J = 19.3 Hz, 1C, C-2Ar), 116.0 (d, J = 3.9 Hz, 1C, C-6Ar), 117.4 (d, J = 18.1 Hz, 1C, C-5Ar), 135.1 (1C, C-1Ar), 150.3 (d, J = 260.6 Hz, 2C, C-3Ar, C-4Ar), 155.9 (1C, C=O). syn-23d:anti-23d = 1:1. Purity (HPLC): 98.2% (tR = 21.73 min).
5.3.38. (11′-syn and 11′-anti)-Spiro ([1,3]dioxolane-2,8′-[4.3.3]propellan)-11′-amine (24)
Pd(OH)2/C (20%, 70 mg) was added to a solution of the benzylpropellanamines 14 (0.7 g, 2.14 mmol) and ammonium formate (0.54 g, 8.56 mmol) in methanol (20 mL). The mixture was heated to reflux for 3 h. After evaporation of the solvent under vacuum, ethyl acetate (30 mL) was added. The mixture was washed with NaOH (1 M, 10 mL) and brine (10 mL), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (3 cm, ethyl acetate:methanol = 1:1, 20 mL, Rf = 0.11(cyclohexane : ethyl acetate : methanol = 6 : 3 : 1)). Pale yellow oil, yield 0.38 g (75%). C14H22NO2 (237.3). MS (ESI): m/z = 238 [M+H]+. Exact mass (APCI): m/z = 238.1834 (calcd. 238.1802 for C14H24NO2 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 2927 (aliphatic ν C-H), 1558 (ν N-H2). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.28–1.58 (m, 8H, 2′-CH2, 3′-CH2, 4′-CH2, 5′-CH2), 1,63 (dd, J = 13.6/6.9 Hz, 2 × 0.5H, 10′-H, 12′-H), 1.71 (dd, J = 13.6/7.5 Hz, 2 × 0.5 H, 10′-H, 12′-H), 1.82 (d, J = 14.3 Hz, 2H, 7′-H, 9′-H), 1.93 (d, J = 14.3 Hz, 2H, 7′-H, 9′-H), 2.05 (dd, J = 13.6/8.8 Hz, 2 × 0.5H, 10′-H, 12′-H), 2.10 (dd, J = 13.6/8.7 Hz, 2 × 0.5H, 10′-H, 12′-H), 3.61–3.67 (m, 0.5 H, 11′-H), 3.68–3.71 (m, 0.5 H, 11′-H), 3.80 (m, 4H, OCH2CH2O). A signal for the NH2 protons is not observed in the spectrum.
5.3.39. 1 -(3,4-Difluorophenyl)-3-(syn- and anti-spiro[1,3]dioxolane-2,8’-[4.3.3]propellan-11’-yl)urea
According to General Procedure A, amine 24 (0.35 g, 1.47 mmol), 3,4-difluorophenyl isocyanate (0.27 g, 1.76 mmol) and Bu2Sn(OAc)2 (52 mg, 0.15 mmol) were dissolved in THF (15 mL) and the mixture was stirred at rt for 48 h. The crude product was purified by fc (3 cm, cyclohexane:ethyl acetate = 7:3, 20 mL, Rf = 0.42) to obtain 0.55 g of a mixture of diastereoisomeric urea and an impurity with the same Rf value. This mixture was used for the next reaction step without further purification.
5.3.40. 1-(3,4-. Difluorophenyl)-3-(8-syn-11-oxo[4.3.3]propellan-8-yl)urea (syn-25) and 1-(3,4-Difluorophenyl)-3-(8-anti-11-oxo[4.3.3]propellan-8-yl)urea (anti-25)
The mixture obtained above (0.50 g, 1.37 mmol) and p-toluenesulfonic acid monohydrate (26 mg, 0.01 mmol) were dissolved in acetone (20 mL) and the mixture was heated to 60 °C for 2 h. The solvent was removed in vacuo and the residue was purified by fc (3 cm, cyclohexane:ethyl acetate = 9:1 to 6:4, 20 mL).
syn-25 (Rf = 0.23, cyclohexane:ethyl acetate = 7:3): Pale yellow solid, mp 161–173 °C, yield 0.20 g (42%). C19H22F2NO2 (348.4). MS (ESI): m/z = 349 [M+H]+. Exact mass (APCI): m/z = 349.1741 (calcd. 349.1722 for C19H23F2N2O2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3340 (ν N-H), 2931 (ν C-H aliphatic), 1735 (ν C=Oketone), 1654 (ν C=Ourea), 1550 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.40–1.58 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2,), (1.75 (dd, J = 14.1/5.5 Hz, 2H, 7-Hsyn, 9-Hsyn), 2.15 (d, J = 18.8 Hz, 2H, 10-H, 12-H), 2.21 (dd, J = 14.1/9.2 Hz, 2H, 7-Hanti, 9-Hanti), 2.26 (d, J = 18.8 Hz, 2H, 10-H, 12-H), 4.35–4.44 (m, 1H, 8-H), 6.84–6.94 (m, 1H, 6-HAr), 7.01–7.08 (m, 1H, 5-HAr), 7.29–7.35 (m, 1H, 2-HAr). Signals for the NH protons are not observed in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.7 (2C, C-3, C-4), 32.5 (2C, C-2, C-5), 44.8 (2C, C-7, C-9), 47.6 (2C, C-10, C-12), 49.1 (1C, C-8), 49.4 (2C, C-1, C-6), 109.8 (d, J = 21.1 Hz, 1C, C-2Ar), 115.8 (d, J = 8.8 Hz, 1C, C-6Ar), 117.5 (d, J = 18.1 Hz, 1C, C-5Ar), 15.2 (1C, C-1Ar), 146.8 (dd, J = 244.8/13.0 Hz, 1C, C-4Ar), 150.4 (dd, J = 247.4/13.3 Hz, 1C, C-3Ar), 155.3 (1C, N(C=O)N), 218.5 (1C, C=O). Purity (HPLC): 92–6% (tR = 18.24 min).
anti-25 (Rf = 0.31, cyclohexane:ethyl acetate = 7:3) Pale yellow solid, mp 100–103 °C, yield 0.17 g (35%). C19H22F2NO2 (348.4). MS (ESI): m/z = 349 [M+H]+. Exact mass (APCI): m/z = 349.1683 (349.1722 calcd. for C19H23F2N2O2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3302 (ν N-H), 2927 (ν C-H aliphatic), 1728 (ν C=Oketone), 1658 (ν C=Ourea), 1527 (δ N-H). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.29–1.54 (m, 10H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(1H), 9-CH2(1H)), 2.19–2.39 (m, 6H, 7-CH2(1H), 9-CH2(1H), 10-CH2, 12-CH2), 4.43–4.52 (m, 1H, 8-H), 6.90–6.94 (m, 1H, 6-HAr), 7.04 (dd, J = 9.4/8.9 Hz, 1H, 5-HAr), 7.32–7.32–7.40 (m, 1H, 2-HAr). Signals for the NH protons are not observed in the spectrum. 13C NMR (100 MHz, CDCl3): δ(ppm) = 21.3 (2C, C-3, C-4), 31.8 (2C, C-2, C-5), 44.9 (2C, C-7, C-9), 47.8 (2C, C-1, C-6), 49.1 (1C, C-8), 50.5 (2C, C-10, C-12), 109.4 (d, J = 21.1 Hz, 1C, C-2Ar), 115.3 (1C, C-6Ar), 117.4 (d, J = 18.1 Hz, C-5Ar), 135.5 (1C, C-1Ar) 146.5 (dd, J = 247.4/12.0 Hz, 1C, C-4Ar), 150.8 (dd, J = 245.6/13.3 Hz, 1C, C-3Ar), 155.4 (1C, N(C=O)N), 220.3 (1C, C=O). Purity (HPLC): 98.7% (tR = 18.43 min).
5.3.41. 1 -(3,4-Difluorophenyl)-3-(8-syn-11-syn- and 8-syn-11-anti-11-hydroxy[4.3.3]-propellan-8-yl)urea (syn,syn-26 and syn,anti-26)
NaBH4 (20 mg, 0.53 mmol) was added to a solution of the ketone syn-25 (0.12 g, 0.34 mmol) in a mixture of THF and methanol (9:1, 10 mL). The mixture was stirred at rt for 30 min, then water (1 mL) was added and the mixture was stirred for additional 10 min. After evaporation of the organic solvent under vacuum, ethyl acetate (10 mL) was added. The mixture was washed with NaOH (1M, 5 mL) and brine (5 mL), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate = 6:4, 5 mL, Rf = 0.49). The mixture could not be separated. Colorless solid, mp 171–178 °C, yield 0.10 g (83%). C19H24F2NO2 (350.4). MS (ESI): m/z = 351 [M+H]+. Exact mass (APCI): m/z = 351.1884 (calcd. 351.1879 for C19H25F2N2O2 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3298 (ν O-H), 2931 (ν C-H aliphatic), 1678 (ν C=Ourea), 1558 (δ N-H). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 1.30–1.48 (m, 10H, 2-CH2, 3-CH2, 4-CH2, 5-CH2, 7-CH2(1H), 9-CH2(1H)), 1.50–1.60 (m, 2H, 10-H, 12-H), 1.77–1.90 (m, 3H, 7-CH2(0.5H), 9-CH2(0.5H), 10-H, 12-H), 2.01 (dd, J = 13.2/8.3 Hz, 2 × 0.5H, 7-H, 9-H), 4.00–4.10 (m, 0.5H, 8-H), 4.14–4.27 (m, 1.5H, 8-H(0.5H), 11-H), 6.95–7.01 (m, 1H, 6-HAr), 7.25 (dd, J = 10.7/9.2 Hz, 1H, 5-HAr), 7.61 (dddd, J = 13.7/7.5/3.5/.26 Hz, 1H, 2-HAr), 8.40 (s, 1H, NH), 8.41 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 19.7, 20.4 (2C, C-3, C-4), 31.9, 32.6 (2C, C-2, C-5), 44.6, 45.5 (2C, C-7, C-9), 47.2, 47.7 (2 × 0.5C, C-8), 47.9, 48.2 (2C, C-10, C-12), 48.4, 48.6 (2C, C-1, C-6), 69.6, 69.9 (2 × 0.5C, C-11), 106.3 (d, J = 21.9 Hz, 1C, C-2Ar), 113.41 (dd, J = 5.7/3.1 Hz, 1C, C-6Ar), 117.4 (d, J = 17.6 Hz, 1C, C-5Ar), 137.7 (dd, J = 9.5/2.8 Hz, 1C, C-1Ar), 143.8 (dd, J = 238.4/12.8 Hz, 1C, C-4Ar), 149.0 (dd, J = 241.7/12.9 Hz, 1C, C-3Ar), 154.4 (1C, N(C=O)N). syn,syn-26:syn,anti-26 = 1:1. Purity (HPLC): 99.2% (tR = 18.26 min).
5.3.42. 1 -(3,4-Difluorophenyl)-3-(8-anti-11-anti-11-hydroxy[4.3.3]propellan-8-yl)urea (anti,anti-26) and 1-(3,4-Difluorophenyl)-3-(8-anti-11-syn-11-hydroxy[4.3.3]propellan-8-yl)urea (anti,syn-26)
NaBH4 (20 mg, 0.53 mmol) was added to a solution of the keto urea anti-25 (0.12 g, 0.34 mmol) in a mixture of THF and methanol (9:1, 10 mL). The mixture was stirred at rt for 30 min, then water (1 mL) was added and the mixture was stirred for additional 10 min. After evaporation of the organic solvent under vacuum, ethyl acetate (10 mL) was added. The mixture was washed with NaOH (1 M, 5 mL) and brine (5 mL), dried (Na2SO4), filtered, the filtrate was concentrated in vacuo and the residue was purified by fc (1 cm, cyclohexane:ethyl acetate = 7:3–5:5, 5 mL).
anti,anti-26 (Rf = 0.22, cyclohexane:ethyl acetate = 7:3): Colorless solid, mp 201–203 °C, yield 32 mg (27%). C19H24F2NO2 (350.4). MS (ESI): m/z = 351 [M+H]+. Exact mass (APCI): m/z = 351.1901 (calcd. 351.1879 for C19H25F2N2O2 [M+H]+). FT-IR (ATR, film): ν (cm−1) = 3232 (ν O-H), 2924 (ν C-H aliphatic), 1670 (ν C=Ourea), 1566 (δ N-H). 1H NMR (400 MHz, CDCl3): 1.11–1.25 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.57 (dd, J = 14.0/4.7 Hz, 2H, 10-Hanti, 12-Hanti), 1.62 (dd, J = 13.6/6.9 Hz, 2H, 7-Hanti, 9-Hanti), 1.98 (dd, J = 14.0/8.2 Hz, 2H, 10-Hsyn,, 12-Hsyn), 2.03 (dd, J = 13.6/9.1 Hz, 2H, 7-Hsyn, 9-Hsyn), 4.23 (tt, J = 9.0/6.9 Hz, 1H, 8-H), 4.34 (tt, J = 8.1/4.7 Hz, 1H, 11-H), 6.76–6.81 (m, 1H, 6-HAr), 6.85 (dd, J = 9.9/8.7 Hz, 1H, 5-HAr), 7.40 (ddd, J = 13.1/7.3/2.5 Hz, 1H, 2-HAr). Signals for the NH protons and the OH proton are not observed in the spectrum. 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.1 (2C, C-3, C-4), 32.1 (2C, C-2, C-5), 45.9 (2C, C-7, C-9), 47.7 (2C, C-10, C-12), 48.8 (1C, C-8), 50.4 (2C, C-1, C-6), 71.9 (1C, C-11), 107.2 (d, J = 21.9 Hz, 1C, C-2Ar), 113.1 (dd, J = 5.6/3.3 Hz, 1C, C-6Ar), 116.5 (d, J = 17.9 Hz, 1C, C-5Ar), 137.0 (dd, J = 9.3/2.8 Hz, C-1Ar), 144.9 (dd, J = 240.6/13.1 Hz, 1C, C-4Ar), 149.8 (dd, J = 242.8/13.1 Hz, 1C, C-3Ar), 155.2 (1C, N(C=O)N). Purity (HPLC): 96.9% (tR = 18.49 min).
anti,syn-26 (Rf = 0.13, cyclohexane:ethyl acetate = 7:3): Colorless solid, mp 186–189 °C, yield 10 mg (8%). C19H24F2NO2 (350.4). MS (ESI): m/z = 351 [M+H]+. Exact mass (APCI): m/z = 351.1907 (calcd. 351.1879 for C19H25F2N2O2 [M+H]+ ). FT-IR (ATR, film): ν (cm−1) = 3383 (ν O-H), 2924 (ν C-H aliphatic), 1654 (δ C=Ourea), 1570 (δ N-H). 1H NMR (400 MHz, DMSO-d6): 1.35 (dd, J = 13.3/8.1 Hz, 2H, 7-Hanti, 9-Hanti), 1.39–1.50 (m, 8H, 2-CH2, 3-CH2, 4-CH2, 5-CH2), 1.56 (dd, J = 13.4/5.4 Hz, 2H, 10-Hsyn, 12-Hsyn), 1.87 (dd, J = 13.6/7.5 Hz, 2H, 10-Hanti, 12-Hanti), 1.95 (dd, J = 13.3/6.0 Hz, 2H, 7-Hsyn, 9-Hsyn), 4.10 (tt, J = 8.1/6.0 Hz, 1H, 8-H), 4.30 (tt, J = 7.3/5.4 Hz, 1H, 11-H), 6.34 (d, J = 7.5 Hz, 1H, NHCONHAr), 6.98 (dddd, J = 9.0/4.1/2.5/1.5 Hz, 1H, 6-HAr), 7.25 (dd, J = 10.6/9.1 Hz, 1H, 5-HAr), 7.60 (ddd, J =13.7/7.5/2.6 Hz, 1H 2-HAr). 8.45 (s, 1H, NHCONHAr). A signal for the OH proton is not observed in the spectrum. 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 19.7 (2C, C-3, C-4), 31.2 (2C, C-2, C-5), 46.2 (2C, C-7, C-9), 47.1 (2C, C-10, C-12), 48.6 (2C, C-1, C-6), 47.7 (1C, C-8), 691 (1C, C-11), 106.3 (d, J = 21.8 Hz, 1C, C-2Ar), 113.38 (1C, C-6Ar), 137.8 (d, J = 6.8 Hz, C-1Ar), 141.8 (d, J = 152.2 Hz, 1C, C-4Ar), 149.1 (d, J = 242.1 Hz, 1C, C-3Ar), 154.5 (1C, N(C=O)N). Purity (HPLC): 93.8% (tR = 17.82 min).


 ,
, 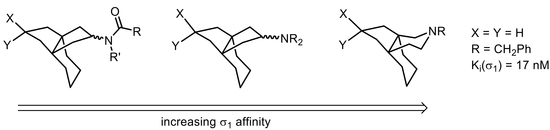
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


