
Tenascin-C in Heart Diseases—The Role of Inflammation

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Features of TNC in the Heart
3. Embryonic Heart Development and TNC
4. Myocardial Infarction
4.1. Acute Myocardial Infarction and TNC
4.2. Post-Infarct Ventricular Remodeling and TNC
5. Hypertensive Cardiac Fibrosis and TNC
6. Myocarditis
6.1. Acute Myocarditis and TNC
6.2. Chronic Myocarditis, Inflammatory DCM(Idcm), Heart Failure and TNC
7. Clinical Application of TNC
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tucker, R.P.; Drabikowski, K.; Hess, J.F.; Ferralli, J.; Chiquet-Ehrismann, R.; Adams, J.C. Phylogenetic analysis of the tenascin gene family: Evidence of origin early in the chordate lineage. BMC Evol. Biol. 2006, 6, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiovaro, F.; Chiquet-Ehrismann, R.; Chiquet, M. Transcriptional regulation of tenascin genes. Cell Adh. Migr. 2015, 9, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Rathjen, F.G.; Hodge, R. Early Days of Tenascin-R Research: Two Approaches Discovered and Shed Light on Tenascin-R. Front. Immunol. 2020, 11, 612482. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Tenascin-X-Discovery and Early Research. Front. Immunol. 2020, 11, 612497. [Google Scholar] [CrossRef]
- Degen, M.; Scherberich, A.; Tucker, R.P. Tenascin-W: Discovery, Evolution, and Future Prospects. Front. Immunol. 2020, 11, 623305. [Google Scholar] [CrossRef] [PubMed]
- Chiquet-Ehrismann, R.; Mackie, E.J.; Pearson, C.A.; Sakakura, T. Tenascin: An extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 1986, 47, 131–139. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960. [Google Scholar] [CrossRef] [Green Version]
- Chiquet, M. Tenascin-C: From Discovery to Structure-Function Relationships. Front. Immunol. 2020, 11, 611789. [Google Scholar] [CrossRef]
- Sakakura, T. Serendipity; Close Encounter of Tenascin C. Front. Immunol. 2020, 11, 620182. [Google Scholar]
- Bornstein, P. Matricellular proteins: An overview. J. Cell Commun. Signal. 2009, 3, 163–165. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K. Tenascin-C in cardiovascular tissue remodeling: From development to inflammation and repair. Circ. J. 2012, 76, 2513–2520. [Google Scholar] [CrossRef] [Green Version]
- Imanaka-Yoshida, K.; Tawara, I.; Yoshida, T. Tenascin-C in cardiac disease: A sophisticated controller of inflammation, repair, and fibrosis. Am. J. Physiol. Cell Physiol. 2020, 319, C781–C796. [Google Scholar] [CrossRef] [PubMed]
- Deligne, C.; Murdamoothoo, D.; Gammage, A.N.; Gschwandtner, M.; Erne, W.; Loustau, T.; Marzeda, A.M.; Carapito, R.; Paul, N.; Velazquez-Quesada, I.; et al. Matrix-Targeting Immunotherapy Controls Tumor Growth and Spread by Switching Macrophage Phenotype. Cancer Immunol. Res. 2020, 8, 368–382. [Google Scholar] [CrossRef]
- Spenle, C.; Loustau, T.; Murdamoothoo, D.; Erne, W.; Beghelli-de la Forest Divonne, S.; Veber, R.; Petti, L.; Bourdely, P.; Morgelin, M.; Brauchle, E.M.; et al. Tenascin-C Orchestrates an Immune-Suppressive Tumor Microenvironment in Oral Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 1122–1138. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Ospelt, C.; Gay, S.; Midwood, K.S. Location, location, location: How the tissue microenvironment affects inflammation in RA. Nat. Rev. Rheumatol. 2021, 17, 195–212. [Google Scholar] [CrossRef]
- Deligne, C.; Midwood, K.S. Macrophages and Extracellular Matrix in Breast Cancer: Partners in Crime or Protective Allies? Front. Oncol. 2021, 11, 620773. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Matsumoto, K.; Hara, M.; Sakakura, T.; Yoshida, T. The dynamic expression of tenascin-C and tenascin-X during early heart development in the mouse. Differentiation 2003, 71, 291–298. [Google Scholar] [CrossRef]
- Yonebayashi, S.; Tajiri, K.; Hara, M.; Saito, H.; Suzuki, N.; Sakai, S.; Kimura, T.; Sato, A.; Sekimoto, A.; Fujita, S.; et al. Generation of Transgenic Mice that Conditionally Overexpress Tenascin-C. Front. Immunol. 2021, 12, 620541. [Google Scholar] [CrossRef]
- Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. [Google Scholar] [CrossRef]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Podesser, B.K.; Kreibich, M.; Dzilic, E.; Santer, D.; Forster, L.; Trojanek, S.; Abraham, D.; Krssak, M.; Klein, K.U.; Tretter, E.V.; et al. Tenascin-C promotes chronic pressure overload-induced cardiac dysfunction, hypertrophy and myocardial fibrosis. J. Hypertens 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, I.F.; Acar, E.; Costantino, S.; Szabo, P.L.; Hamza, O.; Tretter, E.V.; Klein, K.U.; Trojanek, S.; Abraham, D.; Paneni, F.; et al. Epigenetic modulation of tenascin C in the heart: Implications on myocardial ischemia, hypertrophy and metabolism. J. Hypertens 2019, 37, 1861–1870. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Hiroe, M.; Nishikawa, T.; Ishiyama, S.; Shimojo, T.; Ohta, Y.; Sakakura, T.; Yoshida, T. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab. Investig. 2001, 81, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Imanaka-Yoshida, K.; Hiroe, M.; Yasutomi, Y.; Toyozaki, T.; Tsuchiya, T.; Noda, N.; Maki, T.; Nishikawa, T.; Sakakura, T.; Yoshida, T. Tenascin-C is a useful marker for disease activity in myocarditis. J. Pathol. 2002, 197, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Imanaka-Yoshida, K.; Hiramitsu, S.; Kato, S.; Ohtsuki, M.; Uemura, A.; Kato, Y.; Nishikawa, T.; Toyozaki, T.; Hishida, H.; et al. Diagnostic utility of tenascin-C for evaluation of the activity of human acute myocarditis. J. Pathol. 2005, 205, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Suzuki, M.; Onishi, K.; Takakura, N.; Inada, H.; Yoshida, T.; Hiroe, M.; Imanaka-Yoshida, K. Eplerenone attenuates myocardial fibrosis in the angiotensin II-induced hypertensive mouse: Involvement of tenascin-C induced by aldosterone-mediated inflammation. J. Cardiovasc. Pharmacol. 2007, 49, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Shimojo, N.; Terasaki, F.; Otsuka, K.; Hosotani, N.; Kohda, Y.; Tanaka, T.; Nishioka, T.; Yoshida, T.; Hiroe, M.; et al. Atrial natriuretic peptide exerts protective action against angiotensin II-induced cardiac remodeling by attenuating inflammation via endothelin-1/endothelin receptor A cascade. Heart Vessel. 2013, 28, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Hiroe, M.; Yoshida, T. Interaction between cell and extracellular matrix in heart disease: Multiple roles of tenascin-C in tissue remodeling. Histol. Histopathol. 2004, 19, 517–525. [Google Scholar] [PubMed]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adh. Migr. 2015, 9, 48–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Suzuki, H. The Role of Tenascin-C in Tissue Injury and Repair After Stroke. Front. Immunol. 2020, 11, 607587. [Google Scholar] [CrossRef]
- Nakajima, Y.; Imanaka-Yoshida, K. New insights into the developmental mechanisms of coronary vessels and epicardium. Int. Rev. Cell Mol. Biol. 2013, 303, 263–317. [Google Scholar] [PubMed]
- Cao, Y.X.; Duca, S.; Cao, J.L. Epicardium in Heart Development. Cold Spring Harb. Perspect. Biol. 2020, 12, a037143. [Google Scholar] [CrossRef]
- Vrancken Peeters, M.P.; Gittenberger-de Groot, A.C.; Mentink, M.M.; Hungerford, J.E.; Little, C.D.; Poelmann, R.E. Differences in development of coronary arteries and veins. Cardiovasc. Res. 1997, 36, 101–110. [Google Scholar] [CrossRef]
- Vrancken Peeters, M.P.; Gittenberger-de Groot, A.C.; Mentink, M.M.; Hungerford, J.E.; Little, C.D.; Poelmann, R.E. The development of the coronary vessels and their differentiation into arteries and veins in the embryonic quail heart. Dev. Dyn. 1997, 208, 338–348. [Google Scholar] [CrossRef]
- Kattan, J.; Dettman, R.W.; Bristow, J. Formation and remodeling of the coronary vascular bed in the embryonic avian heart. Dev. Dyn. 2004, 230, 34–43. [Google Scholar] [CrossRef]
- Ando, K.; Takahashi, M.; Yamagishi, T.; Miyagawa-Tomita, S.; Imanaka-Yoshida, K.; Yoshida, T.; Nakajima, Y. Tenascin C may regulate the recruitment of smooth muscle cells during coronary artery development. Differentiation 2011, 81, 299–306. [Google Scholar] [CrossRef]
- Ishigaki, T.; Imanaka-Yoshida, K.; Shimojo, N.; Matsushima, S.; Taki, W.; Yoshida, T. Tenascin-C enhances crosstalk signaling of integrin alphavbeta3/PDGFR-beta complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J. Cell Physiol. 2011, 226, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Yoshida, T.; Miyagawa-Tomita, S. Tenascin-C in development and disease of blood vessels. Anat. Rec. 2014, 297, 1747–1757. [Google Scholar] [CrossRef]
- Forsberg, E.; Hirsch, E.; Fröhlich, L.; Meyer, M.; Ekblom, P.; Aszodi, A.; Werner, S.; Fässler, R. Skin wounds and severed nerves heal normally in mice lacking tenascin-C. Proc. Natl. Acad. Sci. USA 1996, 93, 6594–6599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saga, Y.; Yagi, T.; Ikawa, Y.; Sakakura, T.; Aizawa, S. Mice develop normally without tenascin. Genes Dev. 1992, 6, 1821–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, K.; Imanaka-Yoshida, K.; Yoshida, T.; Sugimura, Y. Role of stromal tenascin-C in mouse prostatic development and epithelial cell differentiation. Dev. Biol. 2008, 324, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Roth-Kleiner, M.; Hirsch, E.; Schittny, J.C. Fetal lungs of tenascin-C-deficient mice grow well, but branch poorly in organ culture. Am. J. Respir. Cell Mol. Biol. 2004, 30, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mund, S.I.; Schittny, J.C. Tenascin-C deficiency impairs alveolarization and microvascular maturation during postnatal lung development. J. Appl. Physiol. 2020, 128, 1287–1298. [Google Scholar] [CrossRef]
- Morellini, F.; Schachner, M. Enhanced novelty-induced activity, reduced anxiety, delayed resynchronization to daylight reversal and weaker muscle strength in tenascin-C-deficient mice. Eur. J. Neurosci. 2006, 23, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- De Chevigny, A.; Lemasson, M.; Saghatelyan, A.; Sibbe, M.; Schachner, M.; Lledo, P.M. Delayed onset of odor detection in neonatal mice lacking tenascin-C. Mol. Cell. Neurosci. 2006, 32, 174–186. [Google Scholar] [CrossRef]
- Gurevicius, K.; Kuang, F.; Stoenica, L.; Irintchev, A.; Gureviciene, I.; Dityatev, A.; Schachner, M.; Tanila, H. Genetic ablation of tenascin-C expression leads to abnormal hippocampal CA1 structure and electrical activity in vivo. Hippocampus 2009, 19, 1232–1246. [Google Scholar] [CrossRef]
- Gremlich, S.; Roth-Kleiner, M.; Equey, L.; Fytianos, K.; Schittny, J.C.; Cremona, T.P. Tenascin-C inactivation impacts lung structure and function beyond lung development. Sci. Rep. 2020, 10, 5118. [Google Scholar] [CrossRef] [Green Version]
- Santer, D.; Nagel, F.; Gonçalves, I.F.; Kaun, C.; Wojta, J.; Fagyas, M.; Krššák, M.; Balogh, Á.; Papp, Z.; Tóth, A.; et al. Tenascin-C aggravates ventricular dilatation and angiotensin-converting enzyme activity after myocardial infarction in mice. ESC Heart Fail. 2020, 7, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Perera-Gonzalez, M.; Kiss, A.; Kaiser, P.; Holzweber, M.; Nagel, F.; Watzinger, S.; Acar, E.; Szabo, P.L.; Gonçalves, I.F.; Weber, L.; et al. The Role of Tenascin C in Cardiac Reverse Remodeling Following Banding-Debanding of the Ascending Aorta. Int. J. Mol. Sci. 2021, 22, 2023. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, L.; Li, F.Q.; Yukht, A.; Qin, M.H.; Ruther, H.; Yang, M.J.; Chaux, A.; Shah, P.K.; Sharifi, B.G. Bone Marrow-Derived Tenascin-C Attenuates Cardiac Hypertrophy by Controlling Inflammation. J. Am. Coll. Cardiol. 2017, 70, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Onishi, K.; Shimojo, N.; Nagano, Y.; Matsusaka, H.; Ikeuchi, M.; Ide, T.; Tsutsui, H.; Hiroe, M.; Yoshida, T.; et al. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1072–H1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machino-Ohtsuka, T.; Tajiri, K.; Kimura, T.; Sakai, S.; Sato, A.; Yoshida, T.; Hiroe, M.; Yasutomi, Y.; Aonuma, K.; Imanaka-Yoshida, K. Tenascin-C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J. Am. Heart Assoc. 2014, 3, e001052. [Google Scholar] [CrossRef] [Green Version]
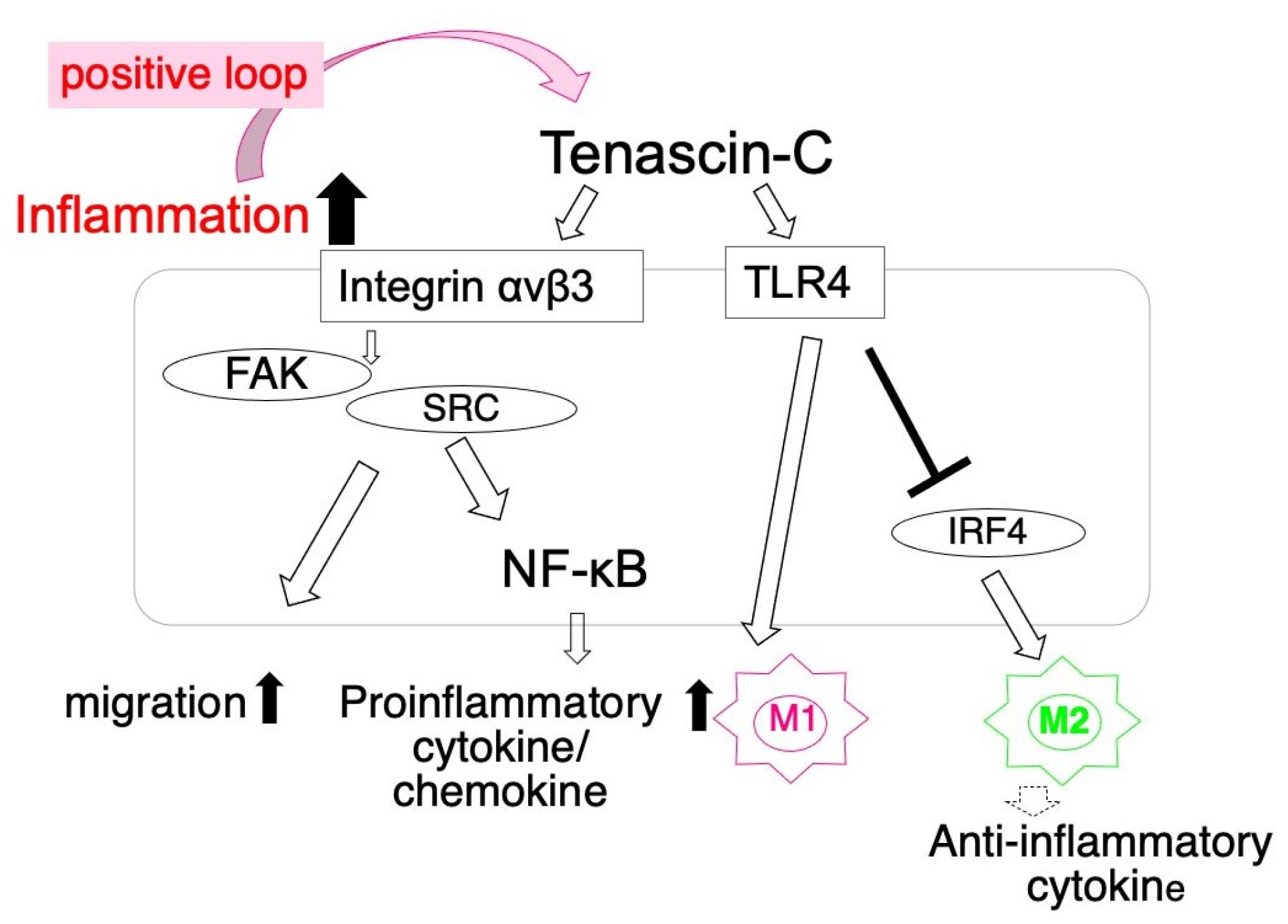
- Shimojo, N.; Hashizume, R.; Kanayama, K.; Hara, M.; Suzuki, Y.; Nishioka, T.; Hiroe, M.; Yoshida, T.; Imanaka-Yoshida, K. Tenascin-C May Accelerate Cardiac Fibrosis by Activating Macrophages via the Integrin alphaVbeta3/Nuclear Factor-kappaB/Interleukin-6 Axis. Hypertension 2015, 66, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Tajiri, K.; Sato, A.; Sakai, S.; Wang, Z.; Yoshida, T.; Uede, T.; Hiroe, M.; Aonuma, K.; Ieda, M.; et al. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc. Res. 2019, 115, 614–624. [Google Scholar] [CrossRef]
- Nagel, F.; Santer, D.; Stojkovic, S.; Kaun, C.; Schaefer, A.K.; Krššák, M.; Abraham, D.; Bencsik, P.; Ferdinandy, P.; Kenyeres, E.; et al. The impact of age on cardiac function and extracellular matrix component expression in adverse post-infarction remodeling in mice. Exp. Gerontol. 2019, 119, 193–202. [Google Scholar] [CrossRef]
- Sato, A.; Aonuma, K.; Imanaka-Yoshida, K.; Yoshida, T.; Isobe, M.; Kawase, D.; Kinoshita, N.; Yazaki, Y.; Hiroe, M. Serum tenascin-C might be a novel predictor of left ventricular remodeling and prognosis after acute myocardial infarction. J. Am. Coll. Cardiol. 2006, 47, 2319–2325. [Google Scholar] [CrossRef] [Green Version]
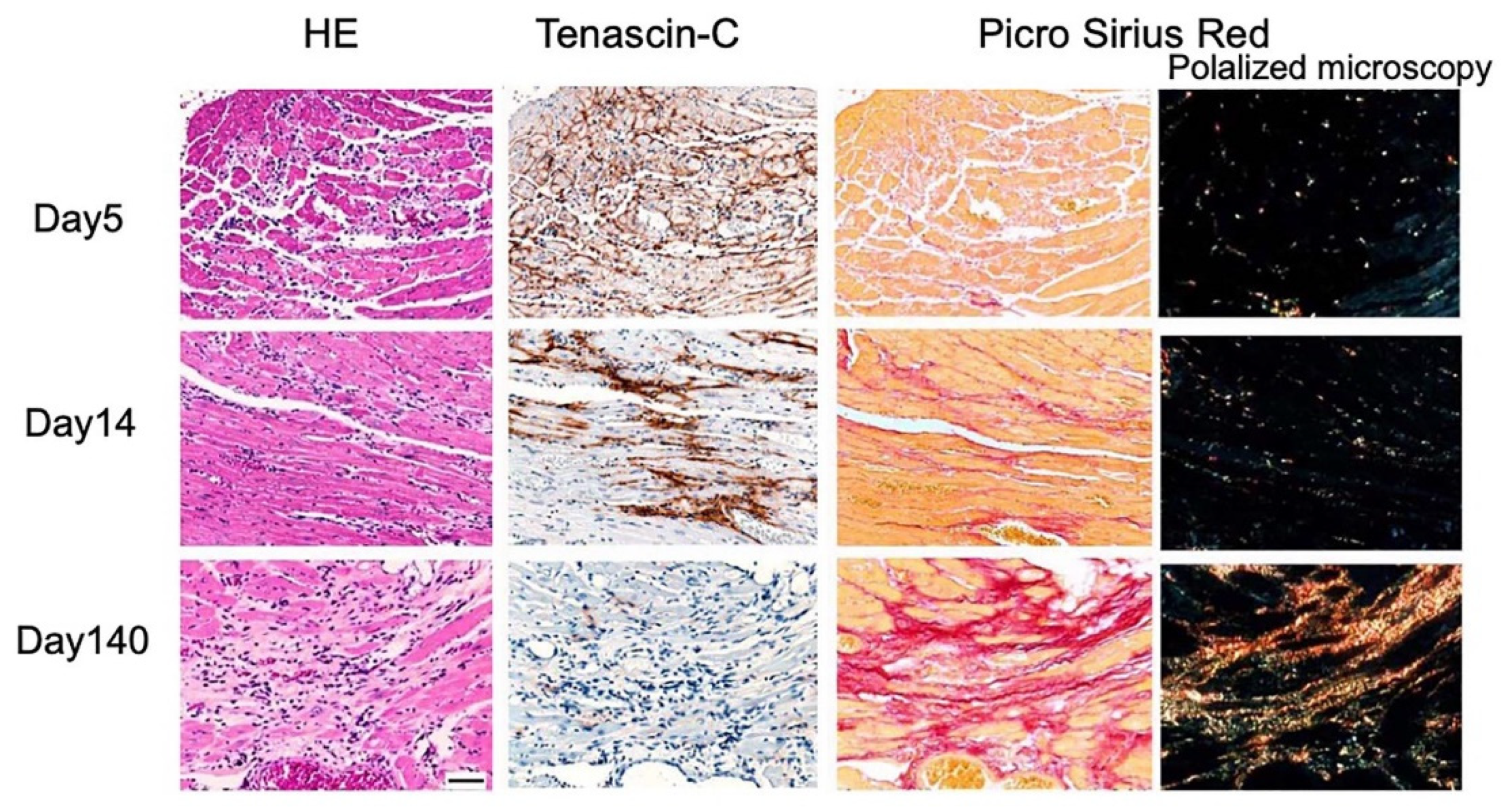
- Willems, I.E.; Arends, J.W.; Daemen, M.J. Tenascin and fibronectin expression in healing human myocardial scars. J. Pathol. 1996, 179, 321–325. [Google Scholar] [CrossRef]
- Tamaoki, M.; Imanaka-Yoshida, K.; Yokoyama, K.; Nishioka, T.; Inada, H.; Hiroe, M.; Sakakura, T.; Yoshida, T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am. J. Pathol. 2005, 167, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Danowski, B.A.; Imanaka-Yoshida, K.; Sanger, J.M.; Sanger, J.W. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 1992, 118, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Imanaka-Yoshida, K.; Danowski, B.A.; Sanger, J.M.; Sanger, J.W. Living adult rat cardiomyocytes in culture: Evidence for dissociation of costameric distribution of vinculin from costameric distributions of attachments. Cell Motil. Cytoskelet. 1996, 33, 263–275. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Enomoto-Iwamoto, M.; Yoshida, T.; Sakakura, T. Vinculin, Talin, Integrin alpha6beta1 and laminin can serve as components of attachment complex mediating contraction force transmission from cardiomyocytes to extracellular matrix. Cell Motil. Cytoskelet. 1999, 42, 1–11. [Google Scholar] [CrossRef]
- Mazhari, R.; Omens, J.H.; Covell, J.W.; McCulloch, A.D. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiovasc. Res. 2000, 47, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Oberhauser, A.F.; Marszalek, P.E.; Erickson, H.P.; Fernandez, J.M. The molecular elasticity of the extracellular matrix protein tenascin. Nature 1998, 393, 181–185. [Google Scholar] [CrossRef]
- Marín, J.L.; Muñiz, J.; Huerta, M.; Trujillo, X. Folding-unfolding of FN-III domains in tenascin: An elastically coupled two-state system. J. Biomech. 2003, 36, 1733–1737. [Google Scholar] [CrossRef]
- Kimura, T.; Shiraishi, K.; Furusho, A.; Ito, S.; Hirakata, S.; Nishida, N.; Yoshimura, K.; Imanaka-Yoshida, K.; Yoshida, T.; Ikeda, Y.; et al. Tenascin C protects aorta from acute dissection in mice. Sci. Rep. 2014, 4, 4051. [Google Scholar] [CrossRef]
- De Haan, J.J.; Smeets, M.B.; Pasterkamp, G.; Arslan, F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediat. Inflamm. 2013, 2013, 206039. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef]
- Marzeda, A.M.; Midwood, K.S. Internal Affairs: Tenascin-C as a Clinically Relevant, Endogenous Driver of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Marzeda, A.M.; Deligne, C.; Schwenzer, A.; McCann, F.E.; Marsden, B.D.; Piccinini, A.M.; Midwood, K.S. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat. Commun. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, A.; Spary, E.J.; Manfield, I.W.; Ruhmann, M.; Zuliani-Alvarez, L.; Gamboa-Esteves, F.O.; Porter, K.E.; Drinkhill, M.J.; Midwood, K.S.; Turner, N.A. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J. Cardiol. 2016, 8, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Zuliani-Alvarez, L.; Lim, J.M.P.; Midwood, K.S. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci. Signal. 2016, 9, ra86. [Google Scholar] [CrossRef] [Green Version]
- Benbow, J.H.; Thompson, K.J.; Cope, H.L.; Brandon-Warner, E.; Culberson, C.R.; Bossi, K.L.; Li, T.; Russo, M.W.; Gersin, K.S.; McKillop, I.H.; et al. Diet-Induced Obesity Enhances Progression of Hepatocellular Carcinoma through Tenascin-C/Toll-Like Receptor 4 Signaling. Am. J. Pathol. 2016, 186, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, N.; Duarte, S.; Hamada, T.; Busuttil, R.W.; Coito, A.J. Tenascin-C: A novel mediator of hepatic ischemia and reperfusion injury. Hepatology 2011, 54, 2125–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef]
- De Couto, G.; Liu, W.; Tseliou, E.; Sun, B.; Makkar, N.; Kanazawa, H.; Arditi, M.; Marbán, E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Investig. 2015, 125, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, S.A.; Dunne, A.; Monaghan, M.G. The Role of Macrophages in the Infarcted Myocardium: Orchestrators of ECM Remodeling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzai, T. Inflammatory Mechanisms of Cardiovascular Remodeling. Circ. J. 2018, 82, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.Y.; Lee, K.W.; Shin, D.; An, S.; Cho, K.H.; Kim, S.H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 3016. [Google Scholar] [CrossRef] [PubMed]
- Hesse, J.; Leberling, S.; Boden, E.; Friebe, D.; Schmidt, T.; Ding, Z.P.; Dieterich, P.; Deussen, A.; Roderigo, C.; Rose, C.R.; et al. CD73-derived adenosine and tenascin-C control cytokine production by epicardium-derived cells formed after myocardial infarction. FASEB J. 2017, 31, 3040–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Midwood, K.S.; Yin, H.; Varga, J. Toll-Like Receptor-4 Signaling Drives Persistent Fibroblast Activation and Prevents Fibrosis Resolution in Scleroderma. Adv. Wound Care 2017, 6, 356–369. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell Mol. Life Sci. 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [Green Version]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin αvβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human Breast Cancer. Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef]
- Sato, A.; Hiroe, M.; Akiyama, D.; Hikita, H.; Nozato, T.; Hoshi, T.; Kimura, T.; Wang, Z.; Sakai, S.; Imanaka-Yoshida, K.; et al. Prognostic value of serum tenascin-C levels on long-term outcome after acute myocardial infarction. J. Card. Fail. 2012, 18, 480–486. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [PubMed]
- Lindsey, M.L.; Iyer, R.P.; Jung, M.; DeLeon-Pennell, K.Y.; Ma, Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J. Mol. Cell Cardiol. 2016, 91, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; López, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; González, A.; Colan, S.D.; Seidman, J.G.; et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, J.; Mlejnek, D.; Sochorova, D.; Nemec, P. Inflammatory Cardiomyopathy: A Current View on the Pathophysiology, Diagnosis, and Treatment. Biomed. Res. Int. 2016, 2016, 4087632. [Google Scholar] [CrossRef] [Green Version]
- Assomull, R.G.; Prasad, S.K.; Lyne, J.; Smith, G.; Burman, E.D.; Khan, M.; Sheppard, M.N.; Poole-Wilson, P.A.; Pennell, D.J. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1977–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Manabe, I. Chronic Inflammation Links Cardiovascular, Metabolic and Renal Diseases. Circ. J. 2011, 75, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 16018. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Elsanhoury, A.; Klein, O.; Sosnowski, M.; Miteva, K.; Lassner, D.; Abou-El-Enein, M.; Pieske, B.; Kuhl, U.; Tschope, C. Telbivudine in chronic lymphocytic myocarditis and human parvovirus B19 transcriptional activity. ESC Heart Fail. 2018, 5, 818–829. [Google Scholar] [CrossRef]
- Hsieh, C.S.; Macatonia, S.E.; O’Garra, A.; Murphy, K.M. T cell genetic background determines default T helper phenotype development in vitro. J. Exp. Med. 1995, 181, 713–721. [Google Scholar] [CrossRef]
- Abbadi, D.; Laroumanie, F.; Bizou, M.; Pozzo, J.; Daviaud, D.; Delage, C.; Calise, D.; Gaits-Iacovoni, F.; Dutaur, M.; Tortosa, F.; et al. Local production of tenascin-C acts as a trigger formonocyte/macrophage recruitment that provokes cardiac dysfunction. Cardiovasc. Res. 2018, 114, 123–137. [Google Scholar] [CrossRef] [PubMed]
- JCS Joint Working Group and treatment of myocarditis (JCS 2009): Digest version. Circ. J. 2011, 75, 734–743. [CrossRef] [Green Version]
- Imanaka-Yoshida, K. Inflammation in myocardial disease: From myocarditis to dilated cardiomyopathy. Pathol. Int. 2020, 70, 1–11. [Google Scholar] [CrossRef]
- Kawada, J.I.; Takeuchi, S.; Imai, H.; Okumura, T.; Horiba, K.; Suzuki, T.; Torii, Y.; Yasuda, K.; Imanaka-Yoshida, K.; Ito, Y. Immune cell infiltration landscapes in pediatric acute myocarditis analyzed by CIBERSORT. J. Cardiol. 2021, 77, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Veronese, G.; Bottiroli, M.; Wang, D.W.; Cipriani, M.; Garascia, A.; Pedrotti, P.; Adler, E.D.; Frigerio, M. Update on acute myocarditis. Trends Cardiovasc. Med. 2020, S1050-1738, 30077–30079. [Google Scholar] [CrossRef]
- Suzuki, H.; Nishikawa, H.; Kawakita, F. Matricellular proteins as possible biomarkers for early brain injury after aneurysmal subarachnoid hemorrhage. Neural Regen Res. 2018, 13, 1175–1178. [Google Scholar] [CrossRef]
- Tajiri, K.; Aonuma, K.; Sekine, I. Immune checkpoint inhibitor-related myocarditis. Jpn J. Clin. Oncol. 2018, 48, 7–12. [Google Scholar] [CrossRef]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; van der Wal, A.C.; Aubry, M.-C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef] [PubMed]
- Halushka, M.K.; Vander Heide, R.S. Myocarditis is rare in COVID-19 autopsies: Cardiovascular findings across 277 postmortem examinations. Cardiovasc. Pathol. 2021, 50, 107300. [Google Scholar] [CrossRef] [PubMed]
- Kawai, C. From myocarditis to cardiomyopathy: Mechanisms of inflammation and cell death: Learning from the past for the future. Circulation 1999, 99, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, J.F.; Woodruff, J.J. Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J. Immunol. 1974, 113, 1726–1734. [Google Scholar]
- Lasrado, N.; Reddy, J. An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 2020, 30, 1–14. [Google Scholar] [CrossRef]
- Gangaplara, A.; Massilamany, C.; Brown, D.M.; Delhon, G.; Pattnaik, A.K.; Chapman, N.; Rose, N.; Steffen, D.; Reddy, J. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-alpha-reactive CD4 T cells in A/J mice. Clin. Immunol. 2012, 144, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Basavalingappa, R.H.; Arumugam, R.; Lasrado, N.; Yalaka, B.; Massilamany, C.; Gangaplara, A.; Riethoven, J.J.; Xiang, S.H.; Steffen, D.; Reddy, J. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol. Immunol. 2020, 124, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Havari, E.; Pinto, S.; Gottumukkala, R.V.; Cornivelli, L.; Raddassi, K.; Matsui, T.; Rosenzweig, A.; Bronson, R.T.; Smith, R.; et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J. Clin. Investig. 2011, 121, 1561–1573. [Google Scholar] [CrossRef]
- Eriksson, U.; Ricci, R.; Hunziker, L.; Kurrer, M.O.; Oudit, G.Y.; Watts, T.H.; Sonderegger, I.; Bachmaier, K.; Kopf, M.; Penninger, J.M. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 2003, 9, 1484–1490. [Google Scholar] [CrossRef]
- Tajiri, K.; Imanaka-Yoshida, K.; Matsubara, A.; Tsujimura, Y.; Hiroe, M.; Naka, T.; Shimojo, N.; Sakai, S.; Aonuma, K.; Yasutomi, Y. Suppressor of cytokine signaling 1 DNA administration inhibits inflammatory and pathogenic responses in autoimmune myocarditis. J. Immunol. 2012, 189, 2043–2053. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.T.; Schwenzer, A.; Marsh, E.K.; Edwards, M.R.; Sabroe, I.; Midwood, K.S.; Parker, L.C. Airway Epithelial Cells Generate Pro-inflammatory Tenascin-C and Small Extracellular Vesicles in Response to TLR3 Stimuli and Rhinovirus Infection. Front. Immunol. 2019, 10, 1987. [Google Scholar] [CrossRef] [Green Version]
- Fouda, G.G.; Jaeger, F.H.; Amos, J.D.; Ho, C.; Kunz, E.L.; Anasti, K.; Stamper, L.W.; Liebl, B.E.; Barbas, K.H.; Ohashi, T.; et al. Tenascin-C is an innate broad-spectrum, HIV-1–neutralizing protein in breast milk. Proc. Natl. Acad. Sci. USA 2013, 110, 18220. [Google Scholar] [CrossRef] [Green Version]
- Mansour, R.G.; Stamper, L.; Jaeger, F.; McGuire, E.; Fouda, G.; Amos, J.; Barbas, K.; Ohashi, T.; Alam, S.M.; Erickson, H.; et al. The Presence and Anti-HIV-1 Function of Tenascin C in Breast Milk and Genital Fluids. PLoS ONE 2016, 11, e0155261. [Google Scholar] [CrossRef]
- Mangan, R.J.; Stamper, L.; Ohashi, T.; Eudailey, J.A.; Go, E.P.; Jaeger, F.H.; Itell, H.L.; Watts, B.E.; Fouda, G.G.; Erickson, H.P.; et al. Determinants of Tenascin-C and HIV-1 envelope binding and neutralization. Mucosal. Immunol. 2019, 12, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Khatun, M.; Steele, R.; Isbell, T.S.; Ray, R.; Ray, R.B. Exosomes from COVID-19 Patients Carry Tenascin-C and Fibrinogen-β in Triggering Inflammatory Signals in Cells of Distant Organ. Int. J. Mol. Sci. 2021, 22, 3184. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.L.; Faal, H.; Moss, D.; Addengast, L.; Fanta, L.; Eyster, K.; Huber, V.C.; Chaussee, M.S. The Streptococcus pyogenes fibronectin/tenascin-binding protein PrtF.2 contributes to virulence in an influenza superinfection. Sci. Rep. 2018, 8, 12126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leipner, C.; Grün, K.; Müller, A.; Buchdunger, E.; Borsi, L.; Kosmehl, H.; Berndt, A.; Janik, T.; Uecker, A.; Kiehntopf, M.; et al. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc. Res. 2008, 79, 118–126. [Google Scholar] [CrossRef]
- Sato, T.; Nakamori, S.; Watanabe, S.; Nishikawa, K.; Inoue, T.; Imanaka-Yoshida, K.; Ishida, M.; Sakuma, H.; Ito, M.; Dohi, K. Monitoring of the Evolution of Immune Checkpoint Inhibitor Myocarditis With Cardiovascular Magnetic Resonance. Circ. Cardiovasc. Imaging 2020, 13, e010633. [Google Scholar] [CrossRef]
- Tajiri, K.; Yonebayashi, S.; Li, S.; Ieda, M. Immunomodulatory Role of Tenascin-C in Myocarditis and Inflammatory Cardiomyopathy. Front. Immunol. 2021, 12, 624703. [Google Scholar] [CrossRef]
- Cihakova, D.; Rose, N.R. Pathogenesis of Myocarditis and Dilated Cardiomyopathy. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2008; Volume 99, pp. 95–114. [Google Scholar]
- Sonderegger, I.; Röhn, T.A.; Kurrer, M.O.; Iezzi, G.; Zou, Y.; Kastelein, R.A.; Bachmann, M.F.; Kopf, M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur. J. Immunol. 2006, 36, 2849–2856. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Orend, G.; Chiquet, M.; Tucker, R.P.; Midwood, K.S. Tenascins in stem cell niches. Matrix Biol. 2014, 37, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Erickson, H.P.; Springer, T.A. Tenascin supports lymphocyte rolling. J. Cell Biol. 1997, 137, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Okuno, Y.; Omatsu, Y.; Okabe, K.; Morimoto, J.; Uede, T.; Nagasawa, T.; Suda, T.; Kubota, Y. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood 2012, 119, 5429–5437. [Google Scholar] [CrossRef] [Green Version]
- Ellis, S.L.; Heazlewood, S.Y.; Williams, B.; Reitsma, A.J.; Grassinger, J.; Borg, J.; Heazlewood, C.K.; Chidgey, A.P.; Nilsson, S.K. The role of Tenascin C in the lymphoid progenitor cell niche. Exp. Hematol. 2013, 41, 1050–1061. [Google Scholar] [CrossRef]
- Nakahara, H.; Gabazza, E.C.; Fujimoto, H.; Nishii, Y.; D’Alessandro-Gabazza, C.N.; Bruno, N.E.; Takagi, T.; Hayashi, T.; Maruyama, J.; Maruyama, K.; et al. Deficiency of tenascin C attenuates allergen-induced bronchial asthma in the mouse. Eur. J. Immunol. 2006, 36, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Kurotaki, D.; Morimoto, J.; Asano, T.; Matsui, Y.; Nakayama, Y.; Saito, Y.; Ito, K.; Kimura, C.; Iwasaki, N.; et al. Alpha9 integrin and its ligands constitute critical joint microenvironments for development of autoimmune arthritis. J. Immunol. 2009, 182, 8015–8025. [Google Scholar] [CrossRef] [Green Version]
- Kanayama, M.; Morimoto, J.; Matsui, Y.; Ikesue, M.; Danzaki, K.; Kurotaki, D.; Ito, K.; Yoshida, T.; Uede, T. alpha9beta1 integrin-mediated signaling serves as an intrinsic regulator of pathogenic Th17 cell generation. J. Immunol. 2011, 187, 5851–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhmann, M.; Piccinini, A.M.; Kong, P.L.; Midwood, K.S. Endogenous activation of adaptive immunity: Tenascin-C drives interleukin-17 synthesis in murine arthritic joint disease. Arthritis Rheum 2012, 64, 2179–2190. [Google Scholar] [CrossRef]
- Matsumoto, K.I.; Aoki, H. The Roles of Tenascins in Cardiovascular, Inflammatory, and Heritable Connective Tissue Diseases. Front. Immunol. 2020, 11, 609752. [Google Scholar] [CrossRef]
- Tajiri, K.; Imanaka-Yoshida, K.; Tsujimura, Y.; Matsuo, K.; Hiroe, M.; Aonuma, K.; Ieda, M.; Yasutomi, Y. A New Mouse Model of Chronic Myocarditis Induced by Recombinant Bacille Calmette-Guèrin Expressing a T-Cell Epitope of Cardiac Myosin Heavy Chain-α. Int. J. Mol. Sci. 2021, 22, 794. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, M.; Dohi, K.; Imanaka-Yoshida, K.; Omori, T.; Moriwaki, K.; Nakamori, S.; Yamada, N.; Ito, M. Fulminant Myocarditis With Prolonged Active Lymphocytic Infiltration After Hemodynamic Recovery. Int. Heart J. 2017, 58, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bültmann, B.; Müller, T.; Lindinger, A.; Böhm, M. Predictors of Outcome in Patients With Suspected Myocarditis. Circulation 2008, 118, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Sugano, Y.; Yokokawa, T.; Nagai, T.; Matsuyama, T.-a.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Ohte, N.; et al. Clinical impact of the presence of macrophages in endomyocardial biopsies of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 490–498. [Google Scholar] [CrossRef]
- Yokokawa, T.; Sugano, Y.; Nakayama, T.; Nagai, T.; Matsuyama, T.A.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Yasuda, S.; et al. Significance of myocardial tenascin-C expression in left ventricular remodelling and long-term outcome in patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2016, 18, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, B.; Terasaki, F.; Shimomura, H.; Otsuka, K.; Otsuka, K.; Katashima, T.; Fujita, S.; Imanaka-Yoshida, K.; Yoshida, T.; Hiroe, M.; et al. High prevalence of chronic myocarditis in dilated cardiomyopathy referred for left ventriculoplasty: Expression of tenascin C as a possible marker for inflammation. Hum. Pathol. 2009, 40, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Odaka, K.; Uehara, T.; Imanaka-Yoshida, K.; Kato, Y.; Oyama, H.; Tadokoro, H.; Akizawa, H.; Tanada, S.; Hiroe, M.; et al. Toward in vivo imaging of heart disease using a radiolabeled single-chain Fv fragment targeting tenascin-C. Anal. Chem. 2011, 83, 9123–9130. [Google Scholar] [CrossRef] [PubMed]
- Dhaouadi, S.; Ben Abderrazek, R.; Loustau, T.; Abou-Faycal, C.; Ksouri, A.; Erne, W.; Murdamoothoo, D.; Mörgelin, M.; Kungl, A.; Jung, A.; et al. Novel Human Tenascin-C Function-Blocking Camel Single Domain Nanobodies. Front. Immunol. 2021, 12, 635166. [Google Scholar] [CrossRef]
- Sato, M.; Toyozaki, T.; Odaka, K.; Uehara, T.; Arano, Y.; Hasegawa, H.; Yoshida, K.; Imanaka-Yoshida, K.; Yoshida, T.; Hiroe, M.; et al. Detection of experimental autoimmune myocarditis in rats by 111In monoclonal antibody specific for tenascin-C. Circulation 2002, 106, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Taki, J.; Inaki, A.; Wakabayashi, H.; Imanaka-Yoshida, K.; Ogawa, K.; Hiroe, M.; Shiba, K.; Yoshida, T.; Kinuya, S. Dynamic expression of tenascin-C after myocardial ischemia and reperfusion: Assessment by 125I-anti-tenascin-C antibody imaging. J. Nucl. Med. 2010, 51, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Taki, J.; Inaki, A.; Wakabayashi, H.; Matsunari, I.; Imanaka-Yoshida, K.; Ogawa, K.; Hiroe, M.; Shiba, K.; Yoshida, T.; Kinuya, S. Effect of postconditioning on dynamic expression of tenascin-C and left ventricular remodeling after myocardial ischemia and reperfusion. EJNMMI Res. 2015, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Ageyama, N.; Kurosawa, H.; Fujimoto, O.; Uehara, T.; Hiroe, M.; Arano, Y.; Yoshida, T.; Yasutomi, Y.; Imanaka-Yoshida, K. Successful Inflammation Imaging of Non-Human Primate Hearts Using an Antibody Specific for Tenascin-C. Int. Heart J. 2019, 60, 151–158. [Google Scholar] [CrossRef]
- Terasaki, F.; Okamoto, H.; Onishi, K.; Sato, A.; Shimomura, H.; Tsukada, B.; Imanaka-Yoshida, K.; Hiroe, M.; Yoshida, T.; Kitaura, Y.; et al. Higher serum tenascin-C levels reflect the severity of heart failure, left ventricular dysfunction and remodeling in patients with dilated cardiomyopathy. Circ. J. 2007, 71, 327–330. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, N.; Onishi, K.; Sato, A.; Terasaki, F.; Tsukada, B.; Nozato, T.; Yamada, T.; Imanaka-Yoshida, K.; Yoshida, T.; Ito, M.; et al. Incremental prognostic values of serum tenascin-C levels with blood B-type natriuretic peptide testing at discharge in patients with dilated cardiomyopathy and decompensated heart failure. J. Card Fail. 2009, 15, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Kotby, A.A.; Abdel Aziz, M.M.; El Guindy, W.M.; Moneer, A.N. Can serum tenascin-C be used as a marker of inflammation in patients with dilated cardiomyopathy? Int. J. Pediatr. 2013, 2013, 608563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Matusiak-Bruckner, M.; Richter, P.; Grun, K.; Ziffels, B.; Neri, D.; Maschek, H.; Schulz, U.; Pfeil, A.; Jung, C.; et al. De novo expression of fetal ED-A(+) fibronectin and B (+) tenascin-C splicing variants in human cardiac allografts: Potential impact for targeted therapy of rejection. J. Mol. Histol. 2014, 45, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Gellen, B.; Thorin-Trescases, N.; Thorin, E.; Gand, E.; Sosner, P.; Brishoual, S.; Rigalleau, V.; Montaigne, D.; Javaugue, V.; Pucheu, Y.; et al. Serum tenascin-C is independently associated with increased major adverse cardiovascular events and death in individuals with type 2 diabetes: A French prospective cohort. Diabetologia 2020, 63, 915–923. [Google Scholar] [CrossRef]
- Kanagala, P.; Arnold, J.R.; Khan, J.N.; Singh, A.; Gulsin, G.S.; Chan, D.C.S.; Cheng, A.S.H.; Yang, J.; Li, Z.; Gupta, P.; et al. Plasma Tenascin-C: A prognostic biomarker in heart failure with preserved ejection fraction. Biomarkers 2020, 25, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2020. in print. [Google Scholar]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Ozmen, C.; Deniz, A.; Deveci, O.S.; Cagliyan, C.E.; Celik, A.I.; Yildiz, İ.; Yildiz, P.; Demir, M.; Kanadasi, M. Association among tenascin-C and NT-proBNP levels and arrhythmia prevalence in heart failure. Clin. Investig. Med. 2017, 40, E219–E227. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, Y.; Yokokawa, M.; Uzui, H.; Hisazaki, K.; Morishita, T.; Ishida, K.; Fukuoka, Y.; Hasegawa, K.; Ikeda, H.; Tama, N.; et al. Serum tenascin-C levels in atrium predict atrial structural remodeling processes in patients with atrial fibrillation. J. Interv. Card Electrophysiol. 2019, 59, 401–406. [Google Scholar] [CrossRef]
- Golledge, J.; Clancy, P.; Maguire, J.; Lincz, L.; Koblar, S. The role of tenascin C in cardiovascular disease. Cardiovasc. Res. 2011, 92, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.; Jung, C.; Lauten, A.; Figulla, H.R.; Berndt, A. Tenascin-C in cardiovascular remodeling: Potential impact for diagnosis, prognosis estimation and targeted therapy. Cell Adh. Migr. 2015, 9, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erer, H.B.; Guvenc, T.S.; Kemik, A.S.; Yilmaz, H.; Kul, S.; Altay, S.; Oz, D.; Zeren, G.; Ekmekci, A.; Zencirci, A.E.; et al. Assessment of tenascin-C levels in ventricular noncompaction/hypertrabeculation patients: A cross-sectional study. Echocardiography 2014, 31, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, H.; Kubo, T.; Baba, Y.; Yamasaki, N.; Matsumura, Y.; Furuno, T.; Doi, Y.L. Serum tenascin-C levels as a prognostic biomarker of heart failure events in patients with hypertrophic cardiomyopathy. J. Cardiol. 2012, 59, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokouchi, Y.; Oharaseki, T.; Enomoto, Y.; Sato, W.; Imanaka-Yoshida, K.; Takahashi, K. Expression of tenascin C in cardiovascular lesions of Kawasaki disease. Cardiovasc. Pathol. 2018, 38, 25–30. [Google Scholar] [CrossRef]
- Karatas, Z.; Baysal, T.; Alp, H.; Toker, A. Serum tenascin-C: A novel biomarker for diagnosis and predicting prognosis of rheumatic carditis? J. Trop Pediatr. 2013, 59, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Karatas, Z.; Baysal, T.; Sap, F.; Altin, H.; Cicekler, H. The role of tenascin-C and oxidative stress in rheumatic and congenital heart valve diseases: An observational study. Anadolu Kardiyol. Derg. 2013, 13, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Sarli, B.; Topsakal, R.; Kaya, E.G.; Akpek, M.; Lam, Y.Y.; Kaya, M.G. Tenascin-C as predictor of left ventricular remodeling and mortality in patients with dilated cardiomyopathy. J. Investig. Med. 2013, 61, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Yoshikane, Y.; Okuma, Y.; Miyamoto, T.; Hashimoto, J.; Fukazawa, R.; Kato, T.; Takeda, A.; Suda, K.; Hiroe, M.; Imanaka-Yoshida, K. Serum tenascin-C predicts resistance to steroid combination therapy in high-risk Kawasaki disease: A multicenter prospective cohort study. Pediatric Rhematol. 2020, in press. [Google Scholar]
- Sun, R.R.; Lu, L.; Liu, M.; Cao, Y.; Li, X.C.; Liu, H.; Wang, J.; Zhang, P.Y. Biomarkers and heart disease. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2927–2935. [Google Scholar]
- Kii, I. Practical Application of Periostin as a Biomarker for Pathological Conditions. Adv. Exp. Med. Biol. 2019, 1132, 195–204. [Google Scholar] [PubMed]
- Abdelaziz Mohamed, I.; Gadeau, A.P.; Hasan, A.; Abdulrahman, N.; Mraiche, F. Osteopontin: A Promising Therapeutic Target in Cardiac Fibrosis. Cells 2019, 8, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.L., Jr.; Kiernan, M.S.; et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e1054–e1091. [Google Scholar] [CrossRef]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probstmeier, R.; Montag, D.; Schachner, M. Galectin-3, a beta-galactoside-binding animal lectin, binds to neural recognition molecules. J. Neurochem. 1995, 64, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fujimoto, M.; Shiba, M.; Kawakita, F.; Liu, L.; Ichikawa, N.; Kanamaru, K.; Imanaka-Yoshida, K.; Yoshida, T. The Role of Matricellular Proteins in Brain Edema after Subarachnoid Hemorrhage. Acta Neurochir. Suppl. 2016, 121, 151–156. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imanaka-Yoshida, K. Tenascin-C in Heart Diseases—The Role of Inflammation. Int. J. Mol. Sci. 2021, 22, 5828. https://doi.org/10.3390/ijms22115828
Imanaka-Yoshida K. Tenascin-C in Heart Diseases—The Role of Inflammation. International Journal of Molecular Sciences. 2021; 22(11):5828. https://doi.org/10.3390/ijms22115828
Chicago/Turabian StyleImanaka-Yoshida, Kyoko. 2021. "Tenascin-C in Heart Diseases—The Role of Inflammation" International Journal of Molecular Sciences 22, no. 11: 5828. https://doi.org/10.3390/ijms22115828
APA StyleImanaka-Yoshida, K. (2021). Tenascin-C in Heart Diseases—The Role of Inflammation. International Journal of Molecular Sciences, 22(11), 5828. https://doi.org/10.3390/ijms22115828