
Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Erythrocytes—Simple, Vital and Fragile Cells
2.1. Main Features and Functions
2.2. Erythrocyte Membranes Exhibit Specific Features
2.3. Erythrocytes Are Subject to Haemolysis
2.3.1. Intra- or Extravascular Haemolysis with Erythrocyte Rupture Lysis
2.3.2. Intracellular Haemolysis or Erythrocyte Phagocytosis
Physiological EP Process and Tissue Macrophages
Erythrocyte Binding and Erythrophagocytosis by Non-Specialised Cells
3. Atherosclerosis, a Pathology in Three Steps
3.1. Fatty Streak Development
3.2. Fibrous Cap Formation
3.3. Atherosclerotic Plaque Complications
4. Involvement of Erythrocytes in the Progression to Atherothrombosis
4.1. Erythrocytes Can Contribute to ATH Progression and Complications
4.1.1. Intraplaque Haemorrhage
4.1.2. Erythrocytes, a Source of Cholesterol That Fuels Plaque Development and Complications
4.1.3. Erythrocytes in the Plaque: Source of Damage-Associated Molecular Pattern with Pathological Consequences
Haemoglobin and Heme
Iron
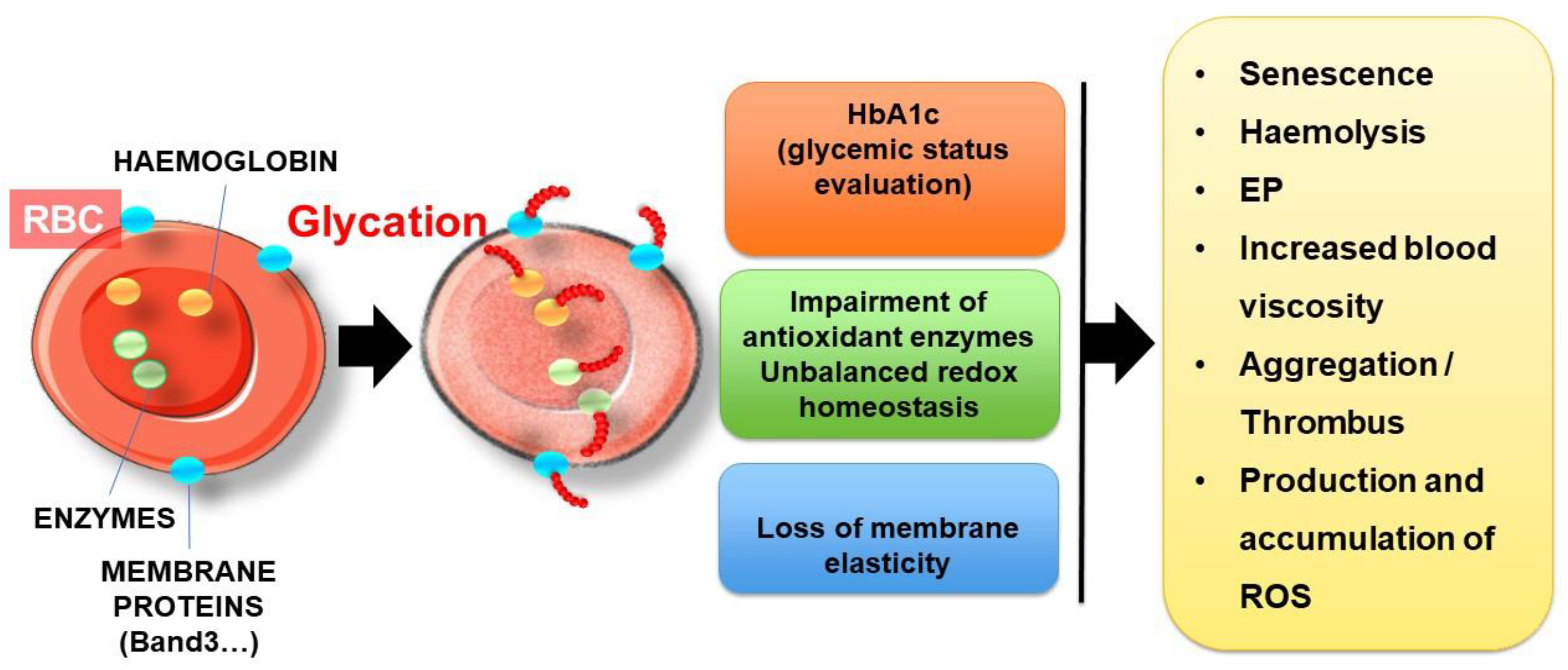
4.2. Enhanced Proatherothrombotic Capacity of Glycated Erythrocytes
4.2.1. Erythrocyte Protein Glycation
Haemoglobin
Membrane Proteins
Erythrocyte Enzymes
4.2.2. Consequences of Glycation: How Erythrocyte Glycation Is Involved in Atherothrombosis
Loss of Deformability and Aggregability
Senescence and Haemolysis of Glycated Erythrocytes
Clearance and Erythrophagocytosis (EP)
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATH | atherosclerosis |
| RBCs | red blood cells |
| EP | erythrophagocytosis |
| LDL | low-density lipoprotein |
| oxLDL | oxidised low-density lipoprotein |
| VSMC | Vascular smooth muscle cells |
| VCAM1 | vascular cell adhesion molecule 1 |
| ICAM1 | Intercellular adhesion molecule1 |
| Hb | haemoglobin |
| oxyHb | oxygenated form of human haemoglobin (or ferro Hb) |
| Hp | haptoglobin |
| Hx | hemopexin |
| HMOX1 | heme oxygenase 1 |
| Fpn | ferroportin |
| Hepc | hepcidin |
| PS | phosphatidylserine |
| DAMPs | Damage-Associated Signalling Molecule Patterns |
| NO | nitric oxide |
| eNOS | endothelial nitric oxide synthase |
| ROS | reactive oxygen species |
| O2 | superoxide anion |
| H2O2 | hydrogen peroxide |
| HO | hydroxyl radical |
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial Infarction-From Atherosclerosis to Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e176–e185. [Google Scholar]
- Michel, J.B.; Martin-Ventura, J.L. Red Blood Cells and Hemoglobin in Human Atherosclerosis and Related Arterial Diseases. Int. J. Mol. Sci. 2020, 21, 6756. [Google Scholar]
- Pasterkamp, G.; Virmani, R. The erythrocyte: A new player in atheromatous core formation. Heart 2002, 88, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F. How do red blood cells know when to die? R. Soc. Open Sci. 2017, 4, 160850. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Markers of oxidative stress in erythrocytes and plasma during aging in humans. Oxid. Med. Cell. Longev. 2010, 3, 2–12. [Google Scholar] [PubMed]
- Hamidi, M.; Tajerzadeh, H. Carrier erythrocytes: An overview. Drug Deliv. 2003, 10, 9–20. [Google Scholar] [PubMed]
- Cimen, M.Y. Free radical metabolism in human erythrocytes. Clin. Chim. Acta 2008, 390, 1–11. [Google Scholar] [CrossRef]
- Sztiller, M.; Puchala, M.; Kowalczyk, A.; Bartosz, G. The influence of ferrylhemoglobin and methemoglobin on the human erythrocyte membrane. Redox Rep. 2006, 11, 263–271. [Google Scholar]
- Dumaswala, U.J.; Zhuo, L.; Jacobsen, D.W.; Jain, S.K.; Sukalski, K.A. Protein and lipid oxidation of banked human erythrocytes: Role of glutathione. Free Radic. Biol. Med. 1999, 27, 1041–1049. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Madrigal-Matute, J.; Martinez-Pinna, R.; Ramos-Mozo, P.; Blanco-Colio, L.M.; Moreno, J.A.; Tarin, C.; Burillo, E.; Fernandez-Garcia, C.E.; Egido, J.; et al. Erythrocytes, leukocytes and platelets as a source of oxidative stress in chronic vascular diseases: Detoxifying mechanisms and potential therapeutic options. Thromb. Haemost. 2012, 108, 435–442. [Google Scholar]
- Baynes, J.W. Oxygen and life. In Medical Biochemistry, 2nd ed.; Baynes, J.W., Dominiczak, M.H., Eds.; Elsevier Mosby: Philadelphia, PA, USA, 2005; pp. 497–506. [Google Scholar]
- Abugo, O.O.; Rifkind, J.M. Oxidation of hemoglobin and the enhancement produced by nitroblue tetrazolium. J. Biol. Chem. 1994, 269, 24845–24853. [Google Scholar] [CrossRef]
- Dei Zotti, F.; Verdoy, R.; Brusa, D.; Lobysheva, I.I.; Balligand, J.L. Redox regulation of nitrosyl-hemoglobin in human erythrocytes. Redox Biol. 2020, 34, 101399. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Whillier, S.; Raftos, J.E.; Kuchel, P.W. Glutathione synthesis by red blood cells in type 2 diabetes mellitus. Redox Rep. 2008, 13, 277–282. [Google Scholar] [CrossRef]
- Aoshiba, K.; Nakajima, Y.; Yasui, S.; Tamaoki, J.; Nagai, A. Red blood cells inhibit apoptosis of human neutrophils. Blood 1999, 93, 4006–4010. [Google Scholar] [CrossRef]
- Lynch, R.E.; Fridovich, I. Permeation of the erythrocyte stroma by superoxide radical. J. Biol. Chem. 1978, 253, 4697–4699. [Google Scholar] [CrossRef]
- Pretorius, E. Erythrocyte deformability and eryptosis during inflammation, and impaired blood rheology. Clin. Hemorheol. Microcirc. 2018, 69, 545–550. [Google Scholar] [CrossRef]
- Nayak, B.S.; Beharry, V.Y.; Armoogam, S.; Nancoo, M.; Ramadhin, K.; Ramesar, K.; Ramnarine, C.; Singh, A.; Singh, A.; Nwachi, K.U.; et al. Determination of RBC membrane and serum lipid composition in Trinidadian type II diabetics with and without nephropathy. Vasc. Health Risk Manag. 2008, 4, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, D.; Williamson, P.; Schlegel, R.A. Bilayer/cytoskeleton interactions in lipid-symmetric erythrocytes assessed by a photoactivable phospholipid analogue. Biochemistry 1991, 30, 7754–7758. [Google Scholar] [CrossRef]
- Sarkar, S.; Bose, D.; Giri, R.P.; Mukhopadhyay, M.K.; Chakrabarti, A. Status of Membrane Asymmetry in Erythrocytes: Role of Spectrin. Adv. Exp. Med. Biol. 2018, 1112, 3–11. [Google Scholar]
- Bakan, E.; Yildirim, A.; Kurtul, N.; Polat, M.F.; Dursun, H.; Cayir, K. Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006, 43, 109–113. [Google Scholar] [CrossRef]
- Bryszewska, M.; Watala, C.; Torzecka, W. Changes in fluidity and composition of erythrocyte membranes and in composition of plasma lipids in type I diabetes. Br. J. Haematol. 1986, 62, 111–116. [Google Scholar] [CrossRef]
- Miossec, P.; Zkhiri, F.; Paries, J.; David-Dufilho, M.; Devynck, M.A.; Valensi, P.E. Effect of pravastatin on erythrocyte rheological and biochemical properties in poorly controlled Type 2 diabetic patients. Diabet Med. 1999, 16, 424–430. [Google Scholar] [CrossRef]
- Podsiedlik, M.; Markowicz-Piasecka, M.; Sikora, J. Erythrocytes as model cells for biocompatibility assessment, cytotoxicity screening of xenobiotics and drug delivery. Chem. Biol. Interact. 2020, 332, 109305. [Google Scholar] [CrossRef]
- Machnicka, B.; Czogalla, A.; Hryniewicz-Jankowska, A.; Boguslawska, D.M.; Grochowalska, R.; Heger, E.; Sikorski, A.F. Spectrins: A structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta 2014, 1838, 620–634. [Google Scholar] [CrossRef]
- Reithmeier, R.A.; Casey, J.R.; Kalli, A.C.; Sansom, M.S.; Alguel, Y.; Iwata, S. Band 3, the human red cell chloride/bicarbonate anion exchanger (AE1, SLC4A1), in a structural context. Biochim. Biophys. Acta 2016, 1858, 1507–1532. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R.; Marino, A. Natural Antioxidants Beneficial Effects on Anion Exchange through Band 3 Protein in Human Erythrocytes. Antioxidants 2019, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Low, P.S. Regulation of the glycophorin C-protein 4.1 membrane-to-skeleton bridge and evaluation of its contribution to erythrocyte membrane stability. J. Biol. Chem. 2001, 276, 22223–22230. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible binding of hemoglobin to band 3 constitutes the molecular switch that mediates O2 regulation of erythrocyte properties. Blood 2016, 128, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Ang, B.; Balne, P.K.; Richards, C.; Smart, T.; Cardoso, J.; Shima, D.; Jones, P.H.; Pavesio, C. Non-Occlusive Retinal Vascular Inflammation and Role of Red Blood Cell Deformability in Birdshot Chorioretinopathy. Ocul. Immunol. Inflamm. 2019, 27, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Smart, T.; Nobre-Cardoso, J.; Richards, C.; Bhatnagar, R.; Tufail, A.; Shima, D.; Jones, P.H.; Pavesio, C. Assessment of red blood cell deformability in type 2 diabetes mellitus and diabetic retinopathy by dual optical tweezers stretching technique. Sci. Rep. 2016, 6, 15873. [Google Scholar] [CrossRef] [PubMed]
- Rebsomen, L.; Tsimaratos, M. Association of reduced red blood cell deformability and diabetic nephropathy. Kidney Int. 2005, 67, 2066; author reply 2066–2067. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; Shin, S. Advances in the measurement of red blood cell deformability: A brief review. J. Cell. Biotechnol. 2015, 1, 63–79. [Google Scholar] [CrossRef]
- Buys, A.V.; Van Rooy, M.J.; Soma, P.; Van Papendorp, D.; Lipinski, B.; Pretorius, E. Changes in red blood cell membrane structure in type 2 diabetes: A scanning electron and atomic force microscopy study. Cardiovasc. Diabetol. 2013, 12, 25. [Google Scholar] [CrossRef]
- Chang, H.Y.; Li, X.; Karniadakis, G.E. Modeling of Biomechanics and Biorheology of Red Blood Cells in Type 2 Diabetes Mellitus. Biophys. J. 2017, 113, 481–490. [Google Scholar] [CrossRef]
- McNamee, A.P.; Richardson, K.; Horobin, J.; Kuck, L.; Simmonds, M.J. Susceptibility of density-fractionated erythrocytes to subhaemolytic mechanical shear stress. Int. J. Artif. Organs 2019, 42, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid. Med. Cell. Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed]
- Nola, M.; Dotlic, S.; Damjanov, I. Chapter 9—The Hematopoietic and Lymphoid Systems. In Pathology Secrets, 3rd ed.; Mosby: Philadelphia, PA, USA, 2009; pp. 161–202. [Google Scholar]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. BioMed Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell. Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Lang, F.; Lang, K.S.; Lang, P.A.; Huber, S.M.; Wieder, T. Mechanisms and significance of eryptosis. Antioxid. Redox Signal. 2006, 8, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Bratosin, D.; Estaquier, J.; Ameisen, J.C.; Montreuil, J. Molecular and cellular mechanisms of erythrocyte programmed cell death: Impact on blood transfusion. Vox Sang. 2002, 83 (Suppl. 1), 307–310. [Google Scholar] [CrossRef]
- Dasgupta, S.K.; Abdel-Monem, H.; Guchhait, P.; Nagata, S.; Thiagarajan, P. Role of lactadherin in the clearance of phosphatidylserine-expressing red blood cells. Transfusion 2008, 48, 2370–2376. [Google Scholar] [CrossRef]
- Kuypers, F.A.; de Jong, K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell. Mol. Biol. 2004, 50, 147–158. [Google Scholar] [PubMed]
- Klei, T.R.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Roy, M.F.; Riendeau, N.; Bedard, C.; Helie, P.; Min-Oo, G.; Turcotte, K.; Gros, P.; Canonne-Hergaux, F.; Malo, D. Pyruvate kinase deficiency confers susceptibility to Salmonella typhimurium infection in mice. J. Exp. Med. 2007, 204, 2949–2961. [Google Scholar] [CrossRef]
- Tucker, R.M.; Williams, P.L.; Arathoon, E.G.; Stevens, D.A. Treatment of mycoses with itraconazole. Ann. N. Y. Acad. Sci. 1988, 544, 451–470. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell. Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell. Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Pilard, N.; Goncalves, A.S.; Beaumont, C.; Canonne-Hergaux, F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005, 106, 3979–3984. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell. Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Ross, S.L.; Tran, L.; Winters, A.; Lee, K.J.; Plewa, C.; Foltz, I.; King, C.; Miranda, L.P.; Allen, J.; Beckman, H.; et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell. Metab. 2012, 15, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.P.; Meynard, D.; Coppin, H. Regulators of hepcidin expression. Vitam. Horm. 2019, 110, 101–129. [Google Scholar] [PubMed]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, S.Y.; Jung, M.Y.; Bae, S.M.; Kim, I.S. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood 2011, 117, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, N.; Ahuja, K.; Selvapandiyan, A.; Dey, R.; Nakhasi, H.; Puri, N. Role of Mast Cells in clearance of Leishmania through extracellular trap formation. Sci. Rep. 2017, 7, 13240. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Puri, N. A new role for mast cells as scavengers for clearance of erythrocytes damaged due to oxidative stress. Immunol. Lett. 2018, 199, 23–35. [Google Scholar] [CrossRef]
- Catan, A.; Turpin, C.; Diotel, N.; Patche, J.; Guerin-Dubourg, A.; Debussche, X.; Bourdon, E.; Ah-You, N.; Le Moullec, N.; Besnard, M.; et al. Aging and glycation promote erythrocyte phagocytosis by human endothelial cells: Potential impact in atherothrombosis under diabetic conditions. Atherosclerosis 2019, 291, 87–98. [Google Scholar] [CrossRef]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiol. 2015, 213, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Bayles, R.G.; Laschet, J.; Ollivier, V.; Ho-Tin-Noe, B.; Touat, Z.; Deschildre, C.; Morvan, M.; Louedec, L.; Gouya, L.; et al. Erythrocyte Efferocytosis by the Arterial Wall Promotes Oxidation in Early-Stage Atheroma in Humans. Front. Cardiovasc. Med. 2017, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Kolb, S.; Vranckx, R.; Huisse, M.G.; Michel, J.B.; Meilhac, O. The phosphatidylserine receptor mediates phagocytosis by vascular smooth muscle cells. J. Pathol. 2007, 212, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: Molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef]
- Wentzel, J.J.; Chatzizisis, Y.S.; Gijsen, F.J.; Giannoglou, G.D.; Feldman, C.L.; Stone, P.H. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: Current understanding and remaining questions. Cardiovasc. Res. 2012, 96, 234–243. [Google Scholar] [CrossRef]
- Plutzky, J. The vascular biology of atherosclerosis. Am. J. Med. 2003, 115 (Suppl. 8A), 55S–61S. [Google Scholar] [CrossRef]
- Čejková, S.; Králová-Lesná, I.; Poledne, R. Monocyte adhesion to the endothelium is an initial stage of atherosclerosis development. Cor Vasa 2016, 58, e419–e425. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Willis, A.I.; Pierre-Paul, D.; Sumpio, B.E.; Gahtan, V. Vascular smooth muscle cell migration: Current research and clinical implications. Vasc. Endovascular. Surg. 2004, 38, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Rudijanto, A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med. Indones. 2007, 39, 86–93. [Google Scholar]
- Ferns, G.A.A.; Heikal, L. Hypoxia in Atherogenesis. Angiology 2017, 68, 472–493. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lu, X.; Shi, G.P. Vasa vasorum in atherosclerosis and clinical significance. Int. J. Mol. Sci. 2015, 16, 11574–11608. [Google Scholar] [CrossRef]
- Ho, T.K.; Abraham, D.J.; Black, C.M.; Baker, D.M. Hypoxia-inducible factor 1 in lower limb ischemia. Vascular 2006, 14, 321–327. [Google Scholar] [CrossRef]
- Cheng, C.; Chrifi, I.; Pasterkamp, G.; Duckers, H.J. Biological mechanisms of microvessel formation in advanced atherosclerosis: The big five. Trends Cardiovasc. Med. 2013, 23, 153–164. [Google Scholar] [CrossRef]
- Iwasaka, C.; Tanaka, K.; Abe, M.; Sato, Y. Ets-1 regulates angiogenesis by inducing the expression of urokinase-type plasminogen activator and matrix metalloproteinase-1 and the migration of vascular endothelial cells. J. Cell. Physiol. 1996, 169, 522–531. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Yeagle, P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Arbustini, E. Total erythrocyte membrane cholesterol: An innocent new marker or an active player in acute coronary syndromes? J. Am. Coll. Cardiol. 2007, 49, 2090–2092. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schroll, A.; Demetz, E.; Tancevski, I.; Theurl, I.; Weiss, G. ‘Ride on the ferrous wheel’—The cycle of iron in macrophages in health and disease. Immunobiology 2015, 220, 280–294. [Google Scholar] [CrossRef]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.L.; Ritter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Kuhn, V.; Diederich, L.; Keller, T.C.S.T.; Kramer, C.M.; Luckstadt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red Blood Cell Function and Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid. Redox Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef]
- Helms, C.C.; Gladwin, M.T.; Kim-Shapiro, D.B. Erythrocytes and Vascular Function: Oxygen and Nitric Oxide. Front. Physiol. 2018, 9, 125. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Zhou, Z.; Mahdi, A.; Tratsiakovich, Y.; Zahoran, S.; Kovamees, O.; Nordin, F.; Uribe Gonzalez, A.E.; Alvarsson, M.; Ostenson, C.G.; Andersson, D.C.; et al. Erythrocytes From Patients With Type 2 Diabetes Induce Endothelial Dysfunction Via Arginase I. J. Am. Coll. Cardiol. 2018, 72, 769–780. [Google Scholar] [CrossRef]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Tan, Y.A.; Gao, Q.F.; Li, S.Q.; Zhang, J.; Chen, Q.G.; Jiang, Y.H.; Zhang, L.; Ying, H.Q.; Wang, X.Z. Circulating fibrinogen to pre-albumin ratio is a promising biomarker for diagnosis of colorectal cancer. J. Clin. Lab. Anal. 2018, 33, e22635. [Google Scholar] [CrossRef]
- Julius, U.; Pietzsch, J. Glucose-induced enhancement of hemin-catalyzed LDL oxidation in vitro and in vivo. Antioxid. Redox Signal. 2005, 7, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Potor, L.; Banyai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid Med. Cell. Longev. 2013, 2013, 676425. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef]
- Smith, C.; Mitchinson, M.J.; Aruoma, O.I.; Halliwell, B. Stimulation of lipid peroxidation and hydroxyl-radical generation by the contents of human atherosclerotic lesions. Biochem J. 1992, 286 Pt 3, 901–905. [Google Scholar] [CrossRef]
- Pang, J.H.; Jiang, M.J.; Chen, Y.L.; Wang, F.W.; Wang, D.L.; Chu, S.H.; Chau, L.Y. Increased ferritin gene expression in atherosclerotic lesions. J. Clin. Investig. 1996, 97, 2204–2212. [Google Scholar] [CrossRef]
- Lee, T.S.; Lee, F.Y.; Pang, J.H.; Chau, L.Y. Erythrophagocytosis and iron deposition in atherosclerotic lesions. Chin. J. Physiol. 1999, 42, 17–23. [Google Scholar]
- Li, W.; Xu, L.H.; Yuan, X.M. Macrophage hemoglobin scavenger receptor and ferritin accumulation in human atherosclerotic lesions. Ann. N. Y. Acad. Sci. 2004, 1030, 196–201. [Google Scholar] [CrossRef]
- Yuan, X.M.; Li, W.; Baird, S.K.; Carlsson, M.; Melefors, O. Secretion of ferritin by iron-laden macrophages and influence of lipoproteins. Free Radic. Res. 2004, 38, 1133–1142. [Google Scholar] [CrossRef]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, A.T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef]
- Sullivan, J.L. Iron in arterial plaque: Modifiable risk factor for atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.C.; McNamara, P.M.; Gordon, T. Menopause and risk of cardiovascular disease: The Framingham study. Ann. Intern. Med. 1976, 85, 447–452. [Google Scholar] [CrossRef]
- Sullivan, J.L. Are menstruating women protected from heart disease because of, or in spite of, estrogen? Relevance to the iron hypothesis. Am. Heart J. 2003, 145, 190–194. [Google Scholar] [CrossRef]
- Ascherio, A.; Rimm, E.B.; Giovannucci, E.; Willett, W.C.; Stampfer, M.J. Blood donations and risk of coronary heart disease in men. Circulation 2001, 103, 52–57. [Google Scholar] [CrossRef]
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211. [Google Scholar] [CrossRef]
- Ponraj, D.; Makjanic, J.; Thong, P.S.; Tan, B.K.; Watt, F. The onset of atherosclerotic lesion formation in hypercholesterolemic rabbits is delayed by iron depletion. FEBS Lett. 1999, 459, 218–222. [Google Scholar] [CrossRef]
- Zheng, H.; Cable, R.; Spencer, B.; Votto, N.; Katz, S.D. Iron stores and vascular function in voluntary blood donors. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Makowski, M.R.; Varma, G.; Wiethoff, A.J.; Smith, A.; Mattock, K.; Jansen, C.H.; Warley, A.; Taupitz, M.; Schaeffter, T.; Botnar, R.M. Noninvasive assessment of atherosclerotic plaque progression in ApoE−/− mice using susceptibility gradient mapping. Circ. Cardiovasc. Imaging 2011, 4, 295–303. [Google Scholar] [CrossRef]
- Sempos, C.T. Do body iron stores increase the risk of developing coronary heart disease? Am. J. Clin. Nutr. 2002, 76, 501–503. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sullivan, J.L. Misconceptions in the debate on the iron hypothesis. J. Nutr. Biochem. 2001, 12, 33–37. [Google Scholar] [CrossRef]
- Sullivan, J.L. Blood donation without adequate iron depletion: An invalid test of the iron hypothesis. Circulation 2001, 104, E149. [Google Scholar] [CrossRef][Green Version]
- Sullivan, J.L. Macrophage iron, hepcidin, and atherosclerotic plaque stability. Exp. Biol. Med. 2007, 232, 1014–1020. [Google Scholar] [CrossRef]
- Xu, S. Iron and Atherosclerosis: The Link Revisited. Trends Mol. Med. 2019, 25, 659–661. [Google Scholar] [CrossRef]
- Saeed, O.; Otsuka, F.; Polavarapu, R.; Karmali, V.; Weiss, D.; Davis, T.; Rostad, B.; Pachura, K.; Adams, L.; Elliott, J.; et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Meng, X.; Si, H.P.; Zhang, C.; Lv, H.X.; Zhao, Y.X.; Yang, J.M.; Dong, M.; Zhang, K.; Liu, S.X.; et al. Hepcidin destabilizes atherosclerotic plaque via overactivating macrophages after erythrophagocytosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Wunderer, F.; Barnes, H.J.; Bagchi, A.; Buswell, M.D.; O’Rourke, C.D.; Slocum, C.L.; Ledsky, C.D.; Peneyra, K.M.; Sigurslid, H.; et al. Hepcidin Deficiency Protects Against Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Marques, L.; Negre-Salvayre, A.; Costa, L.; Canonne-Hergaux, F. Iron gene expression profile in atherogenic Mox macrophages. Biochim. Biophys. Acta 2016, 1862, 1137–1146. [Google Scholar] [CrossRef]
- Xiao, L.; Luo, G.; Guo, X.; Jiang, C.; Zeng, H.; Zhou, F.; Li, Y.; Yu, J.; Yao, P. Macrophage iron retention aggravates atherosclerosis: Evidence for the role of autocrine formation of hepcidin in plaque macrophages. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2020, 1865, 158531. [Google Scholar] [CrossRef]
- Kraml, P.J.; Klein, R.L.; Huang, Y.; Nareika, A.; Lopes-Virella, M.F. Iron loading increases cholesterol accumulation and macrophage scavenger receptor I expression in THP-1 mononuclear phagocytes. Metabolism 2005, 54, 453–459. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, M.; Liu, Y.; Li, H.; Shang, L.; Xu, T.; Chen, Z.; Wang, F.; Qiao, T.; Li, K. Iron accumulation in macrophages promotes the formation of foam cells and development of atherosclerosis. Cell. Biosci. 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Cianetti, L.; Segnalini, P.; Calzolari, A.; Morsilli, O.; Felicetti, F.; Ramoni, C.; Gabbianelli, M.; Testa, U.; Sposi, N.M. Expression of alternative transcripts of ferroportin-1 during human erythroid differentiation. Haematologica 2005, 90, 1595–1606. [Google Scholar]
- Zhang, D.L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell. Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Wu, J.; Shah, B.N.; Greutelaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Muckenthaler, M.U.; Da Silva, M.C.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis and iron: From epidemiology to cellular level. Front. Pharmacol. 2014, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.M.; Anders, W.L.; Olsson, A.G.; Brunk, U.T. Iron in human atheroma and LDL oxidation by macrophages following erythrophagocytosis. Atherosclerosis 1996, 124, 61–73. [Google Scholar] [CrossRef]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Levine, K.B.; Robichaud, T.K.; Hamill, S.; Sultzman, L.A.; Carruthers, A. Properties of the human erythrocyte glucose transport protein are determined by cellular context. Biochemistry 2005, 44, 5606–5616. [Google Scholar] [CrossRef]
- Manno, S.; Mohandas, N.; Takakuwa, Y. ATP-dependent mechanism protects spectrin against glycation in human erythrocytes. J. Biol. Chem. 2010, 285, 33923–33929. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Marini, M.A.; Succurro, E.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; Perticone, F.; Sesti, G. Association between hemoglobin glycation index and hepatic steatosis in non-diabetic individuals. Diabetes Res. Clin. Pract. 2017, 134, 53–61. [Google Scholar] [CrossRef]
- Nayak, A.U.; Singh, B.M.; Dunmore, S.J. Potential Clinical Error Arising From Use of HbA1c in Diabetes: Effects of the Glycation Gap. Endocr. Rev. 2019, 40, 988–999. [Google Scholar] [CrossRef]
- Welsh, K.J.; Kirkman, M.S.; Sacks, D.B. Role of Glycated Proteins in the Diagnosis and Management of Diabetes: Research Gaps and Future Directions. Diabetes Care 2016, 39, 1299–1306. [Google Scholar] [CrossRef]
- Delpierrre, G.; Vertommen, D.; Communi, D.; Rider, M.H.; Van Schaftingen, E. Identification of fructosamine residues deglycated by fructosamine-3-kinase in human hemoglobin. J. Biol. Chem. 2004, 279, 27613–27620. [Google Scholar] [CrossRef]
- Shapiro, R.; McManus, M.J.; Zalut, C.; Bunn, H.F. Sites of nonenzymatic glycosylation of human hemoglobin A. J. Biol. Chem. 1980, 255, 3120–3127. [Google Scholar] [CrossRef]
- Wang, S.H.; Wang, T.F.; Wu, C.H.; Chen, S.H. In-depth comparative characterization of hemoglobin glycation in normal and diabetic bloods by LC-MSMS. J. Am. Soc. Mass Spectrom. 2014, 25, 758–766. [Google Scholar] [CrossRef]
- Muralidharan, M.; Bhat, V.; Bindu, Y.S.; Mandal, A.K. Glycation profile of minor abundant erythrocyte proteome across varying glycemic index in diabetes mellitus. Anal. Biochem. 2019, 573, 37–43. [Google Scholar] [CrossRef]
- Zhang, Q.; Tang, N.; Schepmoes, A.A.; Phillips, L.S.; Smith, R.D.; Metz, T.O. Proteomic profiling of nonenzymatically glycated proteins in human plasma and erythrocyte membranes. J. Proteome Res. 2008, 7, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Grossin, N.; Wautier, M.P.; Picot, J.; Stern, D.M.; Wautier, J.L. Differential effect of plasma or erythrocyte AGE-ligands of RAGE on expression of transcripts for receptor isoforms. Diabetes Metab. 2009, 35, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Monroe, M.E.; Schepmoes, A.A.; Clauss, T.R.; Gritsenko, M.A.; Meng, D.; Petyuk, V.A.; Smith, R.D.; Metz, T.O. Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J. Proteome Res. 2011, 10, 3076–3088. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 1988, 318, 1315–1321. [Google Scholar]
- Awasthi, S.; Gayathiri, S.K.; Ramya, R.; Duraichelvan, R.; Dhason, A.; Saraswathi, N.T. Advanced Glycation-Modified Human Serum Albumin Evokes Alterations in Membrane and Eryptosis in Erythrocytes. Appl. Biochem. Biotechnol. 2015, 177, 1013–1024. [Google Scholar] [CrossRef]
- Ishida, Y.I.; Kayama, T.; Kibune, Y.; Nishimoto, S.; Koike, S.; Suzuki, T.; Horiuchi, Y.; Miyashita, M.; Itokawa, M.; Arai, M.; et al. Identification of an argpyrimidine-modified protein in human red blood cells from schizophrenic patients: A possible biomarker for diseases involving carbonyl stress. Biochem. Biophys. Res. Commun. 2017, 493, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Nagai, R.; Deemer, E.K.; Brock, J.W.; Thorpe, S.R.; Baynes, J.W. Effect of glucose concentration on formation of AGEs in erythrocytes in vitro. Ann. N. Y. Acad. Sci. 2005, 1043, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Beppu, M.; Kikugawa, K.; Nagai, R.; Horiuchi, S. Membrane proteins of human erythrocytes are modified by advanced glycation end products during aging in the circulation. Biochem. Biophys. Res. Commun. 1999, 258, 123–127. [Google Scholar] [CrossRef]
- Bookchin, R.M.; Etzion, Z.; Lew, V.L.; Tiffert, T. Preserved function of the plasma membrane calcium pump of red blood cells from diabetic subjects with high levels of glycated haemoglobin. Cell. Calcium. 2009, 45, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Lim, G. Pyridoxine and pyridoxamine inhibits superoxide radicals and prevents lipid peroxidation, protein glycosylation, and (Na+ + K+)-ATPase activity reduction in high glucose-treated human erythrocytes. Free Radic. Biol. Med. 2001, 30, 232–237. [Google Scholar] [CrossRef]
- Gonzalez Flecha, F.L.; Castello, P.R.; Gagliardino, J.J.; Rossi, J.P.F.C. Molecular Characterization of the Glycated Plasma Membrane Calcium Pump. J. Membr. Biol. 1999, 171, 25–34. [Google Scholar] [CrossRef]
- Turpin, C.; Catan, A.; Guerin-Dubourg, A.; Debussche, X.; Bravo, S.B.; Alvarez, E.; Van Den Elsen, J.; Meilhac, O.; Rondeau, P.; Bourdon, E. Enhanced oxidative stress and damage in glycated erythrocytes. PLoS ONE 2020, 15, e0235335. [Google Scholar] [CrossRef]
- Kucherenko, Y.V.; Bhavsar, S.K.; Grischenko, V.I.; Fischer, U.R.; Huber, S.M.; Lang, F. Increased cation conductance in human erythrocytes artificially aged by glycation. J. Membr. Biol. 2010, 235, 177–189. [Google Scholar] [CrossRef]
- Viskupicova, J.; Blaskovic, D.; Galiniak, S.; Soszynski, M.; Bartosz, G.; Horakova, L.; Sadowska-Bartosz, I. Effect of high glucose concentrations on human erythrocytes in vitro. Redox Biol. 2015, 5, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Nandhini, T.A.; Anuradha, C.V. Inhibition of lipid peroxidation, protein glycation and elevation of membrane ion pump activity by taurine in RBC exposed to high glucose. Clin. Chim. Acta 2003, 336, 129–135. [Google Scholar] [CrossRef]
- Gonzalez Flecha, F.L.; Bermudez, M.C.; Cedola, N.V.; Gagliardino, J.J.; Rossi, J.P. Decreased Ca2(+)-ATPase activity after glycosylation of erythrocyte membranes in vivo and in vitro. Diabetes 1990, 39, 707–711. [Google Scholar] [CrossRef]
- Arai, K.; Maguchi, S.; Fujii, S.; Ishibashi, H.; Oikawa, K.; Taniguchi, N. Glycation and inactivation of human Cu-Zn-superoxide dismutase. Identification of the in vitro glycated sites. J. Biol. Chem. 1987, 262, 16969–16972. [Google Scholar] [CrossRef]
- Manuel y Keenoy, B.; Vertommen, J.; De Leeuw, I. Divergent effects of different oxidants on glutathione homeostasis and protein damage in erythrocytes from diabetic patients: Effects of high glucose. Mol. Cell. Biochem. 2001, 225, 59–73. [Google Scholar] [CrossRef]
- Dincer, Y.; Akcay, T.; Alademir, Z.; Ilkova, H. Effect of oxidative stress on glutathione pathway in red blood cells from patients with insulin-dependent diabetes mellitus. Metabolism 2002, 51, 1360–1362. [Google Scholar] [CrossRef]
- Murakami, K.; Kondo, T.; Ohtsuka, Y.; Fujiwara, Y.; Shimada, M.; Kawakami, Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism 1989, 38, 753–758. [Google Scholar] [CrossRef]
- Yoshida, K.; Hirokawa, J.; Tagami, S.; Kawakami, Y.; Urata, Y.; Kondo, T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: Regulation of glutathione synthesis and efflux. Diabetologia 1995, 38, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Delveaux, J.; Turpin, C.; Veeren, B.; Diotel, N.; Bravo, S.B.; Begue, F.; Alvarez, E.; Meilhac, O.; Bourdon, E.; Rondeau, P. Antirhea borbonica Aqueous Extract Protects Albumin and Erythrocytes from Glycoxidative Damages. Antioxidants 2020, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, M.J.; Bron, A.J.; Peckar, C.O.; Petchey, M.; Ting, H.H.; Howard-Williams, J. NADPH-oxidising activity in lens and erythrocytes in diabetic and nondiabetic patients with cataract. Br. J. Ophthalmol. 1983, 67, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Towhid, S.T.; Schmid, E.; Hoffmann, S.M.; Abed, M.; Munzer, P.; Vogel, S.; Neis, F.; Brucker, S.; Gawaz, M.; et al. Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am. J. Physiol. Cell Physiol. 2014, 306, C291–C297. [Google Scholar] [CrossRef]
- Keymel, S.; Heiss, C.; Kleinbongard, P.; Kelm, M.; Lauer, T. Impaired red blood cell deformability in patients with coronary artery disease and diabetes mellitus. Horm. Metab. Res. 2011, 43, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, L.Z. Hemoglobin A1c level higher than 9.05% causes a significant impairment of erythrocyte deformability in diabetes mellitus. Acta Endocrinol. 2018, 14, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Waczulikova, I.; Sikurova, L.; Carsky, J.; Strbova, L.; Krahulec, B. Decreased fluidity of isolated erythrocyte membranes in type 1 and type 2 diabetes. The effect of resorcylidene aminoguanidine. Gen. Physiol. Biophys. 2000, 19, 381–392. [Google Scholar] [PubMed]
- Morabito, R.; Remigante, A.; Marino, A. Melatonin Protects Band 3 Protein in Human Erythrocytes against H2O2-Induced Oxidative Stress. Molecules 2019, 24, 2741. [Google Scholar] [CrossRef]
- Resmi, H.; Akhunlar, H.; Temiz Artmann, A.; Guner, G. In vitro effects of high glucose concentrations on membrane protein oxidation, G-actin and deformability of human erythrocytes. Cell. Biochem. Funct. 2005, 23, 163–168. [Google Scholar] [CrossRef]
- Lee, H.; Na, W.; Lee, S.B.; Ahn, C.W.; Moon, J.S.; Won, K.C.; Shin, S. Potential Diagnostic Hemorheological Indexes for Chronic Kidney Disease in Patients With Type 2 Diabetes. Front. Physiol. 2019, 10, 1062. [Google Scholar] [CrossRef]
- Sheremet’ev, Y.A.; Popovicheva, A.N.; Rogozin, M.M.; Levin, G.Y. Red blood cell aggregation, disaggregation and aggregate morphology in autologous plasma and serum in diabetic foot disease. Clin. Hemorheol. Microcirc. 2019, 72, 221–227. [Google Scholar] [CrossRef]
- Tripette, J.; Nguyen, L.C.; Allard, L.; Robillard, P.; Soulez, G.; Cloutier, G. In vivo venous assessment of red blood cell aggregate sizes in diabetic patients with a quantitative cellular ultrasound imaging method: Proof of concept. PLoS ONE 2015, 10, e0124712. [Google Scholar] [CrossRef] [PubMed]
- Osmundson, P.J.; O’Fallon, W.M.; Zimmerman, B.R.; Kazmier, F.J.; Langworthy, A.L.; Palumbo, P.J. Course of peripheral occlusive arterial disease in diabetes. Vascular laboratory assessment. Diabetes Care 1990, 13, 143–152. [Google Scholar] [CrossRef]
- Yalcin, O.; Uyuklu, M.; Armstrong, J.K.; Meiselman, H.J.; Baskurt, O.K. Graded alterations of RBC aggregation influence in vivo blood flow resistance. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2644–H2650. [Google Scholar] [CrossRef] [PubMed]
- Budak, Y.; Demirci, H.; Akdogan, M.; Yavuz, D. Erytrocyte membrane anionic charge in type 2 diabetic patients with retinopathy. BMC Ophthalmol. 2004, 4, 14. [Google Scholar] [CrossRef]
- Mahendra, J.V.; Kumar, S.D.; Anuradha, T.S.; Talikoti, P.; Nagaraj, R.S.; Vishali, V. Plasma Fibrinogen in Type 2 Diabetic Patients with Metabolic Syndrome and its Relation with Ischemic Heart Disease (IHD) and Retinopathy. J. Clin. Diagn. Res. 2015, 9, BC18–BC21. [Google Scholar] [PubMed]
- Xue, S.; Lee, B.K.; Shin, S. Disaggregating shear stress: The roles of cell deformability and fibrinogen concentration. Clin. Hemorheol. Microcirc. 2013, 55, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Virtue, M.A.; Furne, J.K.; Nuttall, F.Q.; Levitt, M.D. Relationship between GHb concentration and erythrocyte survival determined from breath carbon monoxide concentration. Diabetes Care 2004, 27, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Jones, R.L.; Koenig, R.J.; Melvin, E.T.; Lehrman, M.L. Reversible hematologic sequelae of diabetes mellitus. Ann. Intern. Med. 1977, 86, 425–429. [Google Scholar] [CrossRef]
- Korot’ko, G.F. The problem of autolytic digestion at amniotropic and lactotropic feeding. Exp. Clin. Gastroenterol. 2015, 9, 75–87. [Google Scholar]
- Chong-Martinez, B.; Buchanan, T.A.; Wenby, R.B.; Meiselman, H.J. Decreased red blood cell aggregation subsequent to improved glycaemic control in Type 2 diabetes mellitus. Diabet Med. 2003, 20, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Foresto, P.; D’Arrigo, M.; Carreras, L.; Cuezzo, R.E.; Valverde, J.; Rasia, R. Evaluation of red blood cell aggregation in diabetes by computerized image analysis. Medicina (B Aires) 2000, 60, 570–572. [Google Scholar] [PubMed]
- Park, K.H.; Kim, U.; Choi, K.U.; Nam, J.H.; Lee, J.H.; Lee, C.H.; Son, J.W.; Park, J.S.; Shin, D.G.; Won, K.C.; et al. Corrigendum: Hemorheologic Alterations in Patients with Type 2 Diabetes Mellitus Presented with an Acute Myocardial Infarction. Diabetes Metab. J. 2018, 42, 254. [Google Scholar] [CrossRef]
- Marar, T. Amelioration of glucose induced hemolysis of human erythrocytes by vitamin E. Chem. Biol. Interact. 2015, 193, 149–153. [Google Scholar] [CrossRef]
- Kiefer, C.R.; Snyder, L.M. Oxidation and erythrocyte senescence. Curr. Opin. Hematol. 2000, 7, 113–116. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Foller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2015, 26, 21–28. [Google Scholar] [CrossRef]
- Bonomini, M.; Sirolli, V.; Settefrati, N.; Dottori, S.; Di Liberato, L.; Arduini, A. Increased erythrocyte phosphatidylserine exposure in chronic renal failure. J. Am. Soc. Nephrol. 1999, 10, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Eda, S.; Sherman, I.W. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell. Physiol. Biochem. 2002, 12, 373–384. [Google Scholar] [CrossRef]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Kempe-Teufel, D.S.; Bissinger, R.; Qadri, S.M.; Wagner, R.; Peter, A.; Lang, F. Cellular markers of eryptosis are altered in type 2 diabetes. Clin. Chem Lab. Med. 2018, 56, e177–e180. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tibrewal, N.; Birge, R.B. Phosphatidylserine recognition by phagocytes: A view to a kill. Trends Cell Biol. 2006, 16, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef]
- Minami, M.; Kume, N.; Shimaoka, T.; Kataoka, H.; Hayashida, K.; Akiyama, Y.; Nagata, I.; Ando, K.; Nobuyoshi, M.; Hanyuu, M.; et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1796–1800. [Google Scholar] [CrossRef]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Burger, P.; Hilarius-Stokman, P.; de Korte, D.; van den Berg, T.K.; van Bruggen, R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 2012, 119, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Shimo, H.; Arjunan, S.N.; Machiyama, H.; Nishino, T.; Suematsu, M.; Fujita, H.; Tomita, M.; Takahashi, K. Particle Simulation of Oxidation Induced Band 3 Clustering in Human Erythrocytes. PLoS Comput. Biol. 2015, 11, e1004210. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Matte, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxid Med. Cell Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef]
- McPherson, R.A.; Sawyer, W.H.; Tilley, L. Rotational diffusion of the erythrocyte integral membrane protein band 3: Effect of hemichrome binding. Biochemistry 1992, 31, 512–518. [Google Scholar] [CrossRef]
- Low, P.S.; Waugh, S.M.; Zinke, K.; Drenckhahn, D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science 1985, 227, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Arese, P.; Gallo, V.; Pantaleo, A.; Turrini, F. Life and Death of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficient Erythrocytes—Role of Redox Stress and Band 3 Modifications. Transfus. Med. Hemother. 2012, 39, 328–334. [Google Scholar] [CrossRef]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Ronzoni, L.; Graziadei, G.; Cappellini, M.D.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014, 99, 570–578. [Google Scholar] [CrossRef]
- Lutz, H.U. Naturally occurring anti-band 3 antibodies in clearance of senescent and oxidatively stressed human red blood cells. Transfus. Med. Hemother. 2012, 39, 321–327. [Google Scholar] [CrossRef]
- Lutz, H.U.; Bussolino, F.; Flepp, R.; Fasler, S.; Stammler, P.; Kazatchkine, M.D.; Arese, P. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc. Natl. Acad. Sci. USA 1987, 84, 7368–7372. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin. Hemorheol. Microcirc. 2013, 53, 11–21. [Google Scholar] [CrossRef]
- Vorchheimer, D.A.; Becker, R. Platelets in atherothrombosis. Mayo Clin. Proc. 2006, 81, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.Y.; Lim, K.M.; Bae, O.N.; Chung, S.M.; Lee, S.W.; Joo, K.M.; Lee, S.D.; Chung, J.H. Procoagulant and prothrombotic activation of human erythrocytes by phosphatidic acid. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H347–H355. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpin, C.; Catan, A.; Meilhac, O.; Bourdon, E.; Canonne-Hergaux, F.; Rondeau, P. Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 5843. https://doi.org/10.3390/ijms22115843
Turpin C, Catan A, Meilhac O, Bourdon E, Canonne-Hergaux F, Rondeau P. Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(11):5843. https://doi.org/10.3390/ijms22115843
Chicago/Turabian StyleTurpin, Chloé, Aurélie Catan, Olivier Meilhac, Emmanuel Bourdon, François Canonne-Hergaux, and Philippe Rondeau. 2021. "Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis" International Journal of Molecular Sciences 22, no. 11: 5843. https://doi.org/10.3390/ijms22115843
APA StyleTurpin, C., Catan, A., Meilhac, O., Bourdon, E., Canonne-Hergaux, F., & Rondeau, P. (2021). Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis. International Journal of Molecular Sciences, 22(11), 5843. https://doi.org/10.3390/ijms22115843