
Molecular Mechanisms of the Deregulation of Muscle Contraction Induced by the R90P Mutation in Tpm3.12 and the Weakening of This Effect by BDM and W7

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
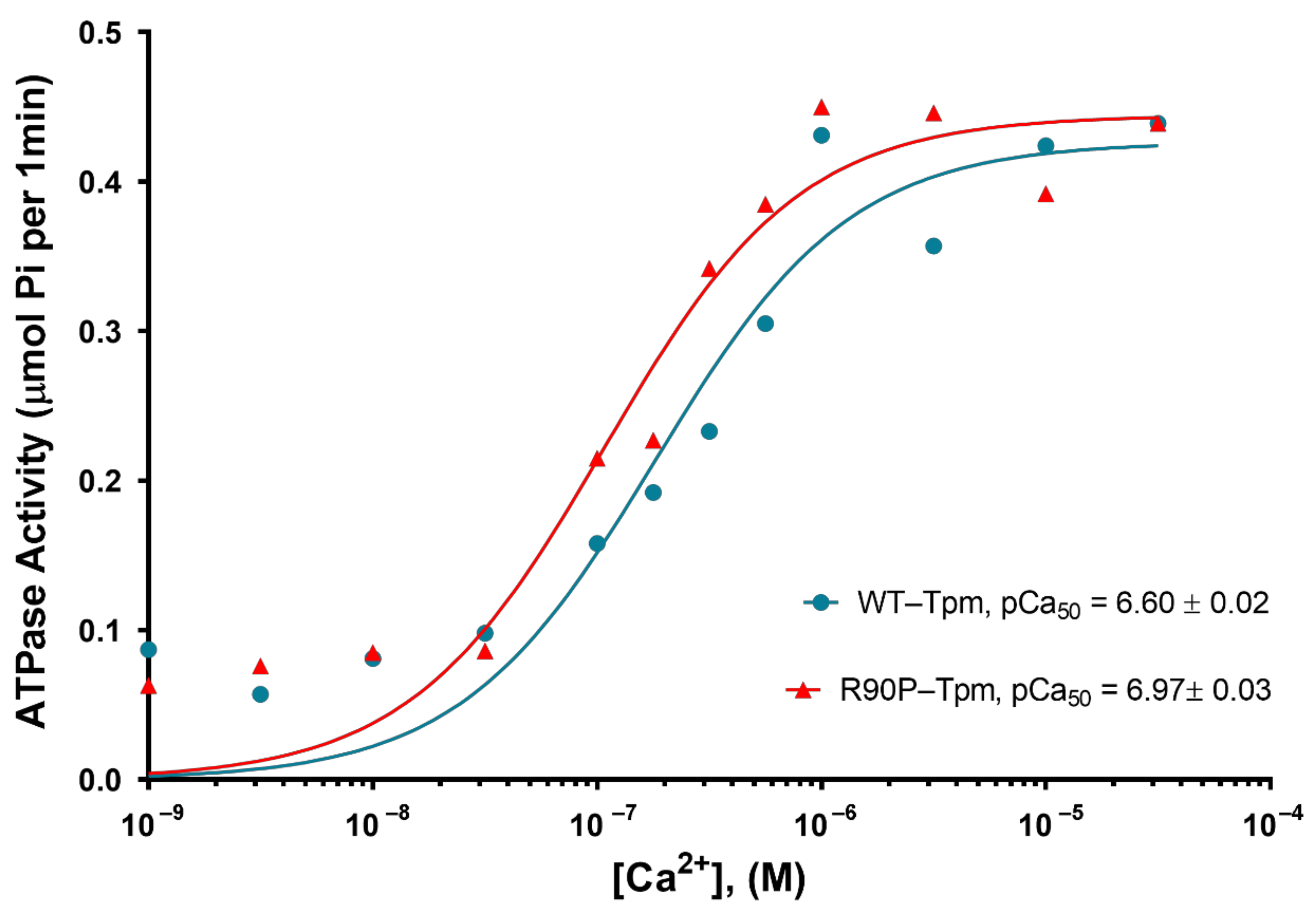
2.1. The R90P Mutation in Tpm3.12 Produces Abnormally High Myofilament Ca2+-Sensitivity in Protein Solution
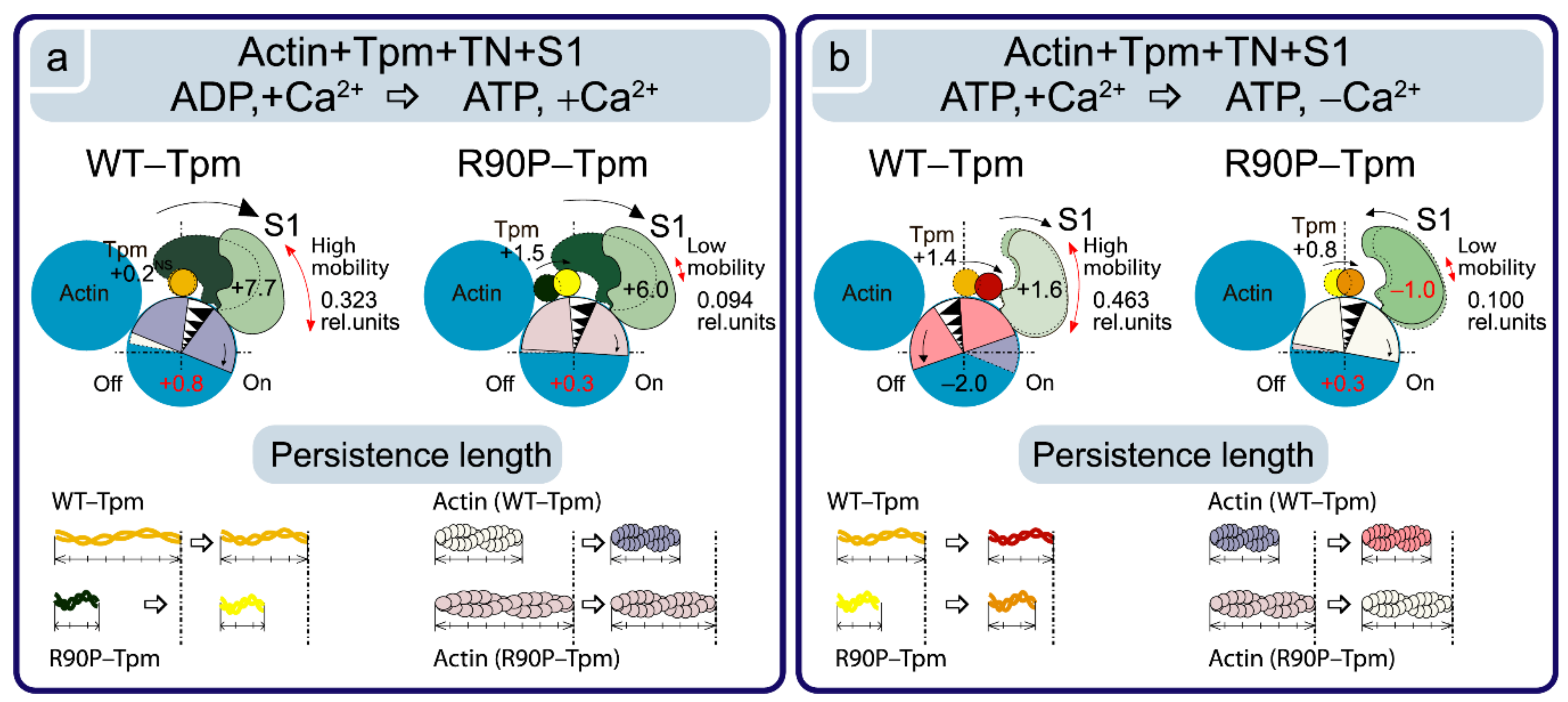
2.2. The R90P Mutation in Tpm3.12 Depresses the Ca2+-Dependent Tpm’s Movement on the Actin Filaments
2.3. The R90P Mutation Inhibits Troponin’s Ability to Switch Actin Monomers off at a Low Ca2+
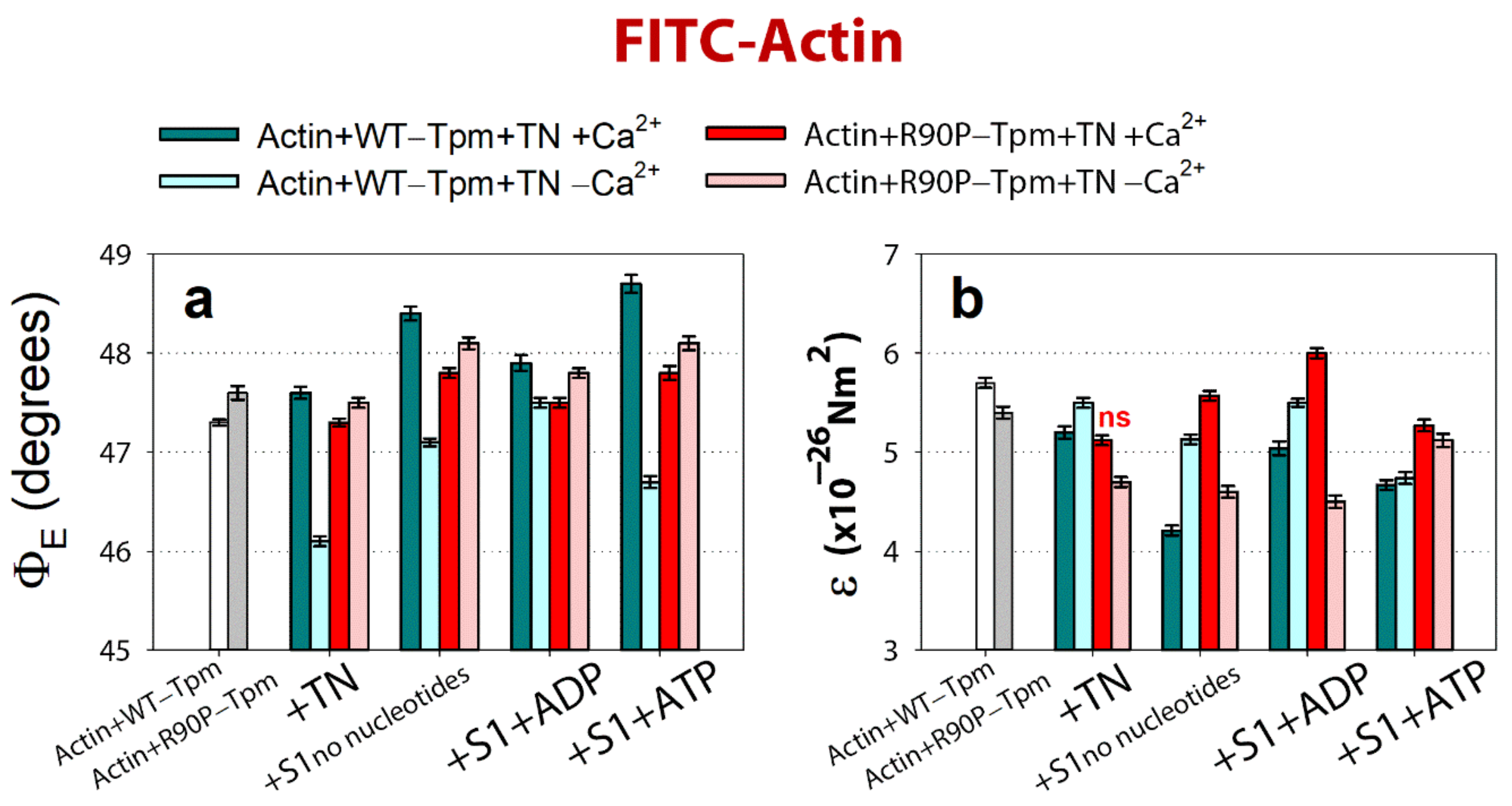
2.4. The R90P Mutation Allows the Strong Binding of the Myosin Heads to F-Actin at a Low Ca2+
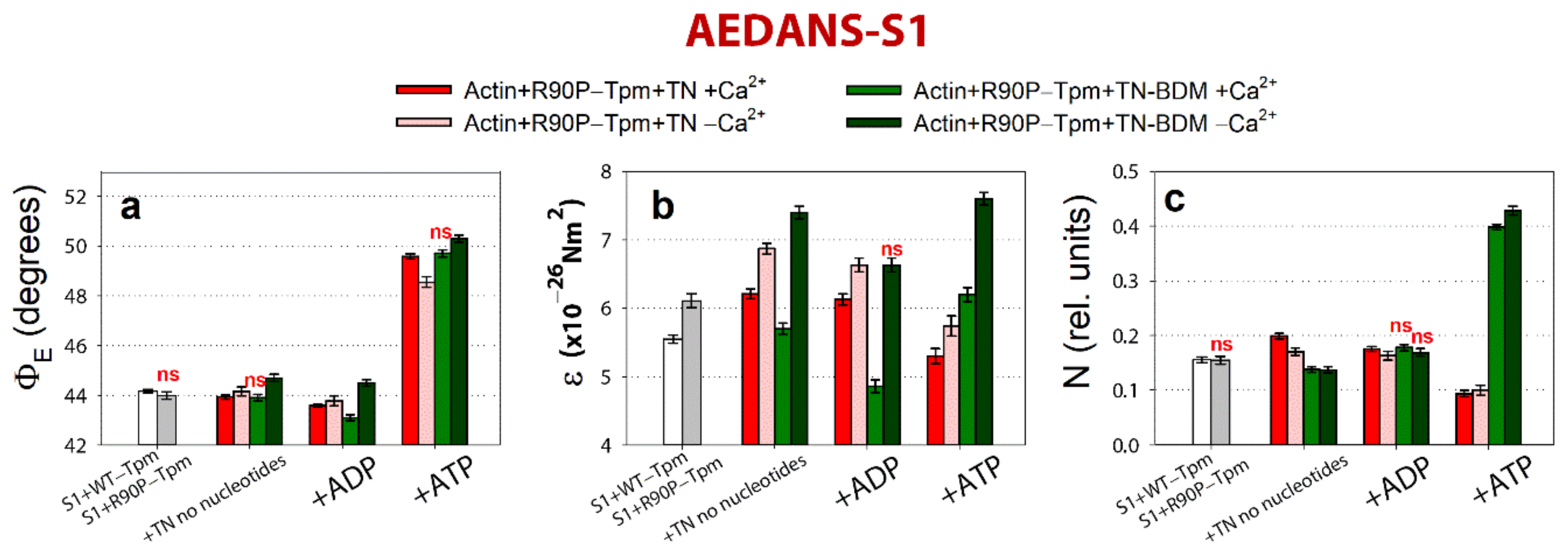
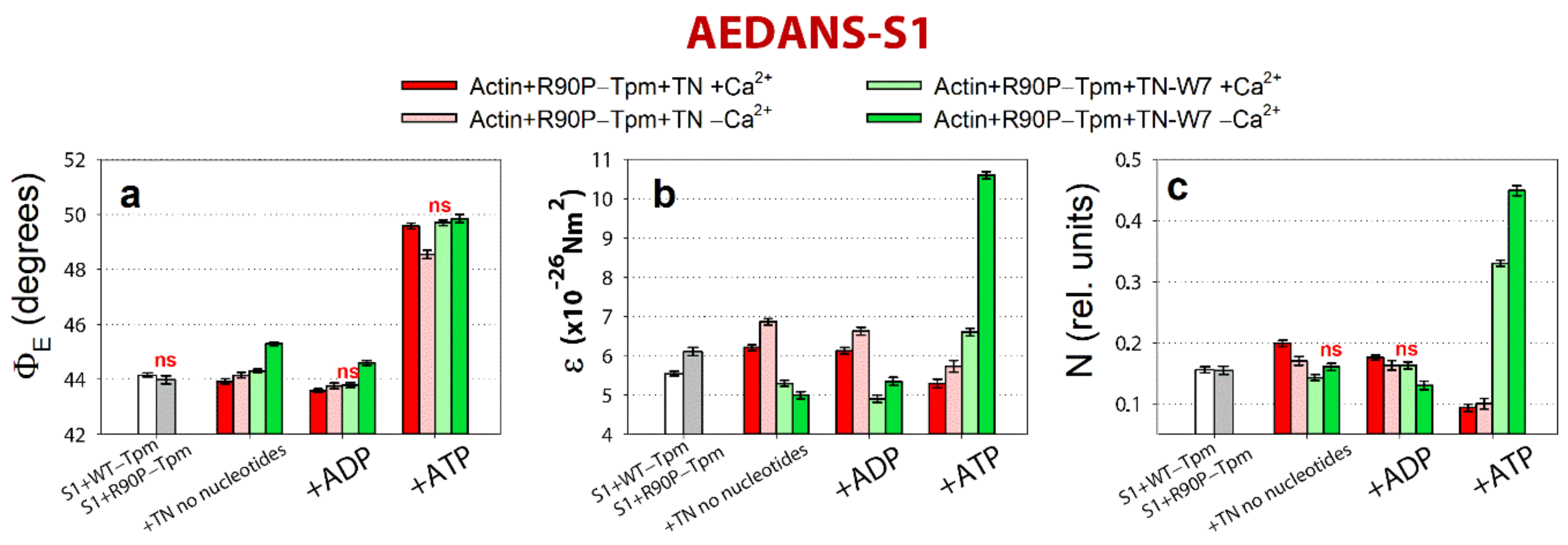
2.5. The Inhibitor of Myosin ATPase Activity, BDM, and the Ca2+-Desensitizer, W7, Are Able to Weaken the Ca2+-Sensitivity of Myofilaments Containing R90P-Tpm in Protein Solution
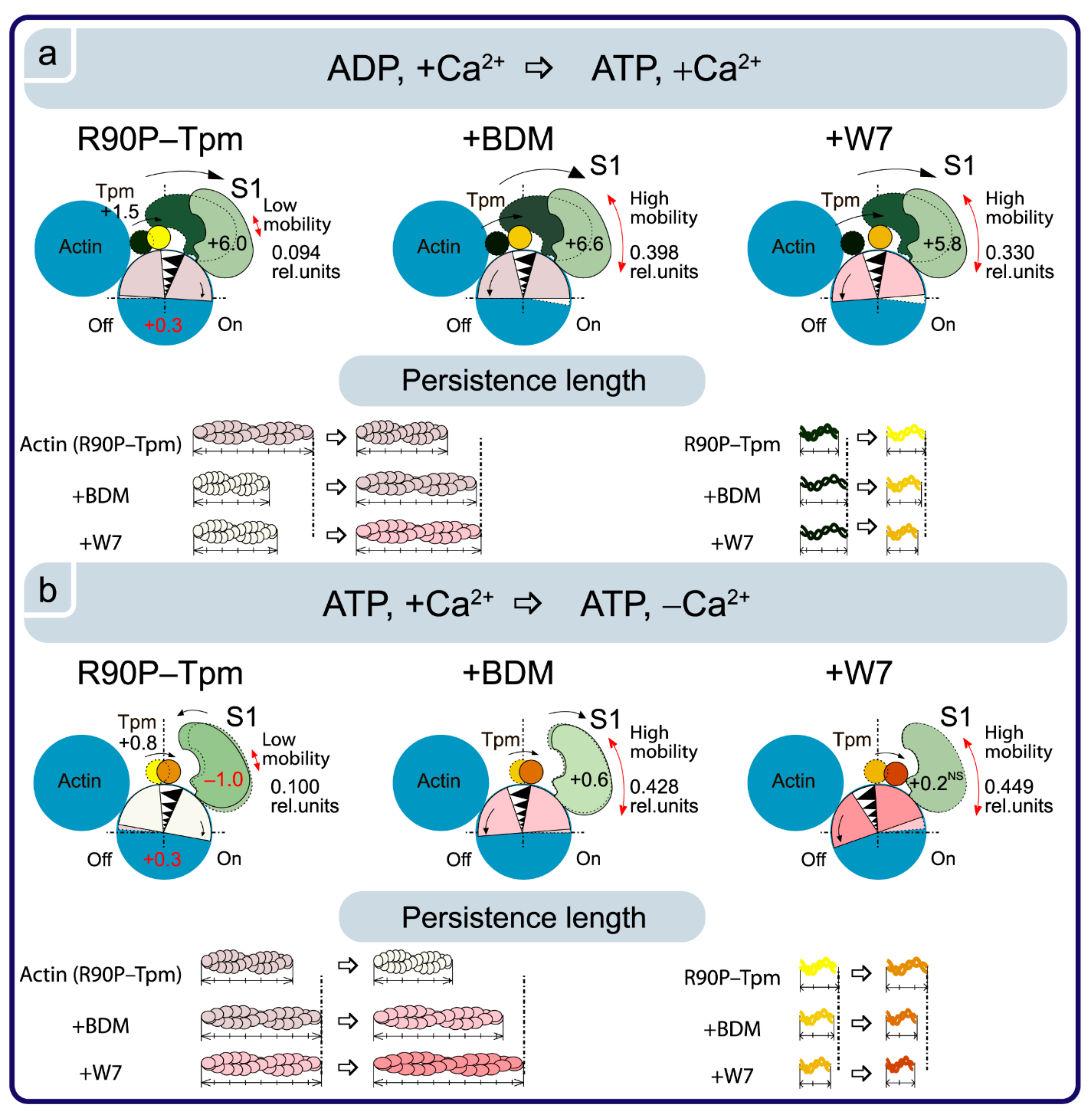
2.6. BDM and W7 Are Able to Weaken the Damaging Effect of the R90P Mutation on the Regulation of Muscle Contractility
3. Materials and Methods
3.1. Using of Experimental Animals
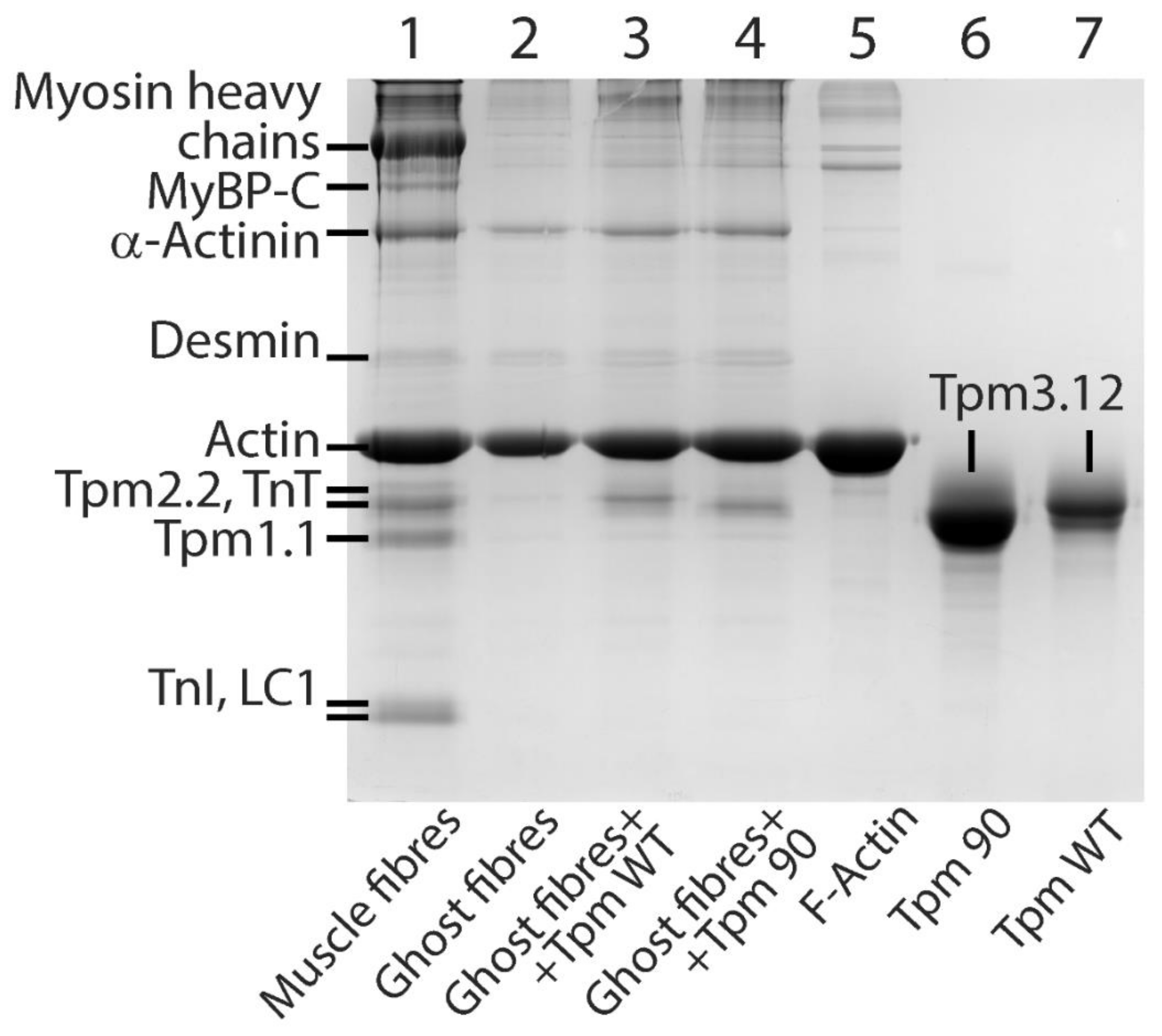
3.2. Preparation of Proteins and Their Labeling by Fluorescent Probes
3.3. Determination of Actin-Activated ATPase of Myosin Subfragment-1
3.4. Preparation and Labeling of Ghost Fibers
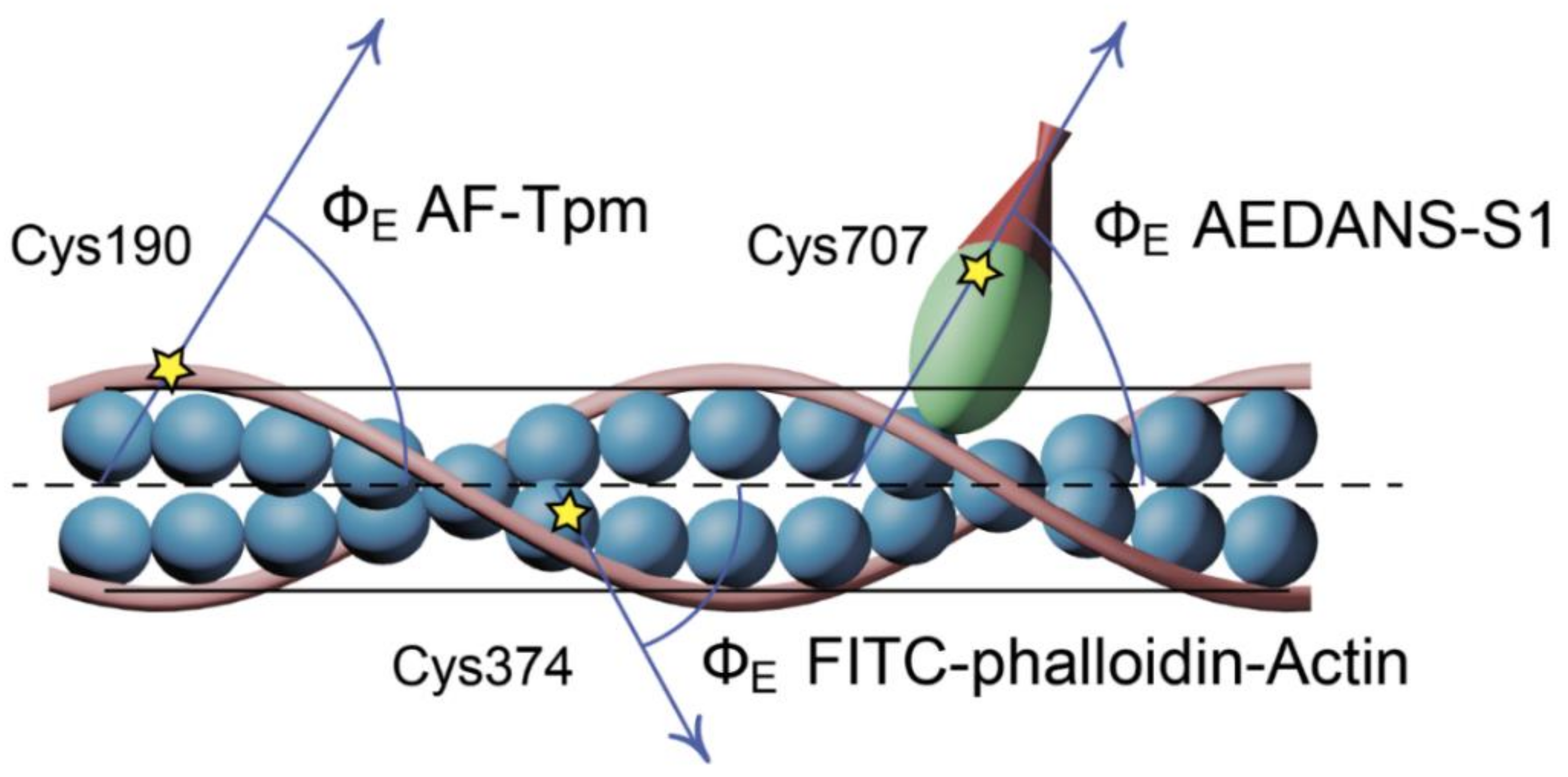
3.5. Polarized Fluorescence Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Tpm | Tropomyosin |
| WT-Tpm | Wild-type tropomyosin |
| R90P-Tpm | Tropomyosin with the Arg90Pro substitution |
| TN | Troponin |
| S1 | Myosin subfragment-1 |
| 1,5-IAEDANS | N-(iodoacetaminoethyl)-1-naphthyl-amine-5-sulfonic acid |
| 5-IAF | 5-Iodoacetamidofluorescein |
| FITC | Fluorescein isothiocyanate |
| W7 | (N-(6-minohexyl) 5-chloro-1-napthalenesulfonamide) |
| BDM | 2, 3-Butanedione 2-monoxime |
References
- Franzini-Armstrong, C. Electron Microscopy: From 2D to 3D Images with Special Reference to Muscle. Eur. J. Transl. Myol. 2015, 25, 4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef] [PubMed]
- Rynkiewicz, M.J.; Schott, V.; Orzechowski, M.; Lehman, W.; Fischer, S. Electrostatic interaction map reveals a new binding position for tropomyosin on F-actin. J. Muscle Res. Cell Motil. 2015, 36, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobacman, L.S. Troponin Revealed: Uncovering the Structure of the Thin Filament On-Off Switch in Striated Muscle. Biophys. J. 2021, 120, 1–9. [Google Scholar] [CrossRef]
- Craig, R.; Lehman, W. Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments. J. Mol. Biol. 2001, 311, 1027–1036. [Google Scholar] [CrossRef]
- Li, X.E.; Lehman, W.; Fischer, S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. J. Struct. Biol. 2010, 170, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Redwood, C.S. Modulation of the effects of tropomyosin on actin and myosin conformational changes by troponin and Ca2+. Biochim. Biophys. Acta 2009, 1794, 985–994. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Rysev, N.A.; Karpicheva, O.E.; Sirenko, V.V.; Avrova, S.V.; Piers, A.; Redwood, C.S. Molecular mechanisms of dysfunction of muscle fibers associated with Glu139 deletion in TPM2 gene. Sci. Rep. 2017, 7, 16797. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Avrova, S.V.; Sirenko, V.V.; Redwood, C.S.; Karpicheva, O.E. Molecular Mechanisms of Muscle Weakness Associated with E173A Mutation in Tpm3.12. Troponin Ca2+ Sensitivity Inhibitor W7 Can Reduce the Damaging Effect of This Mutation. Int. J. Mol. Sci. 2020, 21, 4421. [Google Scholar] [CrossRef]
- Li, X.E.; Tobacman, L.S.; Mun, J.Y.; Craig, R.; Fischer, S.; Lehman, W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 2010, 100, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac Muscle Thin Filament Structures Reveal Calcium Regulatory Mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef]
- Barua, B. Periodicities designed in the tropomyosin sequence and structure define its functions. BioArchitecture 2013, 3, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.V. Vertebrate tropomyosin: Distribution, properties and function. J. Muscle Res. Cell Motil. 2001, 22, 5–49. [Google Scholar] [CrossRef]
- Jin, Y.; Peng, Y.; Lin, Z.; Chen, Y.-C.; Wei, L.; Hacker, T.A.; Larsson, L.; Ge, Y. Comprehensive analysis of tropomyosin isoforms in skeletal muscles by top-down proteomics. J. Muscle Res. Cell Motil. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Marttila, M.; Lehtokari, V.-L.; Marston, S.; Nyman, T.A.; Barnerias, C.; Beggs, A.H.; Bertini, E.; Ceyhan-Birsoy, Ö.; Cintas, P.; Gerard, M.; et al. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum. Mutat. 2014, 35, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; McNamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of TM cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [Green Version]
- Sewry, C.A.; Wallgren-Pettersson, C. Myopathology in congenital myopathies. Neuropathol. Appl. Neurobiol. 2017, 43, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Munot, P.; Lashley, D.; Jungbluth, H.; Feng, L.; Pitt, M.; Robb, S.; Palace, J.; Jayawant, S.; Kennet, R.; Beeson, D.; et al. Congenital fiber type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscul. Disord. 2010, 20, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.W.; Dechene, E.T.; Roumm, E.; Geggel, A.S.; Moghadaszadeh, B.; Beggs, A.H. Mutations of tropomyosin 3 (TPM3) are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion. Hum. Mutat. 2010, 31, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borejdo, J.; Muthu, P.; Talent, J.; Akopova, I.; Burghardt, T.P. Rotation of actin monomers during isometric contraction of skeletal muscle. J. Biomed. Opt. 2007, 12, 14013. [Google Scholar] [CrossRef]
- Borovikov, Y.S. Conformational changes of contractile proteins and their role in muscle contraction. Int. Rev. Cytol. 1999, 189, 267–301. [Google Scholar]
- Borovikov, Y.S.; Rysev, N.A.; Avrova, S.V.; Karpicheva, O.E.; Borys, D.; Moraczewska, J. Molecular mechanisms of deregulation of the thin filament associated with the R167H and K168E substitutions in tropomyosin Tpm1.1. Arch. Biochem. Biophys. 2017, 614, 28–40. [Google Scholar] [CrossRef]
- Lamkin, M.; Tao, T.; Lehrer, S.S. Tropomyosin-troponin and tropomyosin-actin interactions: A fluorescence quenching study. Biochemistry 1983, 22, 3053–3058. [Google Scholar] [CrossRef]
- Gonchar, A.D.; Kopylova, G.V.; Kochurova, A.M.; Berg, V.Y.; Shchepkin, D.V.; Koubasova, N.A.; Tsaturyan, A.K.; Kleymenov, S.Y.; Matyushenko, A.M.; Levitsky, D.I. Effects of myopathy-causing mutations R91P and R245G in the TPM3 gene on structural and functional properties of slow skeletal muscle tropomyosin. Biochem. Biophys. Res. Commun. 2021, 534, 8–13. [Google Scholar] [CrossRef]
- Lorenz, M.; Popp, D.; Holmes, K.C. Refinement of the F-actin model against X-ray fiber diffraction data by the use of a directed mutation algorithm. J. Mol. Biol. 1993, 234, 826–836. [Google Scholar] [CrossRef]
- Dancker, P.; Low, I.; Hasselbach, W.; Wieland, T. Interaction of actin with phalloidin: Polymerization and stabilization of F-actin. Biochim. Biophys. Acta 1975, 400, 407–414. [Google Scholar] [CrossRef]
- Bukatina, A.E.; Fuchs, F. Effect of phalloidin on the ATPase activity of striated muscle myofibrils. J. Muscle Res. Cell Motil. 1994, 15, 29–36. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Dedova, I.V.; dos Remedios, C.G.; Vikhoreva, N.N.; Vikhorev, P.G.; Avrova, S.V.; Hazlett, T.L.; Van Der Meer, B.W. Fluorescence depolarization of actin filaments in reconstructed myofibers: The effect of S1 or pPDM-S1 on movements of distinct areas of actin. Biophys. J. 2004, 86, 3020–3029. [Google Scholar] [CrossRef] [Green Version]
- Rysev, N.A.; Nevzorov, I.A.; Avrova, S.V.; Karpicheva, O.E.; Redwood, C.S.; Levitsky, D.I.; Borovikov, Y.S. Gly126Arg substitution causes anomalous behaviour of α-skeletal and β-smooth tropomyosins during the ATPase cycle. Arch. Biochem. Biophys. 2014, 543, 57–66. [Google Scholar] [CrossRef]
- Prochniewicz-Nakayama, E.; Yanagida, T.; Oosawa, F. Studies on conformation of F-actin in muscle fibers in the relaxed state, rigor, and during contraction using fluorescent phalloidin. J. Cell Biol. 1983, 97, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Mudalige, W.A.; Tao, T.C.; Lehrer, S.S. Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157-163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. J. Mol. Biol. 2009, 389, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Isambert, H.; Venier, P.; Maggs, A.C.; Fattoum, A.; Kassab, R.; Pantaloni, D.; Carlier, M.F. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 1995, 270, 11437–11444. [Google Scholar] [CrossRef] [Green Version]
- Titus, M.A.; Ashiba, G.; Szent-Gyorgyi, A.G. SH-1 modification of rabbit myosin interferes with calcium regulation. J. Muscle Res. Cell Motil. 1989, 10, 25–33. [Google Scholar] [CrossRef]
- Bobkov, A.A.; Bobkova, E.A.; Homsher, E.; Reisler, E. Activation of regulated actin by SH1-modified myosin subfragment 1. Biochemistry 1997, 36, 7733–7738. [Google Scholar] [CrossRef]
- Onishi, H.; Nitanai, Y. Thiol reactivity as a sensor of rotation of the converter in myosin. Biochem. Biophys. Res. Commun. 2008, 369, 115–123. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Simonyan, A.O.; Chernev, A.A.; Karpicheva, O.E.; Piers, A.; Redwood, C.S. Aberrant movement of β-tropomyosin associated with congenital myopathy causes defective response of myosin heads and actin during the ATPase cycle. Arch. Biochem. Biophys. 2015, 577, 13–23. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Simonyan, A.O.; Sirenko, V.V.; Redwood, C.S. The molecular mechanism of muscle dysfunction associated with the R133W mutation in Tpm2.2. Biochem. Biophys. Res. Commun. 2020, 523, 258–262. [Google Scholar] [CrossRef]
- Fujita, H.; Lu, X.; Suzuki, M.; Ishiwata, S.; Kawai, M. The effect of tropomyosin on force and elementary steps of the cross-bridge cycle in reconstituted bovine myocardium. J. Physiol. 2004, 556, 637–649. [Google Scholar] [CrossRef]
- Chitose, R.; Watanabe, A.; Asano, M.; Hanashima, A.; Sasano, K.; Bao, Y.; Maruyama, K.; Kimura, S. Isolation of nebulin from rabbit skeletal muscle and its interaction with actin. J. Biomed. Biotechnol. 2010, 2010, 108495. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Karpicheva, O.E.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Piers, A.; Redwood, C.S. The reason for a high Ca(2+)-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 2017, 494, 681–686. [Google Scholar] [CrossRef]
- Karpicheva, O.E.; Sirenko, V.V.; Rysev, N.A.; Simonyan, A.O.; Borys, D.; Moraczewska, J.; Borovikov, Y.S. Deviations in conformational rearrangements of thin filaments and myosin caused by the Ala155Thr substitution in hydrophobic core of tropomyosin. Biochim. Biophys. Acta 2017, 1865, 1790–1799. [Google Scholar] [CrossRef]
- Bond, L.M.; Tumbarello, D.A.; Kendrick-Jones, J.; Buss, F. Small-molecule inhibitors of myosin proteins. Future Med. Chem. 2013, 5, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Horiuti, K.; Higuchi, H.; Umazume, Y.; Konishi, M.; Okazaki, O.; Kurihara, S. Mechanism of action of 2, 3-butanedione 2-monoxime on contraction of frog skeletal muscle fibers. J. Muscle Res. Cell Motil. 1988, 9, 156–164. [Google Scholar] [CrossRef]
- Higuchi, H.; Takemori, S. Butanedione monoxime suppresses contraction and ATPase activity of rabbit skeletal muscle. J. Biochem. 1989, 105, 638–643. [Google Scholar] [CrossRef]
- Ostap, E.M. 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J. Muscle Res. Cell Motil. 2002, 23, 305–308. [Google Scholar] [CrossRef]
- Herrmann, C.; Wray, J.; Travers, F.; Barman, T. Effect of 2,3-butanedione monoxime on myosin and myofibrillar ATPases. An example of an uncompetitive inhibitor. Biochemistry 1992, 31, 12227–12232. [Google Scholar] [CrossRef]
- Iwamoto, H. Effects of myosin inhibitors on the X-ray diffraction patterns of relaxed and calcium-activated rabbit skeletal muscle fibers. Biophys. Phys. 2018, 15, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidaka, H.; Yamaki, T.; Naka, M.; Tanaka, T.; Hayashi, H.; Kobayashi, R. Calcium-regulated modulator protein interacting agents inhibit smooth muscle calcium-stimulated protein kinase and ATPase. Mol. Pharmacol. 1980, 17, 66–72. [Google Scholar] [PubMed]
- Ogawa, Y.; Kurebayashi, N. Modulations by drugs of the relationship between calcium binding to troponin C and tension. Prog. Clin. Biol. Res. 1989, 31, 75–86. [Google Scholar]
- Adhikari, B.B.; Wang, K. Interplay of troponin- and Myosin-based pathways of calcium activation in skeletal and cardiac muscle: The use of W7 as an inhibitor of thin filament activation. Biophys. J. 2004, 86, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Margossian, S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzym. 1982, 85, 55–71. [Google Scholar]
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. J. Biol. Chem. 1971, 246, 4866. [Google Scholar] [CrossRef]
- Okamoto, Y.; Sekine, T. A streamlined method of subfragment one preparation from myosin. J. Biochem. 1985, 98, 1143–1145. [Google Scholar] [CrossRef]
- Borejdo, J.; Putnam, S. Polarization of flourescence from single skinned glycerinated rabbit psoas fibers in rigor and relaxation. Biochim. Biophys. Acta 1977, 459, 578–595. [Google Scholar] [CrossRef]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C.S. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007, 21, 896–905. [Google Scholar] [CrossRef]
- Karpicheva, O.E.; Simonyan, A.O.; Kuleva, N.V.; Redwood, C.S.; Borovikov, Y.S. Myopathy-causing Q147P TPM2 mutation shifts tropomyosin strands further towards the open position and increases the proportion of strong-binding cross-bridges during the ATPase cycle. Biochim. Biophys. Acta 2016, 1864, 260–267. [Google Scholar] [CrossRef]
- Fiske, C.H.; Subbarow, Y. Determination of inorganic phosphate. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar] [CrossRef]
- Yanagida, T.; Oosawa, F. Polarized fluorescence from epsilon-ADP incorporated into F-actin in a myosin-free single fiber: Conformation of F-actin and changes induced in it by heavy meromyosin. J. Mol. Biol. 1978, 126, 507–524. [Google Scholar] [CrossRef]
- Kakol, I.; Borovikov, Y.S.; Szczesna, D.; Kirillina, V.P.; Levitsky, D.I. Conformational changes of F-actin in myosin-free ghost single fiber induced by either phosphorylated or dephosphorylated heavy meromyosin. Biochim. Biophys. Acta 1987, 913, 1–9. [Google Scholar] [CrossRef]
- Goody, R.S.; Hofmann, W. Stereochemical aspects of the interaction of myosin and actomyosin with nucleotides. J. Muscle Res. Cell Motil. 1980, 1, 101–115. [Google Scholar] [CrossRef]
- Kawas, R.F.; Anderson, R.L.; Ingle, S.R.B.; Song, Y.; Sran, A.S.; Rodriguez, H.M. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J. Biol. Chem. 2017, 292, 16571–16577. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borovikov, Y.S.; Andreeva, D.D.; Avrova, S.V.; Sirenko, V.V.; Simonyan, A.O.; Redwood, C.S.; Karpicheva, O.E. Molecular Mechanisms of the Deregulation of Muscle Contraction Induced by the R90P Mutation in Tpm3.12 and the Weakening of This Effect by BDM and W7. Int. J. Mol. Sci. 2021, 22, 6318. https://doi.org/10.3390/ijms22126318
Borovikov YS, Andreeva DD, Avrova SV, Sirenko VV, Simonyan AO, Redwood CS, Karpicheva OE. Molecular Mechanisms of the Deregulation of Muscle Contraction Induced by the R90P Mutation in Tpm3.12 and the Weakening of This Effect by BDM and W7. International Journal of Molecular Sciences. 2021; 22(12):6318. https://doi.org/10.3390/ijms22126318
Chicago/Turabian StyleBorovikov, Yurii S., Daria D. Andreeva, Stanislava V. Avrova, Vladimir V. Sirenko, Armen O. Simonyan, Charles S. Redwood, and Olga E. Karpicheva. 2021. "Molecular Mechanisms of the Deregulation of Muscle Contraction Induced by the R90P Mutation in Tpm3.12 and the Weakening of This Effect by BDM and W7" International Journal of Molecular Sciences 22, no. 12: 6318. https://doi.org/10.3390/ijms22126318