
Skullcapflavone II Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Production in HaCaT Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
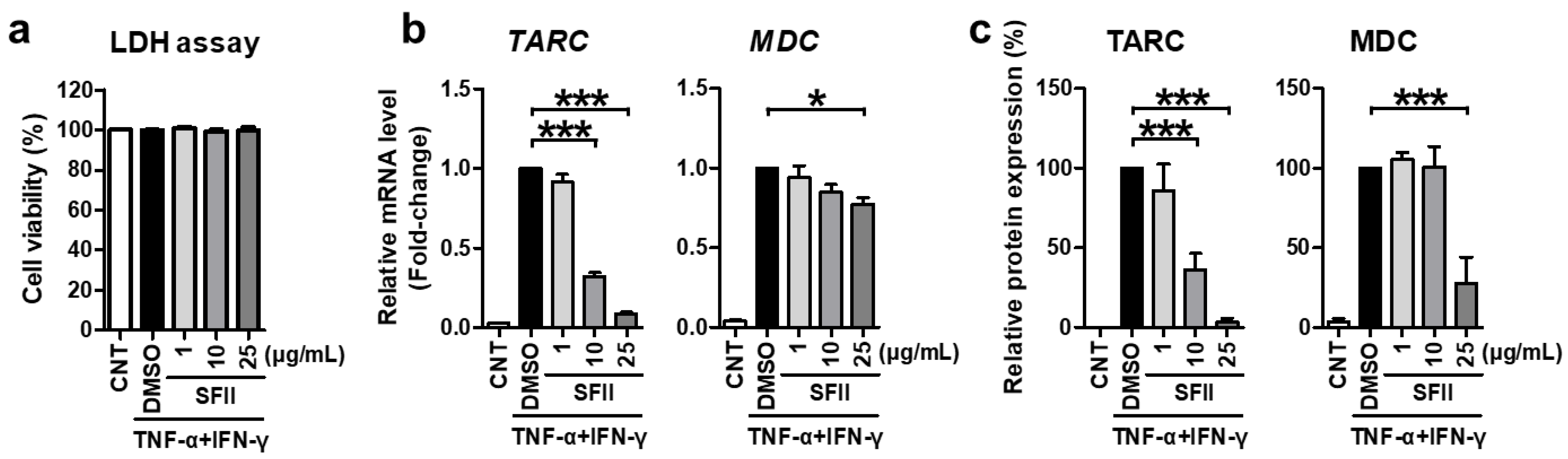
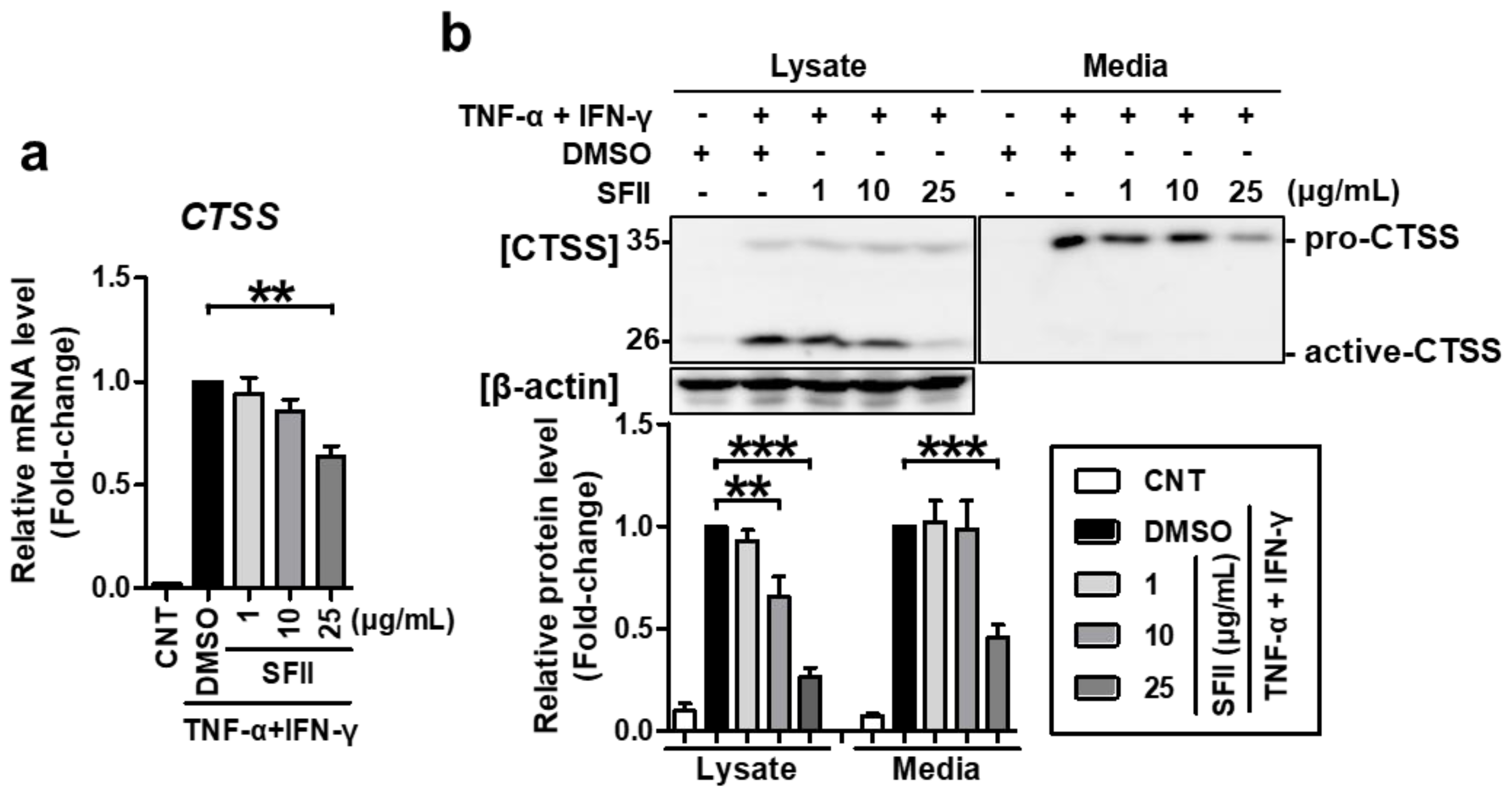
2.1. SFII Significantly Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Expression
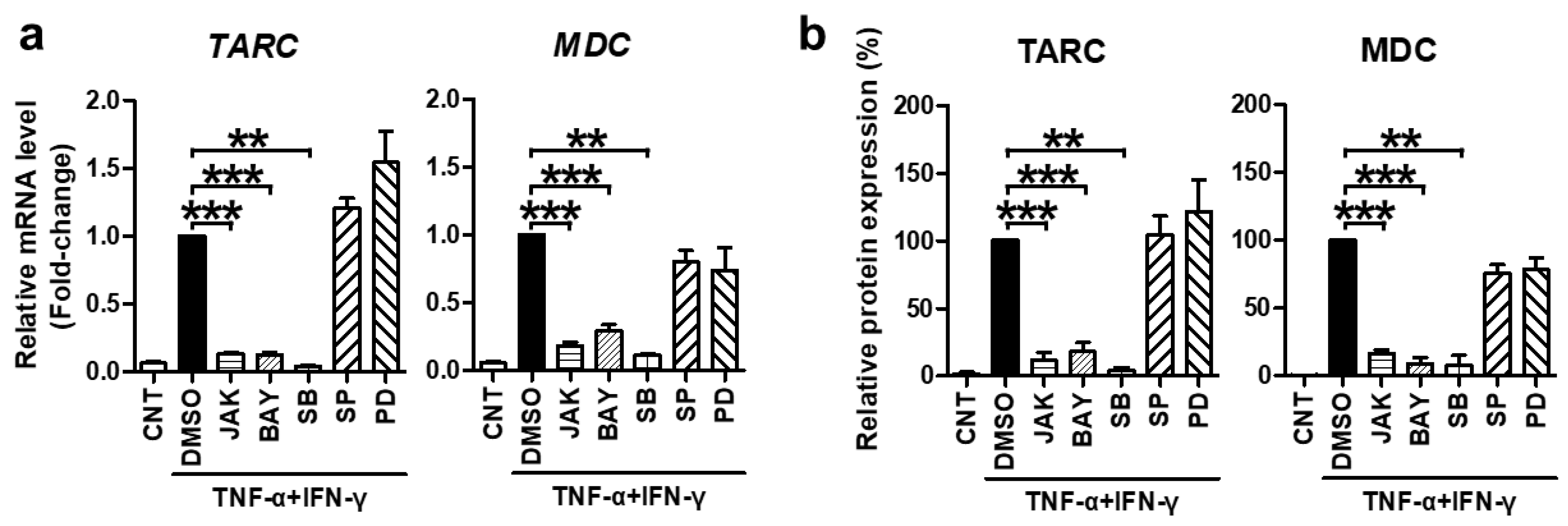
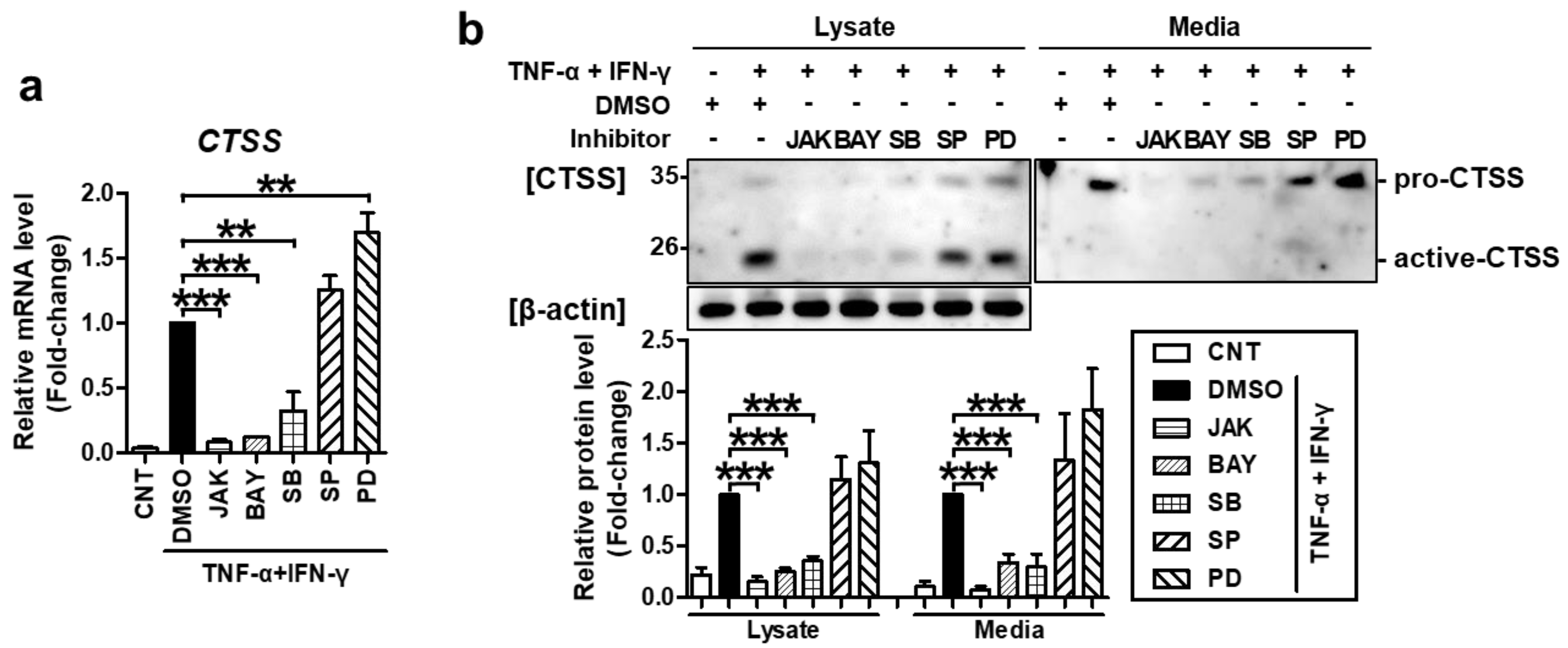
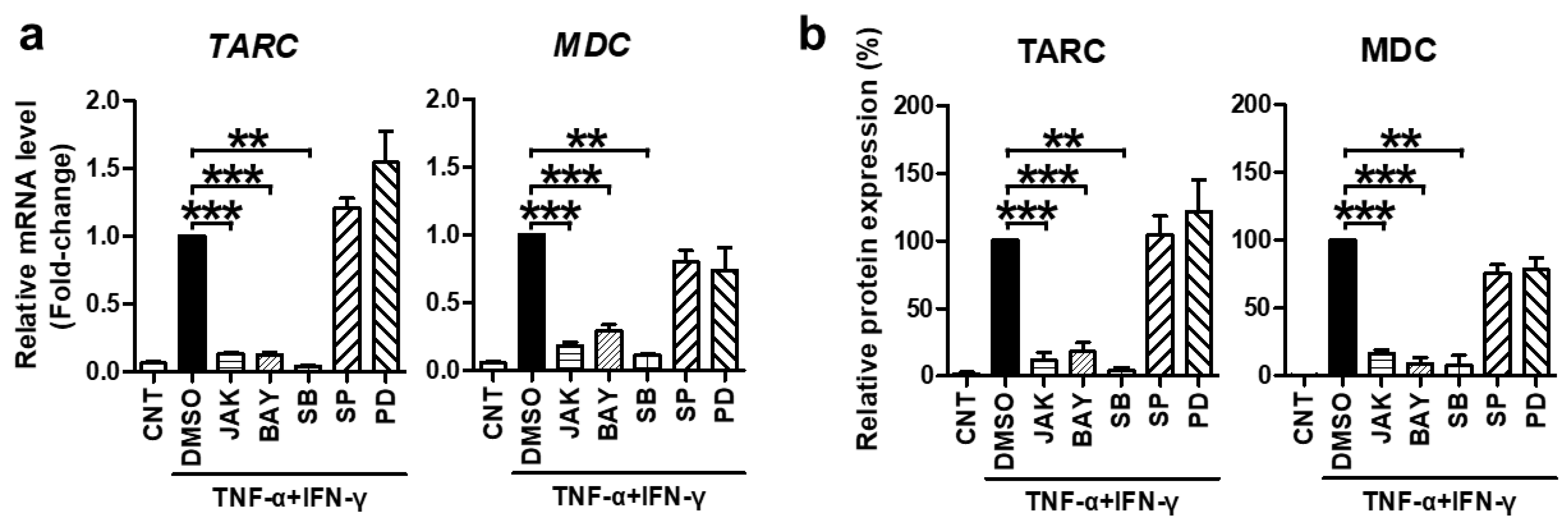
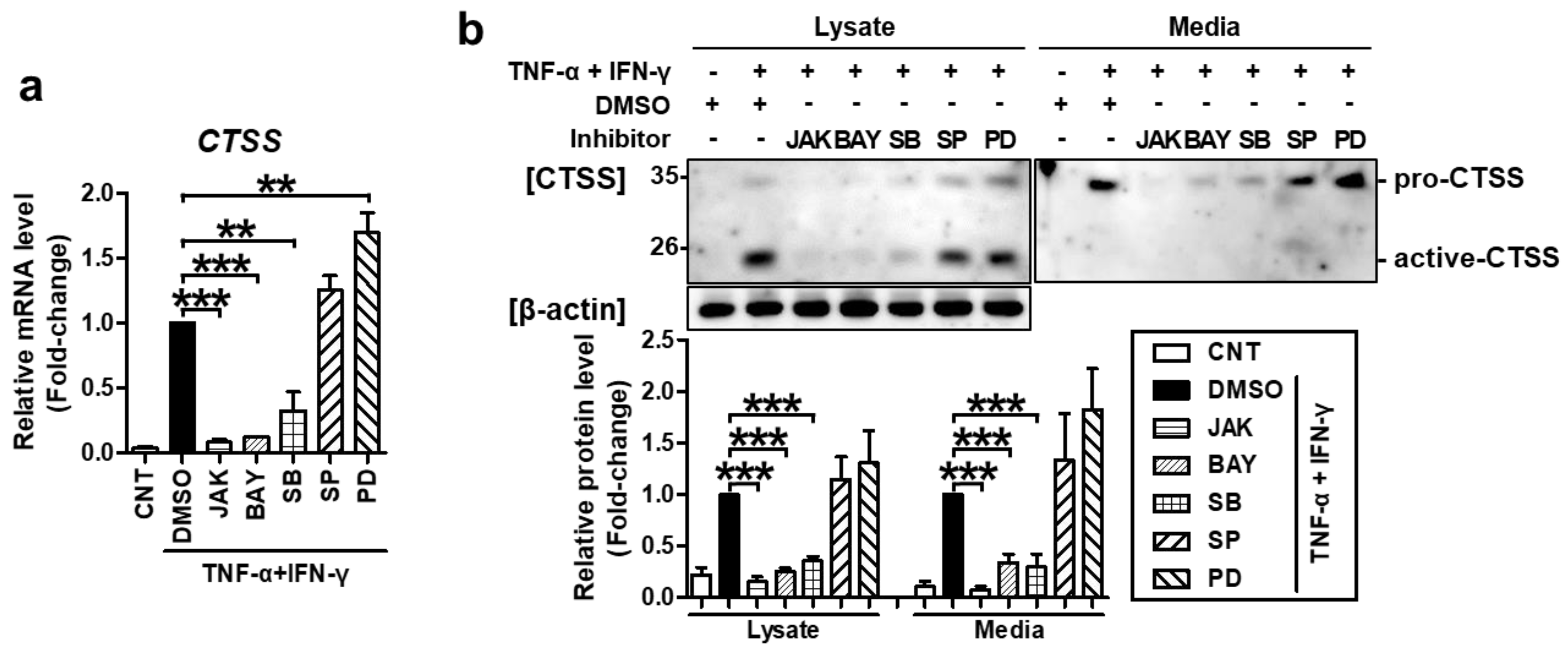
2.2. TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Expression Is Mediated by STAT1, NF-κB, and p38 MAPK Activation
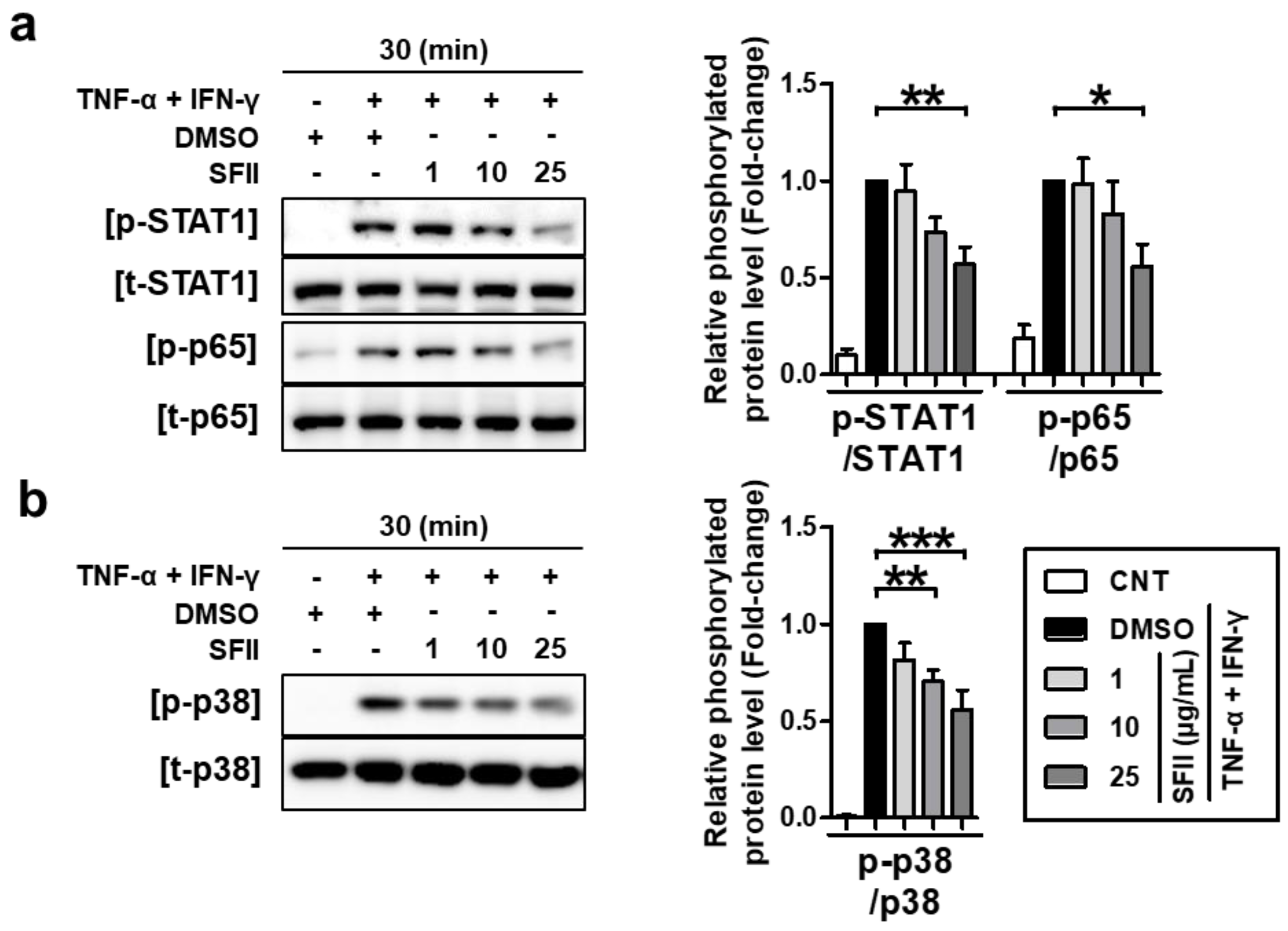
2.3. SFII Suppresses TARC, MDC, and CTSS by Inhibiting TNF-α/IFN-γ-Induced Phosphorylation of STAT1, p65, and p38 MAPK
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Cell Cytotoxicity
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Quantitative Real-Time PCR
4.6. Western Blot Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SFII | Skullcapflavone II |
| TNF-α | tumor necrosis factor-α |
| IFN-γ | interferon-γ |
| TARC | thymus- and activation-regulated chemokine |
| MDC | macrophage-derived chemokine |
| CTSS | cathepsin S |
| AD | atopic dermatitis |
References
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef]
- Leung, D.Y.; Guttman-Yassky, E. Deciphering the complexities of atopic dermatitis: Shifting paradigms in treatment approaches. J. Allergy Clin. Immunol. 2014, 134, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Prim. 2018, 4, 1. [Google Scholar] [CrossRef]
- Imai, T.; Nagira, M.; Takagi, S.; Kakizaki, M.; Nishimura, M.; Wang, J.; Gray, P.W.; Matsushima, K.; Yoshie, O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 1999, 11, 81–88. [Google Scholar] [CrossRef]
- Kataoka, Y. Thymus and activation-regulated chemokine as a clinical biomarker in atopic dermatitis. J. Dermatol. 2014, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Stander, S. Atopic Dermatitis. N. Engl. J. Med. 2021, 384, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, A.K.; Szafraniec, K.; Zuber, Z.; Wojas-Pelc, A.; Jaworek, J. Interleukin 25, thymic stromal lymphopoietin and house dust mites in pathogenesis of atopic dermatitis. J. Physiol. Pharmacol. 2020, 71, 291–297. [Google Scholar]
- Kakinuma, T.; Nakamura, K.; Wakugawa, M.; Mitsui, H.; Tada, Y.; Saeki, H.; Torii, H.; Asahina, A.; Onai, N.; Matsushima, K.; et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J. Allergy Clin. Immunol. 2001, 107, 535–541. [Google Scholar] [CrossRef]
- Horikawa, T.; Nakayama, T.; Hikita, I.; Yamada, H.; Fujisawa, R.; Bito, T.; Harada, S.; Fukunaga, A.; Chantry, D.; Gray, P.W.; et al. IFN-gamma-inducible expression of thymus and activation-regulated chemokine/CCL17 and macrophage-derived chemokine/CCL22 in epidermal keratinocytes and their roles in atopic dermatitis. Int. Immunol. 2002, 14, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijs, J.; Krastev, T.; Weidinger, S.; Buckens, C.F.; de Bruin-Weller, M.; Bruijnzeel-Koomen, C.; Flohr, C.; Hijnen, D. Biomarkers for atopic dermatitis: A systematic review and meta-analysis. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedoszytko, B.; Sokolowska-Wojdylo, M.; Ruckemann-Dziurdzinska, K.; Roszkiewicz, J.; Nowicki, R.J. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: Atopic dermatitis, psoriasis and skin mastocytosis. Postep. Dermatol. Alergol. 2014, 31, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.B.; Sivaprasad, U. Th2 Cytokines and Atopic Dermatitis. J. Clin. Cell. Immunol. 2011, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, C.; Yoneyama, H.; Murai, M.; Nakamura, K.; Tamaki, K.; Terashima, Y.; Imai, T.; Yoshie, O.; Irimura, T.; Mizutani, H.; et al. Overproduction of Th2-specific chemokines in NC/Nga mice exhibiting atopic dermatitis-like lesions. J. Clin. Investig. 1999, 104, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, C.; Bang, K.; Gesser, B.; Yoneyama, H.; Matsushima, K.; Larsen, C.G. A Th-2 chemokine, TARC, produced by keratinocytes may recruit CLA(+)CCR4(+) lymphocytes into lesional atopic dermatitis skin. J. Investig. Dermatol. 2000, 115, 640–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, C.; Saeki, H.; Komine, M.; Kagami, S.; Tsunemi, Y.; Ohtsuki, M.; Nakagawa, H. Mechanism of Macrophage-Derived Chemokine/CCL22 Production by HaCaT Keratinocytes. Ann. Dermatol. 2015, 27, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Komine, M.; Kakinuma, T.; Kagami, S.; Hanakawa, Y.; Hashimoto, K.; Tamaki, K. Mechanism of thymus- and activation-regulated chemokine (TARC)/CCL17 production and its modulation by roxithromycin. J. Investig. Dermatol. 2005, 125, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Mo, X.; Yan, F.; Liu, J.; Ye, S.; Zhang, Y.; Lin, Y.; Li, H.; Chen, D. Role of YAP-related T cell imbalance and epidermal keratinocyte dysfunction in the pathogenesis of atopic dermatitis. J. Dermatol. Sci. 2021, 101, 164–173. [Google Scholar] [CrossRef]
- Kim, S.M.; Ha, S.E.; Vetrivel, P.; Kim, H.H.; Bhosale, P.B.; Park, J.E.; Heo, J.D.; Kim, Y.S.; Kim, G.S. Cellular Function of Annexin A1 Protein Mimetic Peptide Ac2-26 in Human Skin Keratinocytes HaCaT and Fibroblast Detroit 551 Cells. Nutrients 2020, 12, 3261. [Google Scholar] [CrossRef]
- Reddy, V.B.; Shimada, S.G.; Sikand, P.; LaMotte, R.H.; Lerner, E.A. Cathepsin S Elicits Itch and Signals via Protease-Activated Receptors. J. Investig. Dermatol. 2010, 130, 1468–1470. [Google Scholar] [CrossRef] [Green Version]
- Steinhoff, M.; Neisius, U.; Ikoma, A.; Fartasch, M.; Heyer, G.; Skov, P.S.; Luger, T.A.; Schmelz, M. Proteinase-activated receptor-2 mediates itch: A novel pathway for pruritus in human skin. J. Neurosci. 2003, 23, 6176–6180. [Google Scholar] [CrossRef]
- Schwarz, G.; Boehncke, W.H.; Braun, M.; Schroter, C.J.; Burster, T.; Flad, T.; Dressel, D.; Weber, E.; Schmid, H.; Kalbacher, H. Cathepsin S activity is detectable in human keratinocytes and is selectively upregulated upon stimulation with interferon-gamma. J. Investig. Dermatol. 2002, 119, 44–49. [Google Scholar] [PubMed] [Green Version]
- Schonefuss, A.; Wendt, W.; Schattling, B.; Schulten, R.; Hoffmann, K.; Stuecker, M.; Tigges, C.; Lubbert, H.; Stichel, C. Upregulation of cathepsin S in psoriatic keratinocytes. Exp. Dermatol. 2010, 19, e80–e88. [Google Scholar] [CrossRef]
- Kim, N.; Bae, K.B.; Kim, M.O.; Yu, D.H.; Kim, H.J.; Yuh, H.S.; Ji, Y.R.; Park, S.J.; Kim, S.; Son, K.H.; et al. Overexpression of cathepsin S induces chronic atopic dermatitis in mice. J. Investig. Dermatol. 2012, 132, 1169–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, K.; Nishikawa, K.; Omoto, T.; Asai, I.; Yoshihira, K.; Shimomura, K. Two flavone 2’-glucosides from Scutellaria baicalensis. Phytochemistry 1995, 40, 279–281. [Google Scholar] [CrossRef]
- Bui, T.T.; Piao, C.H.; Song, C.H.; Chai, O.H. Skullcapflavone II attenuates ovalbumin-induced allergic rhinitis through the blocking of Th2 cytokine production and mast cell histamine release. Int. Immunopharmacol. 2017, 52, 77–84. [Google Scholar] [CrossRef]
- Salaritabar, A.; Darvishi, B.; Hadjiakhoondi, F.; Manayi, A.; Sureda, A.; Nabavi, S.F.; Fitzpatrick, L.R.; Nabavi, S.M.; Bishayee, A. Therapeutic potential of flavonoids in inflammatory bowel disease: A comprehensive review. World J. Gastroenterol. 2017, 23, 5097–5114. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Busselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.J.; Huang, W.C.; Hsieh, M.C.; Sung, P.J.; Kuo, Y.H.; Wu, W.H. Flavones Isolated from Scutellariae radix Suppress Propionibacterium Acnes-Induced Cytokine Production In Vitro and In Vivo. Molecules 2015, 21, 15. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.Y.; Ahn, K.S.; Park, M.J.; Kwon, O.K.; Lee, H.K.; Oh, S.R. Skullcapflavone II inhibits ovalbumin-induced airway inflammation in a mouse model of asthma. Int. Immunopharmacol. 2012, 12, 666–674. [Google Scholar] [CrossRef]
- Lee, J.; Son, H.S.; Lee, H.I.; Lee, G.R.; Jo, Y.J.; Hong, S.E.; Kim, N.; Kwon, M.; Kim, N.Y.; Kim, H.J.; et al. Skullcapflavone II inhibits osteoclastogenesis by regulating reactive oxygen species and attenuates the survival and resorption function of osteoclasts by modulating integrin signaling. FASEB J. 2019, 33, 2026–2036. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Seo, E.K.; Lee, S.T. Skullcapflavone II Inhibits Degradation of Type I Collagen by Suppressing MMP-1 Transcription in Human Skin Fibroblasts. Int. J. Mol. Sci. 2019, 20, 2734. [Google Scholar] [CrossRef] [Green Version]
- Ju, S.M.; Song, H.Y.; Lee, S.J.; Seo, W.Y.; Sin, D.H.; Goh, A.R.; Kang, Y.H.; Kang, I.J.; Won, M.H.; Yi, J.S.; et al. Suppression of thymus- and activation-regulated chemokine (TARC/CCL17) production by 1,2,3,4,6-penta-O-galloyl-beta-D-glucose via blockade of NF-kappaB and STAT1 activation in the HaCaT cells. Biochem. Biophys. Res. Commun. 2009, 387, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Hieshima, K.; Nagakubo, D.; Sato, E.; Nakayama, M.; Kawa, K.; Yoshie, O. Selective induction of Th2-attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein-Barr virus. J. Virol. 2004, 78, 1665–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Lee, H.S.; Lim, Y.; Paik, J.H.; Kwon, O.K.; Kim, J.H.; Paryanto, I.; Yunianto, P.; Choi, S.; Oh, S.R.; et al. Rhododendron album Blume extract inhibits TNF-/IFN--induced chemokine production via blockade of NF-B and JAK/STAT activation in human epidermal keratinocytes. Int. J. Mol. Med. 2018, 41, 3642–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.J.; Bae, Y.S.; Ju, S.M.; Goh, A.R.; Youn, G.S.; Choi, S.Y.; Park, J. Casuarinin suppresses TARC/CCL17 and MDC/CCL22 production via blockade of NF-kappaB and STAT1 activation in HaCaT cells. Biochem. Biophys. Res. Commun. 2012, 417, 1254–1259. [Google Scholar] [CrossRef]
- Baradaran Rahimi, V.; Askari, V.R.; Hosseinzadeh, H. Promising influences of Scutellaria baicalensis and its two active constituents, baicalin, and baicalein, against metabolic syndrome: A review. Phytother. Res. PTR 2021. [Google Scholar] [CrossRef]
- Dinda, B.; Dinda, S.; DasSharma, S.; Banik, R.; Chakraborty, A.; Dinda, M. Therapeutic potentials of baicalin and its aglycone, baicalein against inflammatory disorders. Eur. J. Med. Chem 2017, 131, 68–80. [Google Scholar] [CrossRef]
- Yun, M.Y.; Yang, J.H.; Kim, D.K.; Cheong, K.J.; Song, H.H.; Kim, D.H.; Cheong, K.J.; Kim, Y.I.; Shin, S.C. Therapeutic effects of Baicalein on atopic dermatitis-like skin lesions of NC/Nga mice induced by dermatophagoides pteronyssinus. Int. Immunopharmacol. 2010, 10, 1142–1148. [Google Scholar] [CrossRef]
- Kim, J.; Lee, I.; Park, S.; Choue, R. Effects of Scutellariae radix and Aloe vera gel extracts on immunoglobulin E and cytokine levels in atopic dermatitis NC/Nga mice. J. Ethnopharmacol. 2010, 132, 529–532. [Google Scholar] [CrossRef]
- Kubo, M.; Matsuda, H.; Tanaka, M.; Kimura, Y.; Okuda, H.; Higashino, M.; Tani, T.; Namba, K.; Arichi, S. Studies on Scutellariae radix. VII. Anti-arthritic and anti-inflammatory actions of methanolic extract and flavonoid components from Scutellariae radix. Chem. Pharm. Bull. 1984, 32, 2724–2729. [Google Scholar] [CrossRef] [Green Version]
- Parsafar, S.; Nayeri, Z.; Aliakbari, F.; Shahi, F.; Mohammadi, M.; Morshedi, D. Multiple neuroprotective features of Scutellaria pinnatifida-derived small molecule. Heliyon 2020, 6, e04737. [Google Scholar] [CrossRef]
- Wang, Z.L.; Wang, S.; Kuang, Y.; Hu, Z.M.; Qiao, X.; Ye, M. A comprehensive review on phytochemistry, pharmacology, and flavonoid biosynthesis of Scutellaria baicalensis. Pharm. Biol. 2018, 56, 465–484. [Google Scholar] [CrossRef] [Green Version]
- Tak, P.P.; Firestein, G.S. NF-kappa B: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 map kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Saccani, S.; Pantano, S.; Natoli, G. p38-dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat. Immunol. 2002, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Lahesmaa, R.; Vahedi, G.; Laurence, A.; Kanno, Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat. Rev. Immunol. 2011, 11, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhang, H.; Chan, L.S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAK-STAT 2013, 2, e24137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Melendo, C.; Cubiro, X.; Puig, L. Janus Kinase Inhibitors in Dermatology: Part 2: Applications in Psoriasis, Atopic Dermatitis, and Other Dermatoses. Actas Dermosifiliogr. 2021. [Google Scholar] [CrossRef]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Ramana, C.V.; Gil, M.P.; Schreiber, R.D.; Stark, G.R. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002, 23, 96–101. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Shimada, Y.; Takehara, K.; Sato, S. Both Th2 and Th1 chemokines (TARC/CCL17, MDC/CCL22, and Mig/CXCL9) are elevated in sera from patients with atopic dermatitis. J. Dermatol. Sci. 2004, 34, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Thijs, J.L.; de Bruin-Weller, M.S.; Hijnen, D. Current and Future Biomarkers in Atopic Dermatitis. Immunol. Allergy Clin. N. Am. 2017, 37, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wohlmann, A.; Sebastian, K.; Borowski, A.; Krause, S.; Friedrich, K. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol. Chem. 2010, 391, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; Peterson, D.; Ramseier, J.; Al-Bawardy, B.; Chun, H.; Proctor, D.; Strand, V.; Flavell, R.A.; King, B. The emerging role of Janus kinase inhibitors in the treatment of autoimmune and inflammatory diseases. J. Allergy Clin. Immunol. 2021, 147, 814–826. [Google Scholar] [CrossRef]
- Qi, X.F.; Kim, D.H.; Yoon, Y.S.; Li, J.H.; Song, S.B.; Jin, D.; Huang, X.Z.; Teng, Y.C.; Lee, K.J. The adenylyl cyclase-cAMP system suppresses TARC/CCL17 and MDC/CCL22 production through p38 MAPK and NF-kappaB in HaCaT keratinocytes. Mol. Immunol. 2009, 46, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Do, H.J.; Lee, E.; Yim, N.H.; Cho, W.K.; Park, K.I.; Ma, J.Y. Jageum-Jung improves 2,4-dinitrochlorobenzene-induced atopic dermatitis-like skin lesions in mice and suppresses pro-inflammatory chemokine production by inhibiting TNF-alpha/IFN-gamma-induced STAT-1 and NFkappaB signaling in HaCaT cells. J. Ethnopharmacol. 2018, 221, 48–55. [Google Scholar] [CrossRef]
- Kee, J.Y.; Jeon, Y.D.; Kim, D.S.; Han, Y.H.; Park, J.; Youn, D.H.; Kim, S.J.; Ahn, K.S.; Um, J.Y.; Hong, S.H. Korean Red Ginseng improves atopic dermatitis-like skin lesions by suppressing expression of proinflammatory cytokines and chemokines in vivo and in vitro. J. Ginseng Res. 2017, 41, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samukawa, K.; Izumi, Y.; Shiota, M.; Nakao, T.; Osada-Oka, M.; Miura, K.; Iwao, H. Red ginseng inhibits scratching behavior associated with atopic dermatitis in experimental animal models. J. Pharmacol. Sci. 2012, 118, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.H.; Kim, H.S.; Lee, W.; Han, E.J.; Kim, S.Y.; Fernando, I.P.S.; Ahn, G.; Kim, K.N. Eckol from Ecklonia cava ameliorates TNF-alpha/IFN-gamma-induced inflammatory responses via regulating MAPKs and NF-kappaB signaling pathway in HaCaT cells. Int. Immunopharmacol. 2020, 82, 106146. [Google Scholar] [CrossRef]
- Han, E.J.; Fernando, I.P.S.; Kim, H.S.; Lee, D.S.; Kim, A.; Je, J.G.; Seo, M.J.; Jee, Y.H.; Jeon, Y.J.; Kim, S.Y.; et al. (-)-Loliolide Isolated from Sargassum horneri Suppressed Oxidative Stress and Inflammation by Activating Nrf2/HO-1 Signaling in IFN-gamma/TNF-alpha-Stimulated HaCaT Keratinocytes. Antioxidants 2021, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Baek, J.; Lee, J.R.; Roh, J.Y.; Jung, Y. Optimization of Cytokine Milieu to Reproduce Atopic Dermatitis-related Gene Expression in HaCaT Keratinocyte Cell Line. Immune Netw. 2018, 18, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lim, J.; Oh, J.H.; Cho, S.; Chung, J.H. IGF-1 Upregulates Biglycan and Decorin by Increasing Translation and Reducing ADAMTS5 Expression. Int. J. Mol. Sci. 2021, 22, 1403. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Lee, D.H.; Oh, J.-H.; Chung, J.H. Skullcapflavone II Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Production in HaCaT Cells. Int. J. Mol. Sci. 2021, 22, 6428. https://doi.org/10.3390/ijms22126428
Lee H, Lee DH, Oh J-H, Chung JH. Skullcapflavone II Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Production in HaCaT Cells. International Journal of Molecular Sciences. 2021; 22(12):6428. https://doi.org/10.3390/ijms22126428
Chicago/Turabian StyleLee, Hanon, Dong Hun Lee, Jang-Hee Oh, and Jin Ho Chung. 2021. "Skullcapflavone II Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Production in HaCaT Cells" International Journal of Molecular Sciences 22, no. 12: 6428. https://doi.org/10.3390/ijms22126428
APA StyleLee, H., Lee, D. H., Oh, J. -H., & Chung, J. H. (2021). Skullcapflavone II Suppresses TNF-α/IFN-γ-Induced TARC, MDC, and CTSS Production in HaCaT Cells. International Journal of Molecular Sciences, 22(12), 6428. https://doi.org/10.3390/ijms22126428