
Gastritis, Gastric Polyps and Gastric Cancer



{kind=link}
Abstract
1. Introduction
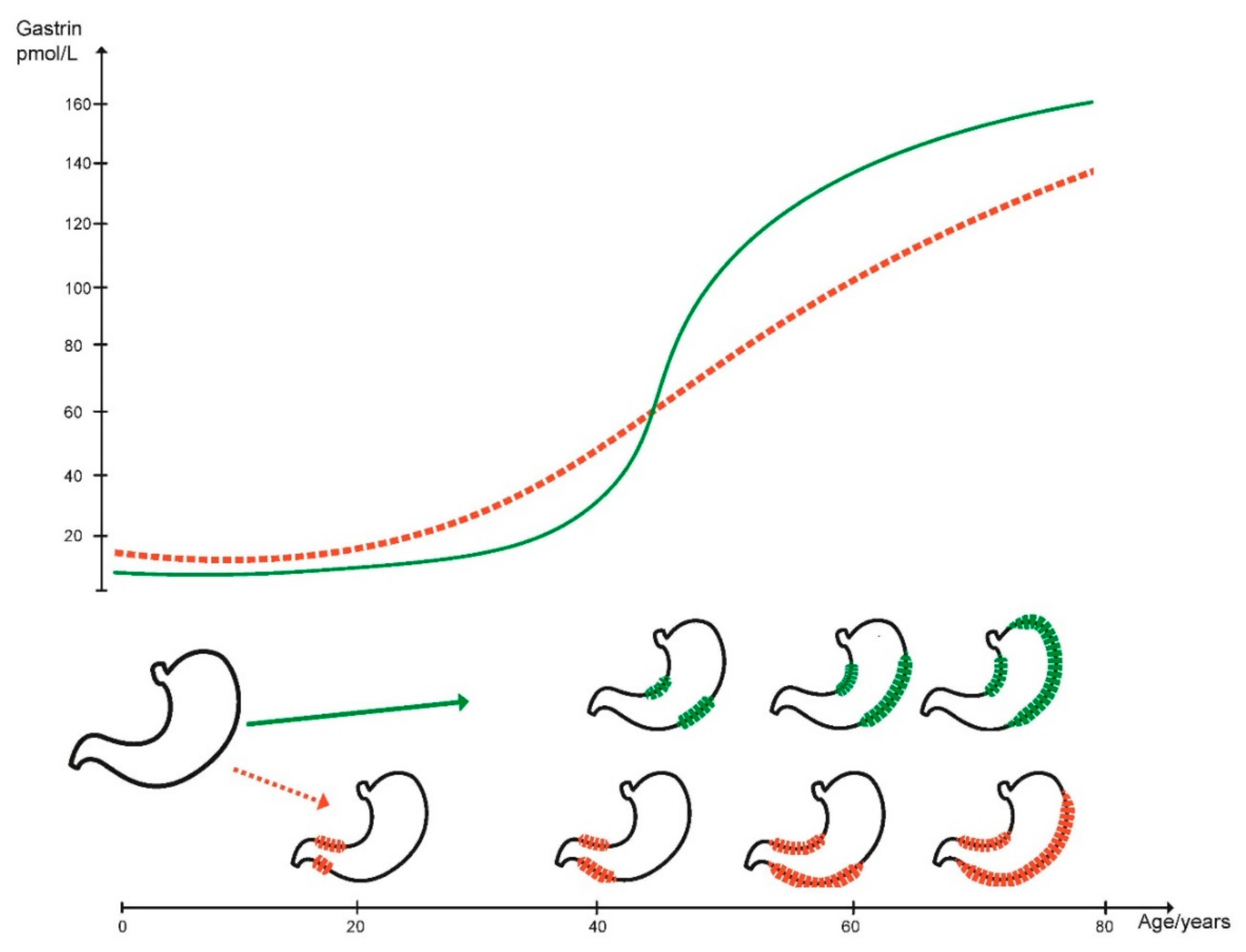
2. Gastric Cancer
3. Gastric Polyps
3.1. Hyperplastic Polyps
3.2. Adenomatous Polyps
3.3. Fundic Gland Polyps
3.4. Gastric Polyps in Patients with Portal Hypertension
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Waldum, H.L.; Hauso, O.; Sordal, O.F.; Fossmark, R. Gastrin May Mediate the Carcinogenic Effect of Helicobacter pylori Infection of the Stomach. Dig. Dis. Sci. 2015, 60, 1522–1527. [Google Scholar] [CrossRef]
- Waldum, H.L.; Fossmark, R. Types of Gastric Carcinomas. Int. J. Mol. Sci. 2018, 19, 4109. [Google Scholar] [CrossRef]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attemt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.T.; Zeng, L.; Yang, J.; Zeng, C.; Chen, Y. Analysis of the Incidence and Survival of Gastric Cancer Based on the Lauren Classification: A Large Population-Based Study Using SEER. Front. Oncol. 2020, 10, 1212. [Google Scholar] [CrossRef] [PubMed]
- Schauer, M.; Peiper, M.; Theisen, J.; Knoefel, W. Prognostic factors in patients with diffuse type gastric cancer (linitis plastica) after operative treatment. Eur. J. Med. Res. 2011, 16, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Sheweita, S.A.; Alsamghan, A.S. Molecular Mechanisms Contributing Bacterial Infections to the Incidence of Various Types of Cancer. Mediators Inflamm. 2020, 2020, 4070419. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Forman, D.; Waskito, L.A.; Yamaoka, Y.; Crabtree, J.E. Epidemiology of Helicobacter pylori and CagA-Positive Infections and Global Variations in Gastric Cancer. Toxins 2018, 10, 163. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Miller, G.G.; Tham, K.T.; Perez-Perez, G.I.; Zhao, X.; Atherton, J.C.; Blaser, M.J. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab. Investig. 1995, 73, 760–770. [Google Scholar] [PubMed]
- Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. Int. J. Mol. Sci. 2020, 21, 7430. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Nascakova, Z.; Mihai, A.I.; Cheng, P.F.; Levesque, M.P.; Lampart, S.; Hurwitz, R.; Pfannkuch, L.; Dobrovolna, J.; Jacobs, M.; et al. The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat. Commun. 2020, 11, 5117. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Negrini, R.; Savio, A.; Poiesi, C.; Appelmelk, B.J.; Buffoli, F.; Paterlini, A.; Cesari, P.; Graffeo, M.; Vaira, D.; Franzin, G. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology 1996, 111, 655–665. [Google Scholar] [CrossRef]
- Faller, G.; Steininger, H.; Appelmelk, B.; Kirchner, T. Evidence of novel pathogenic pathways for the formation of antigastric autoantibodies in Helicobacter pylori gastritis. J. Clin. Pathol. 1998, 51, 244–245. [Google Scholar] [CrossRef]
- Rowland, M.; Daly, L.; Vaughan, M.; Higgins, A.; Bourke, B.; Drumm, B. Age-specific incidence of Helicobacter pylori. Gastroenterology 2006, 130, 65–72. [Google Scholar] [CrossRef]
- Haruma, K.; Kamada, T.; Kawaguchi, H.; Okamoto, S.; Yoshihara, M.; Sumii, K.; Inoue, M.; Kishimoto, S.; Kajiyama, G.; Miyoshi, A. Effect of age and Helicobacter pylori infection on gastric acid secretion. J. Gastroenterol. Hepatol. 2000, 15, 277–283. [Google Scholar] [CrossRef]
- Waldum, H.; Mjønes, P. Towards Understanding of Gastric Cancer Based upon Physiological Role of Gastrin and ECL Cells. Cancers 2020, 12, 3477. [Google Scholar] [CrossRef] [PubMed]
- Strickland, R.G.; Bhathal, P.S.; Korman, M.G.; Hansky, J. Serum gastrin and the antral mucosa in atrophic gastritis. Br. Med. J. 1971, 4, 451–453. [Google Scholar] [CrossRef]
- Borch, K.; Renvall, H.; Liedberg, G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985, 88, 638–648. [Google Scholar] [CrossRef]
- Zamcheck, N.; Grable, E.; Ley, A.; Norman, L. Occurrence of gastric cancer among patients with pernicious anemia at the Boston City Hospital. N. Engl. J. Med. 1955, 252, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Bizzaro, N.; Antico, A.; Villalta, D. Autoimmunity and Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 377. [Google Scholar] [CrossRef]
- Sato, Y.; Iwafuchi, M.; Ueki, J.; Yoshimura, A.; Mochizuki, T.; Motoyama, H.; Sugimura, K.; Honma, T.; Narisawa, R.; Ichida, T.; et al. Gastric carcinoid tumors without autoimmune gastritis in Japan: A relationship with Helicobacter pylori infection. Dig. Dis. Sci. 2002, 47, 579–585. [Google Scholar] [CrossRef]
- Antonodimitrakis, P.; Tsolakis, A.; Welin, S.; Kozlovacki, G.; Oberg, K.; Granberg, D. Gastric carcinoid in a patient infected with Helicobacter pylori: A new entity? World J. Gastroenterol. 2011, 17, 3066–3068. [Google Scholar] [CrossRef]
- Hansson, L.R.; Engstrand, L.; Nyren, O.; Lindgren, A. Prevalence of Helicobacter pylori infection in subtypes of gastric cancer. Gastroenterology 1995, 109, 885–888. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, H.S.; Kim, N.; Shin, C.M.; Lee, S.H.; Park, Y.S.; Hwang, J.H.; Kim, J.W.; Jeong, S.H.; Lee, D.H.; et al. Prevalence and clinicopathologic characteristics of gastric cardia cancer in South Korea. Helicobacter 2012, 17, 358–368. [Google Scholar] [CrossRef]
- Sjöblom, S.M.; Sipponen, P.; Miettinen, M.; Karonen, S.L.; Jrvinen, H.J. Gastroscopic screening for gastric carcinoids and carcinoma in pernicious anemia. Endoscopy 1988, 20, 52–56. [Google Scholar] [CrossRef]
- Qvigstad, G.; Qvigstad, T.; Westre, B.; Sandvik, A.K.; Brenna, E.; Waldum, H.L. Neuroendocrine differentiation in gastric adenocarcinomas associated with severe hypergastrinemia and/or pernicious anemia. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2002, 110, 132–139. [Google Scholar] [CrossRef]
- Bakke, I.; Qvigstad, G.; Brenna, E.; Sandvik, A.K.; Waldum, H.L. Gastrin has a specific proliferative effect on the rat enterochromaffin-like cell, but not on the parietal cell: A study by elutriation centrifugation. Acta Physiol. Scand. 2000, 169, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tielemans, Y.; Chen, D.; Sundler, F.; Håkanson, R.; Willems, G. Reversibility of the cell kinetic changes induced by omeprazole in the rat oxyntic mucosa. An autoradiographic study using tritiated thymidine. Scand. J. Gastroenterol. 1992, 27, 155–160. [Google Scholar] [CrossRef]
- Fukui, H.; Kinoshita, Y.; Maekawa, T.; Okada, A.; Waki, S.; Hassan, S.; Okamoto, H.; Chiba, T. Regenerating gene protein may mediate gastric mucosal proliferation induced by hypergastrinemia in rats. Gastroenterology 1998, 115, 1483–1493. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Kadowaki, Y.; Ishihara, S.; Ose, T.; Fukuhara, H.; Kazumori, H.; Takasawa, S.; Okamoto, H.; Chiba, T.; Kinoshita, Y. Transgenic overexpression of Reg protein caused gastric cell proliferation and differentiation along parietal cell and chief cell lineages. Oncogene 2004, 23, 3572–3579. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Spence, A.D.; Cardwell, C.R.; McMenamin, Ú.C.; Hicks, B.M.; Johnston, B.T.; Murray, L.J.; Coleman, H.G. Adenocarcinoma risk in gastric atrophy and intestinal metaplasia: A systematic review. BMC Gastroenterol. 2017, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hayakawa, Y.; Koike, K. Metaplasia in the Stomach-Precursor of Gastric Cancer? Int. J. Mol. Sci. 2017, 18, 2063. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Zou, W.Y. Guilt by association: Intestinal metaplasia does not progress to gastric cancer. Curr. Opin. Gastroenterol. 2018, 34, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.J., 3rd; Richardson, C.T.; Walsh, J.H.; Feldman, M. Variable contribution of gastrin to gastric acid secretion after a meal in humans. Gastroenterology 1987, 92, 944–949. [Google Scholar] [CrossRef]
- Jianu, C.S.; Fossmark, R.; Viset, T.; Qvigstad, G.; Sordal, O.; Marvik, R.; Waldum, H.L. Gastric carcinoids after long-term use of a proton pump inhibitor. Aliment. Pharmacol. Ther. 2012, 36, 644–649. [Google Scholar] [CrossRef]
- Cavalcoli, F.; Zilli, A.; Conte, D.; Ciafardini, C.; Massironi, S. Gastric neuroendocrine neoplasms and proton pump inhibitors: Fact or coincidence? Scand. J. Gastroenterol. 2015, 50, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Nandy, N.; Hanson, J.A.; Strickland, R.G.; McCarthy, D.M. Solitary Gastric Carcinoid Tumor Associated with Long-Term Use of Omeprazole: A Case Report and Review of the Literature. Dig. Dis. Sci. 2016, 61, 708–712. [Google Scholar] [CrossRef]
- Lamberts, R.; Creutzfeldt, W.; Stockmann, F.; Jacubaschke, U.; Maas, S.; Brunner, G. Long-term omeprazole treatment in man: Effects on gastric endocrine cell populations. Digestion 1988, 39, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Jianu, C.S.; Lange, O.J.; Viset, T.; Qvigstad, G.; Martinsen, T.C.; Fougner, R.; Kleveland, P.M.; Fossmark, R.; Hauso, Ø.; Waldum, H.L. Gastric neuroendocrine carcinoma after long-term use of proton pump inhibitor. Scand. J. Gastroenterol. 2012, 47, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Calvete, O.; Reyes, J.; Zuniga, S.; Paumard-Hernandez, B.; Fernandez, V.; Bujanda, L.; Rodriguez-Pinilla, M.S.; Palacios, J.; Heine-Suner, D.; Banka, S.; et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum. Mol. Genet. 2015, 24, 2914–2922. [Google Scholar] [CrossRef]
- Fossmark, R.; Calvete, O.; Mjones, P.; Benitez, J.; Waldum, H.L. ECL-cell carcinoids and carcinoma in patients homozygous for an inactivating mutation in the gastric H(+) K(+) ATPase alpha subunit. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2016, 124, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Solcia, E.; Rindi, G.; Silini, E.; Villani, L. Enterochromaffin-like (ECL) cells and their growths: Relationships to gastrin, reduced acid secretion and gastritis. Baillieres Clin. Gastroenterol. 1993, 7, 149–165. [Google Scholar] [CrossRef]
- Solcia, E.; Capella, C.; Buffa, R.; Usellini, L.; Frigerio, B.; Fontana, P. Endocrine cells of the gastrointestinal tract and related tumors. Pathobiol. Annu. 1979, 9, 163–204. [Google Scholar]
- Moayyedi, P.; Eikelboom, J.W.; Bosch, J.; Connolly, S.J.; Dyal, L.; Shestakovska, O.; Leong, D.; Anand, S.S.; Stork, S.; Branch, K.R.H.; et al. Safety of Proton Pump Inhibitors Based on a Large, Multi-Year, Randomized Trial of Patients Receiving Rivaroxaban or Aspirin. Gastroenterology 2019, 157, 682–691. [Google Scholar] [CrossRef]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Wong, I.C.K.; Leung, W.K. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: A population-based study. Gut 2018, 67, 28–35. [Google Scholar] [CrossRef]
- Cheung, K.S.; Leung, W.K. Long-term use of proton-pump inhibitors and risk of gastric cancer: A review of the current evidence. Ther. Adv. Gastroenterol. 2019, 12, 1756284819834511. [Google Scholar] [CrossRef]
- Brusselaers, N.; Wahlin, K.; Engstrand, L.; Lagergren, J. Maintenance therapy with proton pump inhibitors and risk of gastric cancer: A nationwide population-based cohort study in Sweden. BMJ Open 2017, 7, e017739. [Google Scholar] [CrossRef]
- Bergquist, J.R.; Leiting, J.L.; Habermann, E.B.; Cleary, S.P.; Kendrick, M.L.; Smoot, R.L.; Nagorney, D.M.; Truty, M.J.; Grotz, T.E. Early-onset gastric cancer is a distinct disease with worrisome trends and oncogenic features. Surgery 2019, 166, 547–555. [Google Scholar] [CrossRef]
- Waldum, H.L. The increase in early-onset gastric carcinomas from 1995 is probably due to the introduction of proton pump inhibitors. Surgery 2020, 168, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Lundell, L.; Klinkenberg-Knol, E.C.; Havu, N.; Festen, H.P.; Liedman, B.; Lamers, C.B.; Jansen, J.B.; Dalenback, J.; Snel, P.; et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N. Engl. J. Med. 1996, 334, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, S.G.; Craanen, M.E.; Kuipers, E.J. Gastric mucosal morphological consequences of acid suppression: A balanced view. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.E.; Kuipers, E.J.; Klinkenberg-Knol, E.C.; Bloemena, E.; Nelis, G.F.; Festen, H.P.; Jansen, E.H.; Biemond, I.; Lamers, C.B.; Meuwissen, S.G. Hypergastrinaemia during long-term omeprazole therapy: Influences of vagal nerve function, gastric emptying and Helicobacter pylori infection. Aliment. Pharmacol. Ther. 1998, 12, 605–612. [Google Scholar] [CrossRef]
- Havu, N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion 1986, 35 (Suppl. 1), 42–55. [Google Scholar] [CrossRef]
- Poynter, D.; Selway, S.A.; Papworth, S.A.; Riches, S.R. Changes in the gastric mucosa of the mouse associated with long lasting unsurmountable histamine H2 blockade. Gut 1986, 27, 1338–1346. [Google Scholar] [CrossRef]
- Martinsen, T.C.; Kawase, S.; Hakanson, R.; Torp, S.H.; Fossmark, R.; Qvigstad, G.; Sandvik, A.K.; Waldum, H.L. Spontaneous ECL cell carcinomas in cotton rats: Natural course and prevention by a gastrin receptor antagonist. Carcinogenesis 2003, 24, 1887–1896. [Google Scholar] [CrossRef]
- Cadiot, G.; Vissuzaine, C.; Potet, F.; Mignon, M. Fundic argyrophil carcinoid tumor in a patient with sporadic-type Zollinger-Ellison syndrome. Dig. Dis. Sci. 1995, 40, 1275–1278. [Google Scholar] [CrossRef]
- Solcia, E.; Capella, C.; Fiocca, R.; Rindi, G.; Rosai, J. Gastric argyrophil carcinoidosis in patients with Zollinger-Ellison syndrome due to type 1 multiple endocrine neoplasia. A newly recognized association. Am. J. Surg. Pathol. 1990, 14, 503–513. [Google Scholar] [CrossRef]
- Bordi, C.; Senatore, S.; Missale, G. Gastric carcinoid following gastrojejunostomy. Am. J. Dig. Dis. 1976, 21, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Moore, A.R.; Sagatun, L.; Parsons, B.N.; Varro, A.; Campbell, F.; Fossmark, R.; Waldum, H.L.; Pritchard, D.M. Netazepide, a gastrin/cholecystokinin-2 receptor antagonist, can eradicate gastric neuroendocrine tumours in patients with autoimmune chronic atrophic gastritis. Br. J. Clin. Pharmacol. 2017, 83, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Fossmark, R.; Sordal, O.; Jianu, C.S.; Qvigstad, G.; Nordrum, I.S.; Boyce, M.; Waldum, H.L. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment. Pharmacol. Ther. 2012, 36, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.H.; Christensen, S.; McLaughlin, J.K.; Thomsen, R.W.; Sørensen, H.T.; Olsen, J.H.; Friis, S. Proton pump inhibitors and risk of gastric cancer: A population-based cohort study. Br. J. Cancer 2009, 100, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.I.; Park, C.H.; You, S.C.; Kim, J.Y.; Lee, K.J.; Kim, J.; Kim, Y.; Yoo, J.J.; Seo, W.W.; Lee, H.S.; et al. Association between proton pump inhibitor use and gastric cancer: A population-based cohort study using two different types of nationwide databases in Korea. Gut 2021. [Google Scholar] [CrossRef]
- Waldum, H.L.; Sørdal, Ø.; Fossmark, R. Proton pump inhibitors (PPIs) may cause gastric cancer—Clinical consequences. J. Gastroenterol. 2018, 53, 639–642. [Google Scholar] [CrossRef]
- Qvigstad, G.; Arnestad, J.S.; Brenna, E.; Waldum, H.L. Treatment with proton pump inhibitors induces tolerance to histamine-2 receptor antagonists in Helicobacter pylori-negative patients. Scand. J. Gastroenterol. 1998, 33, 1244–1248. [Google Scholar]
- Wölffling, S.; Daddi, A.A.; Imai-Matsushima, A.; Fritsche, K.; Goosmann, C.; Traulsen, J.; Lisle, R.; Schmid, M.; Del Mar Reines-Benassar, M.; Pfannkuch, L.; et al. EGF and BMPs govern differentiation and patterning in human gastric glands. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Kishikawa, H.; Ojiro, K.; Nakamura, K.; Katayama, T.; Arahata, K.; Takarabe, S.; Miura, S.; Kanai, T.; Nishida, J. Previous Helicobacter pylori infection-induced atrophic gastritis: A distinct disease entity in an understudied population without a history of eradication. Helicobacter 2020, 25, e12669. [Google Scholar] [CrossRef]
- Okada, K.; Suzuki, S.; Naito, S.; Yamada, Y.; Haruki, S.; Kubota, M.; Nakajima, Y.; Shimizu, T.; Ando, K.; Uchida, Y.; et al. Incidence of metachronous gastric cancer in patients whose primary gastric neoplasms were discovered after Helicobacter pylori eradication. Gastrointest. Endosc. 2019, 89, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Algin, E.; Baykara, M.; Yilmaz, G.; Cetin, B.; Ekinci, O.; Uner, A.; Ozet, A. Is there any relationship between Helicobacter pylori infection and human epidermal growth factor receptor 2 expression in gastric cancer? J. Cancer Res. Ther. 2020, 16, S128–S132. [Google Scholar] [CrossRef]
- Leushacke, M.; Tan, S.H.; Wong, A.; Swathi, Y.; Hajamohideen, A.; Tan, L.T.; Goh, J.; Wong, E.; Denil, S.; Murakami, K.; et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat. Cell Biol. 2017, 19, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Take, S.; Mizuno, M.; Ishiki, K.; Kusumoto, C.; Imada, T.; Hamada, F.; Yoshida, T.; Yokota, K.; Mitsuhashi, T.; Okada, H. Risk of gastric cancer in the second decade of follow-up after Helicobacter pylori eradication. J. Gastroenterol. 2020, 55, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ness-Jensen, E.; Bringeland, E.A.; Mattsson, F.; Mjønes, P.; Lagergren, J.; Grønbech, J.E.; Waldum, H.L.; Fossmark, R. Hypergastrinemia is associated with an increased risk of gastric adenocarcinoma with proximal location: A prospective population-based nested case-control study. Int. J. Cancer 2021, 148, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Roland, J.T.; Barlow, B.J.; O’Neal, R.; Rich, A.E.; Nam, K.T.; Shi, C.; Goldenring, J.R. Cell lineage distribution atlas of the human stomach reveals heterogeneous gland populations in the gastric antrum. Gut 2014, 63, 1711–1720. [Google Scholar] [CrossRef]
- Waldum, H.L.; Oberg, K.; Sordal, O.F.; Sandvik, A.K.; Gustafsson, B.I.; Mjones, P.; Fossmark, R. Not only stem cells, but also mature cells, particularly neuroendocrine cells, may develop into tumours: Time for a paradigm shift. Ther. Adv. Gastroenterol. 2018, 11, 1756284818775054. [Google Scholar] [CrossRef]
- Alexandraki, K.I.; Spyroglou, A.; Kykalos, S.; Daskalakis, K.; Kyriakopoulos, G.; Sotiropoulos, G.C.; Kaltsas, G.A.; Grossman, A.B. Changing biological behaviour of NETs during the evolution of the disease: Progress on progression. Endocr. Relat. Cancer 2021, 28, R121–R140. [Google Scholar] [CrossRef]
- Vos, S.; van der Post, R.S.; Brosens, L.A.A. Gastric Epithelial Polyps: When to Ponder, When to Panic. Surg. Pathol. Clin. 2020, 13, 431–452. [Google Scholar] [CrossRef] [PubMed]
- Carmack, S.W.; Genta, R.M.; Schuler, C.M.; Saboorian, M.H. The current spectrum of gastric polyps: A 1-year national study of over 120,000 patients. Am. J. Gastroenterol. 2009, 104, 1524–1532. [Google Scholar] [CrossRef]
- Abraham, S.C.; Singh, V.K.; Yardley, J.H.; Wu, T.T. Hyperplastic polyps of the stomach: Associations with histologic patterns of gastritis and gastric atrophy. Am. J. Surg. Pathol. 2001, 25, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, E.; Lahner, E.; Micheletti, A.; Milione, M.; D’Ambra, G.; Bordi, C.; Delle Fave, G.; Annibale, B. Occurrence and risk factors for benign epithelial gastric polyps in atrophic body gastritis on diagnosis and follow-up. Aliment. Pharmacol. Ther. 2005, 21, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Goddard, A.F.; Badreldin, R.; Pritchard, D.M.; Walker, M.M.; Warren, B. The management of gastric polyps. Gut 2010, 59, 1270–1276. [Google Scholar] [CrossRef]
- Fan, N.N.; Yang, J.; Sun, G.; Lu, Z.S.; Ling Hu, E.Q.; Wang, X.D.; Yang, Y.S. Changes in the spectrum of gastric polyps in the Chinese population. World J. Gastroenterol. 2015, 21, 9758–9764. [Google Scholar] [CrossRef]
- Suzuki, S.; Ohkusa, T.; Shimoi, K.; Horiuchi, T.; Fujiki, K.; Takashimizu, I. Disappearance of multiple hyperplastic polyps after the eradication of Helicobacter pylori. Gastrointest. Endosc. 1997, 46, 566–568. [Google Scholar] [CrossRef]
- Terada, T. Malignant transformation of foveolar hyperplastic polyp of the stomach: A histopathological study. Med. Oncol. 2011, 28, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Melton, S.D.; Genta, R.M. Gastric cardiac polyps: A clinicopathologic study of 330 cases. Am. J. Surg. Pathol. 2010, 34, 1792–1797. [Google Scholar] [CrossRef]
- Rich, A.; Toro, T.Z.; Tanksley, J.; Fiske, W.H.; Lind, C.D.; Ayers, G.D.; Piessevaux, H.; Washington, M.K.; Coffey, R.J. Distinguishing Ménétrier’s disease from its mimics. Gut 2010, 59, 1617–1624. [Google Scholar] [CrossRef]
- Ohkusa, T.; Miwa, H.; Hojo, M.; Kumagai, J.; Tanizawa, T.; Asaoka, D.; Terai, T.; Ohkura, R.; Sato, N. Endoscopic, histological and serologic findings of gastric hyperplastic polyps after eradication of Helicobacter pylori: Comparison between responder and non-responder cases. Digestion 2003, 68, 57–62. [Google Scholar] [CrossRef]
- Chetty, R.; Gill, P.; Mugon, P.; Shrimankar, J.; Hughes, C. Gastric neuroendocrine cell hyperplasia and type 1 tumours occurring within gastric hyperplastic polyps. Virchows Arch. 2012, 461, 483–487. [Google Scholar] [CrossRef]
- Hongo, M.; Fujimoto, K. Incidence and risk factor of fundic gland polyp and hyperplastic polyp in long-term proton pump inhibitor therapy: A prospective study in Japan. J. Gastroenterol. 2010, 45, 618–624. [Google Scholar] [CrossRef]
- Bordi, C.; Bertelè, A.; Davighi, M.C.; Pilato, F.; Carfagna, G.; Missale, G. Clinical and pathological associations of argyrophil cell hyperplasias of the gastric mucosa. Appl. Pathol. 1984, 2, 282–291. [Google Scholar] [PubMed]
- Hu, H.; Zhang, Q.; Chen, G.; Pritchard, D.M.; Zhang, S. Risk factors and clinical correlates of neoplastic transformation in gastric hyperplastic polyps in Chinese patients. Sci. Rep. 2020, 10, 2582. [Google Scholar] [CrossRef]
- Banks, M.; Graham, D.; Jansen, M.; Gotoda, T.; Coda, S.; di Pietro, M.; Uedo, N.; Bhandari, P.; Pritchard, D.M.; Kuipers, E.J.; et al. British Society of Gastroenterology guidelines on the diagnosis and management of patients at risk of gastric adenocarcinoma. Gut 2019, 68, 1545–1575. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Oh, T.H.; Seo, J.Y.; Joen, T.J.; Seo, D.D.; Shin, W.C.; Choi, W.C.; Kim, J.Y. Clinical factors predicting for neoplastic transformation of gastric hyperplastic polyps. Korean J. Gastroenterol. 2011, 58, 184–189. [Google Scholar] [CrossRef]
- Daibo, M.; Itabashi, M.; Hirota, T. Malignant transformation of gastric hyperplastic polyps. Am. J. Gastroenterol. 1987, 82, 1016–1025. [Google Scholar]
- Evans, J.A.; Chandrasekhara, V.; Chathadi, K.V.; Decker, G.A.; Early, D.S.; Fisher, D.A.; Foley, K.; Hwang, J.H.; Jue, T.L.; Lightdale, J.R.; et al. The role of endoscopy in the management of premalignant and malignant conditions of the stomach. Gastrointest. Endosc. 2015, 82, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Na, K.; Sung, J.Y.; Jang, J.Y.; Kim, Y.W. Adenocarcinoma and Neuroendocrine Collision Tumor in a Giant Gastric Hyperplastic Polyp. J. Nippon. Med. Sch. 2020, 87, 157–161. [Google Scholar] [CrossRef]
- Zea-Iriarte, W.L.; Itsuno, M.; Makiyama, K.; Hara, K.; Haraguchi, M.; Ajioka, Y. Signet ring cell carcinoma in hyperplastic polyp. Scand. J. Gastroenterol. 1995, 30, 604–608. [Google Scholar] [CrossRef]
- Carneiro, F.; David, L.; Seruca, R.; Castedo, S.; Nesland, J.M.; Sobrinho-Simões, M. Hyperplastic polyposis and diffuse carcinoma of the stomach. A study of a family. Cancer 1993, 72, 323–329. [Google Scholar] [CrossRef]
- Anjiki, H.; Mukaisho, K.I.; Kadomoto, Y.; Doi, H.; Yoshikawa, K.; Nakayama, T.; Vo, D.T.; Hattori, T.; Sugihara, H. Adenocarcinoma arising in multiple hyperplastic polyps in a patient with Helicobacter pylori infection and hypergastrinemia during long-term proton pump inhibitor therapy. Clin. J. Gastroenterol. 2017, 10, 128–136. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kato, M.; Matsuda, K.; Abiko, S.; Tsuda, M.; Mizushima, T.; Yamamoto, K.; Ono, S.; Kudo, T.; Shimizu, Y.; et al. Gastric Hyperplastic Polyps Associated with Proton Pump Inhibitor Use in a Case without a History of Helicobacter pylori Infection. Intern. Med. 2017, 56, 1825–1829. [Google Scholar] [CrossRef]
- Kara, D.; Hüsing-Kabar, A.; Schmidt, H.; Grünewald, I.; Chandhok, G.; Maschmeier, M.; Kabar, I. Portal Hypertensive Polyposis in Advanced Liver Cirrhosis: The Unknown Entity? Can. J. Gastroenterol. Hepatol. 2018, 2018, 2182784. [Google Scholar] [CrossRef]
- Yasugi, K.; Haruma, K.; Kawanaka, M.; Suehiro, M.; Nakamura, J.; Urata, N.; Tanikawa, T.; Oka, T.; Monobe, Y.; Fujita, T.; et al. Disappearance of Gastric Hyperplastic Polyps after the Discontinuation of Proton Pump Inhibitor in a Patient with Liver Cirrhosis. Case Rep. Gastroenterol. 2021, 15, 202–209. [Google Scholar] [CrossRef]
- Saito, K.; Arai, K.; Mori, M.; Kobayashi, R.; Ohki, I. Effect of Helicobacter pylori eradication on malignant transformation of gastric adenoma. Gastrointest. Endosc. 2000, 52, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.C.; Park, S.J.; Lee, J.H.; Mugartegui, L.; Wu, T.T. Genetic alterations in gastric adenomas of intestinal and foveolar phenotypes. Mod. Pathol. 2003, 16, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Borch, K.; Skarsgård, J.; Franzén, L.; Mårdh, S.; Rehfeld, J.F. Benign gastric polyps: Morphological and functional origin. Dig. Dis. Sci. 2003, 48, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Odze, R.D.; Marcial, M.A.; Antonioli, D. Gastric fundic gland polyps: A morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum. Pathol. 1996, 27, 896–903. [Google Scholar] [CrossRef]
- Zwick, A.; Munir, M.; Ryan, C.K.; Gian, J.; Burt, R.W.; Leppert, M.; Spirio, L.; Chey, W.Y. Gastric adenocarcinoma and dysplasia in fundic gland polyps of a patient with attenuated adenomatous polyposis coli. Gastroenterology 1997, 113, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Koornstra, J.J.; Götz, J.M.; van der Waaij, L.A.; de Jong, S.; Zwart, N.; Karrenbeld, A.; Kleibeuker, J.H. High-grade dysplasia in sporadic fundic gland polyps: A case report and review of the literature. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1229–1233. [Google Scholar] [CrossRef]
- Sekine, S.; Shibata, T.; Yamauchi, Y.; Nakanishi, Y.; Shimoda, T.; Sakamoto, M.; Hirohashi, S. Beta-catenin mutations in sporadic fundic gland polyps. Virchows Arch. 2002, 440, 381–386. [Google Scholar] [CrossRef]
- Straub, S.F.; Drage, M.G.; Gonzalez, R.S. Comparison of dysplastic fundic gland polyps in patients with and without familial adenomatous polyposis. Histopathology 2018, 72, 1172–1179. [Google Scholar] [CrossRef]
- Iwama, T.; Mishima, Y.; Utsunomiya, J. The impact of familial adenomatous polyposis on the tumorigenesis and mortality at the several organs. Its rational treatment. Ann. Surg. 1993, 217, 101–108. [Google Scholar] [CrossRef]
- Mankaney, G.; Leone, P.; Cruise, M.; LaGuardia, L.; O’Malley, M.; Bhatt, A.; Church, J.; Burke, C.A. Gastric cancer in FAP: A concerning rise in incidence. Fam. Cancer 2017, 16, 371–376. [Google Scholar] [CrossRef]
- Walton, S.J.; Frayling, I.M.; Clark, S.K.; Latchford, A. Gastric tumours in FAP. Fam. Cancer 2017, 16, 363–369. [Google Scholar] [CrossRef]
- Li, J.; Woods, S.L.; Healey, S.; Beesley, J.; Chen, X.; Lee, J.S.; Sivakumaran, H.; Wayte, N.; Nones, K.; Waterfall, J.J.; et al. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am. J. Hum. Genet. 2016, 98, 830–842. [Google Scholar] [CrossRef]
- Worthley, D.L.; Phillips, K.D.; Wayte, N.; Schrader, K.A.; Healey, S.; Kaurah, P.; Shulkes, A.; Grimpen, F.; Clouston, A.; Moore, D.; et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): A new autosomal dominant syndrome. Gut 2012, 61, 774–779. [Google Scholar] [CrossRef]
- De Boer, W.B.; Ee, H.; Kumarasinghe, M.P. Neoplastic Lesions of Gastric Adenocarcinoma and Proximal Polyposis Syndrome (GAPPS) Are Gastric Phenotype. Am. J. Surg. Pathol. 2018, 42, 1–8. [Google Scholar] [CrossRef]
- Genta, R.M.; Schuler, C.M.; Robiou, C.I.; Lash, R.H. No association between gastric fundic gland polyps and gastrointestinal neoplasia in a study of over 100,000 patients. Clin. Gastroenterol. Hepatol. 2009, 7, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Dickey, W.; Kenny, B.D.; McConnell, J.B. Prevalence of fundic gland polyps in a western European population. J. Clin. Gastroenterol. 1996, 23, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, I.E.; Kohlmann, W.K.; Gligorich, K.; Hall, A.; Lyon, E.; Downs-Kelly, E.; Samowitz, W.S.; Bronner, M.P. A Clinicopathologic Evaluation of Incidental Fundic Gland Polyps With Dysplasia: Implications for Clinical Management. Am. J. Gastroenterol. 2017, 112, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Fossmark, R.; Jianu, C.S.; Martinsen, T.C.; Qvigstad, G.; Syversen, U.; Waldum, H.L. Serum gastrin and chromogranin A levels in patients with fundic gland polyps caused by long-term proton-pump inhibition. Scand. J. Gastroenterol. 2008, 43, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.C.; Chenevix-Trench, G.; Yeomans, N.D. Systematic review with meta-analysis: Fundic gland polyps and proton pump inhibitors. Aliment. Pharmacol. Ther. 2016, 44, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Koornstra, J.J.; Wesseling, J.; Boezen, H.M.; De Jong, S.; Kleibeuker, J.H. Increased risk of fundic gland polyps during long-term proton pump inhibitor therapy. Aliment. Pharmacol. Ther. 2006, 24, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Seno, H.; Nakajima, T.; Yazumi, S.; Miyamoto, S.; Matsumoto, S.; Itoh, T.; Kawanami, C.; Okazaki, K.; Chiba, T. Regression of fundic gland polyps following acquisition of Helicobacter pylori. Gut 2002, 51, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H. Proton Pump Inhibitor-Related Gastric Mucosal Changes. Gut Liver 2020, 15, 1976–2283. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Chae, H.S.; Kim, H.K.; Cho, Y.S.; Park, Y.W.; Son, H.S.; Han, S.W.; Choi, K.Y. Spontaneous resolution of multiple fundic gland polyps after cessation of treatment with omeprazole. Korean J. Gastroenterol. 2008, 51, 305–308. [Google Scholar]
- Hamada, K.; Takeuchi, Y.; Akasaka, T.; Iishi, H. Fundic Gland Polyposis Associated with Proton-Pump Inhibitor Use. Eur. J. Case Rep. Intern. Med. 2017, 4, 000607. [Google Scholar] [CrossRef]
- Kazantsev, G.B.; Schwesinger, W.H.; Heim-Hall, J. Spontaneous resolution of multiple fundic gland polyps after cessation of treatment with lansoprazole and Nissen fundoplication: A case report. Gastrointest. Endosc. 2002, 55, 600–602. [Google Scholar] [CrossRef]
- Fukuda, M.; Ishigaki, H.; Sugimoto, M.; Mukaisho, K.I.; Matsubara, A.; Ishida, H.; Moritani, S.; Itoh, Y.; Sugihara, H.; Andoh, A.; et al. Histological analysis of fundic gland polyps secondary to PPI therapy. Histopathology 2019, 75, 537–545. [Google Scholar] [CrossRef]
- Shibukawa, N.; Wakahara, Y.; Ouchi, S.; Wakamatsu, S.; Kaneko, A. Synchronous Three Gastric Fundic Gland Polyps with Low-grade Dysplasia Treated with Endoscopic Mucosal Resection after Being Diagnosed to Be Tubular Adenocarcinoma Based on a Biopsy Specimen. Intern. Med. 2019, 58, 1871–1875. [Google Scholar] [CrossRef]
- Martín Domínguez, V.; Díaz Méndez, A.; Santander, C.; García-Buey, L. Portal hypertensive polyps, a new entity? Rev. Esp. Enferm. Dig. 2016, 108, 279–280. [Google Scholar] [PubMed]
- Topal, F.; Akbulut, S.; Karahanlı, C.; Günay, S.; Sarıtaş Yüksel, E.; Topal, F.E. Portal Hypertensive Polyps as Gastroscopic Finding in Liver Cirrhosis. Gastroenterol. Res. Pract. 2020, 2020, 9058909. [Google Scholar] [CrossRef] [PubMed]
- Seleem, W.M.; Hanafy, A.S. Management of a Portal Hypertensive Polyp: Case Report of a Rare Entity. Gastrointest. Tumors 2019, 6, 137–141. [Google Scholar] [CrossRef]
- Andersson, T.; Olsson, R.; Regårdh, C.G.; Skånberg, I. Pharmacokinetics of [14C]omeprazole in patients with liver cirrhosis. Clin. Pharmacokinet. 1993, 24, 71–78. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldum, H.; Fossmark, R. Gastritis, Gastric Polyps and Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 6548. https://doi.org/10.3390/ijms22126548
Waldum H, Fossmark R. Gastritis, Gastric Polyps and Gastric Cancer. International Journal of Molecular Sciences. 2021; 22(12):6548. https://doi.org/10.3390/ijms22126548
Chicago/Turabian StyleWaldum, Helge, and Reidar Fossmark. 2021. "Gastritis, Gastric Polyps and Gastric Cancer" International Journal of Molecular Sciences 22, no. 12: 6548. https://doi.org/10.3390/ijms22126548
APA StyleWaldum, H., & Fossmark, R. (2021). Gastritis, Gastric Polyps and Gastric Cancer. International Journal of Molecular Sciences, 22(12), 6548. https://doi.org/10.3390/ijms22126548