
Effects of Incretin-Related Diabetes Drugs on Bone Formation and Bone Resorption

{kind=link}
Abstract
:1. Introduction
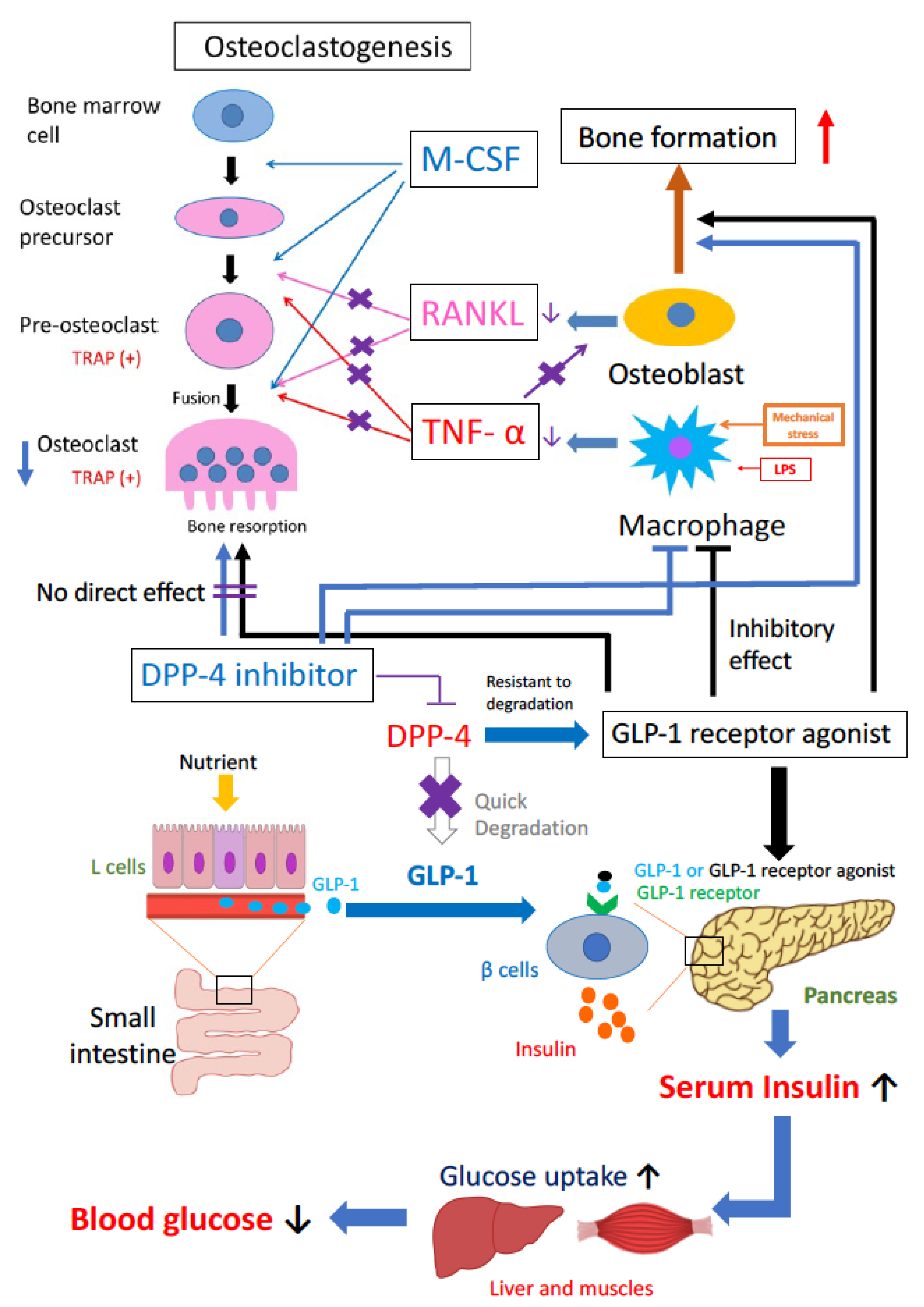
2. Effects of Incretin on Bone Formation
3. Effects of Incretin on Bone Resorption
4. Effects of Incretin-Related Drugs on Bone Formation and Bone Resorption
4.1. GLP-1R Agonists
4.2. DPP-4 Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| BMD | bone mineral density |
| HbA1c | hemoglobin A1c |
| AGEs | advanced glycation end products |
| RANKL | receptor activator of nuclear factor-κB ligand |
| M-CSF | macrophage colony-stimulating factor |
| RANK | activator of nuclear factor-κB |
| TNF-α | tumor necrosis factor-α |
| PPAR-g | peroxisome proliferator-activated receptor-g |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP-1 | Glucagon-like peptide-1 |
| DPP-4 | dipeptidyl peptidase-4 |
| GLP-1R | GLP-1 receptor |
| ADA | American Diabetes Association |
| EASD | European Association for the Study of Diabetes |
| LPS | lipopolysaccharide |
| CTX | carboxy-terminal collagen crosslinks |
| μCT | micro-computed tomography |
| KO | knockout |
| BMP-2 | bone morphogenetic protein-2 |
| Runx2 | Runt-related transcription factor 2 |
| TBPf | trabecular bone pattern factor |
| OVX | ovariectomized |
| OPG | osteoprotegerin |
| SOST | sclerostin |
| SAH | subarachnoid hemorrhage |
| COX-2 | cyclooxygenase-2 |
| iNOS | inducible nitric oxide synthase |
| BMSCs | bone marrow stromal cells |
| BMMs | bone marrow-derived macrophages |
| NFATc1 | nuclear factor of activated T cells cytoplasmic 1 |
| siRNA | small interfering RNA |
| OTM | Orthodontic tooth movement |
| FDA | Food and Drug Administration |
| CPRD | Clinical Practice Research Datalink |
| GPRD | General Practice Research Database |
| SU | sulfonylurea |
| TZD | thiazolidinedione |
| TRAP | tartrate-resistant acid phosphatase |
References
- Sozen, T.; Basaran, N.C.; Tinazli, M.; Ozisik, L. Musculoskeletal problems in diabetes mellitus. Eur. J. Rheumatol. 2018, 5, 258–265. [Google Scholar] [CrossRef]
- Courties, A.; Sellam, J. Osteoarthritis and type 2 diabetes mellitus: What are the links? Diabetes Res. Clin. Pract. 2016, 122, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Eitner, A.; Wildemann, B. Diabetes—Osteoarthritis and joint pain. Bone Jt. Res. 2021, 10, 307–309. [Google Scholar] [CrossRef]
- Williams, M.F.; London, D.A.; Husni, E.M.; Navaneethan, S.; Kashyap, S.R. Type 2 diabetes and osteoarthritis: A systematic review and meta-analysis. J. Diabetes Complicat. 2016, 30, 944–950. [Google Scholar] [CrossRef]
- Farooqui, K.J.; Mithal, A.; Kerwen, A.K.; Chandran, M. Type 2 diabetes and bone fragility- An under-recognized association. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.I.; Liu, C.S.; Lin, W.Y.; Meng, N.H.; Chen, C.C.; Yang, S.Y.; Chen, H.J.; Lin, C.C.; Li, T.C. Glycated Hemoglobin Level and Risk of Hip Fracture in Older People with Type 2 Diabetes: A Competing Risk Analysis of Taiwan Diabetes Cohort Study. J. Bone Miner. Res. 2015, 30, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef]
- Janghorbani, M.; Van Dam, R.M.; Willett, W.C.; Hu, F.B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 2007, 166, 495–505. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Vittinghoff, E.; Bauer, D.C.; Hillier, T.A.; Strotmeyer, E.S.; Ensrud, K.E.; Donaldson, M.G.; Cauley, J.A.; Harris, T.B.; Koster, A.; et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA 2011, 305, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, A.; Rozas-Moreno, P.; Reyes-Garcia, R.; Morales-Santana, S.; Garcia-Fontana, B.; Garcia-Salcedo, J.A.; Munoz-Torres, M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Nicodemus, K.K.; Folsom, A.R. Iowa Women’s Health Study. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care 2001, 24, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Oei, L.; Zillikens, M.C.; Dehghan, A.; Buitendijk, G.H.; Castano-Betancourt, M.C.; Estrada, K.; Stolk, L.; Oei, E.H.; van Meurs, J.B.; Janssen, J.A.; et al. High bone mineral density and fracture risk in type 2 diabetes as skeletal complications of inadequate glucose control: The Rotterdam Study. Diabetes Care 2013, 36, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.L.; Williams, E.K.; Brancati, F.L.; Blecker, S.; Coresh, J.; Selvin, E. Diabetes and risk of fracture-related hospitalization: The Atherosclerosis Risk in Communities Study. Diabetes Care 2013, 36, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.V.; Vittinghoff, E.; Margolis, K.L.; Scibora, L.M.; Palermo, L.; Ambrosius, W.T.; Hue, T.F.; Ensrud, K.E. Intensive glycemic control and thiazolidinedione use: Effects on cortical and trabecular bone at the radius and tibia. Calcif. Tissue Int. 2013, 92, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, M. Clinical aspects and management of osteoporosis and fragility fractures in patients with diabetes. Osteoporos. Sarcopenia 2017, 3, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L.; Bone, I.O.F.; Diabetes Working, G. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Xiao, E.; Graves, D.T. Diabetes and Its Effect on Bone and Fracture Healing. Curr. Osteoporos. Rep. 2015, 13, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Kume, S.; Kato, S.; Yamagishi, S.; Inagaki, Y.; Ueda, S.; Arima, N.; Okawa, T.; Kojiro, M.; Nagata, K. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J. Bone Miner. Res. 2005, 20, 1647–1658. [Google Scholar] [CrossRef]
- Sanguineti, R.; Storace, D.; Monacelli, F.; Federici, A.; Odetti, P. Pentosidine effects on human osteoblasts in vitro. Ann. N. Y. Acad. Sci. 2008, 1126, 166–172. [Google Scholar] [CrossRef]
- McCarthy, A.D.; Uemura, T.; Etcheverry, S.B.; Cortizo, A.M. Advanced glycation endproducts interefere with integrin-mediated osteoblastic attachment to a type-I collagen matrix. Int. J. Biochem. Cell Biol. 2004, 36, 840–848. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Valenti, R.; Ceccarelli, E.; Ruvio, M.; Pietrini, M.G.; Capodarca, C.; Franci, M.B.; Campagna, M.S.; Calabro, A.; et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudio, A.; Privitera, F.; Battaglia, K.; Torrisi, V.; Sidoti, M.H.; Pulvirenti, I.; Canzonieri, E.; Tringali, G.; Fiore, C.E. Sclerostin levels associated with inhibition of the Wnt/beta-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 3744–3750. [Google Scholar] [CrossRef] [PubMed]
- Heilmeier, U.; Carpenter, D.R.; Patsch, J.M.; Harnish, R.; Joseph, G.B.; Burghardt, A.J.; Baum, T.; Schwartz, A.V.; Lang, T.F.; Link, T.M. Volumetric femoral BMD, bone geometry, and serum sclerostin levels differ between type 2 diabetic postmenopausal women with and without fragility fractures. Osteoporos. Int. 2015, 26, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Fujii, K.; Mori, Y.; Marumo, K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos. Int. 2006, 17, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Vashishth, D.; Gibson, G.J.; Khoury, J.I.; Schaffler, M.B.; Kimura, J.; Fyhrie, D.P. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 2001, 28, 195–201. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamaguchi, T.; Yamauchi, M.; Sugimoto, T. Low serum level of the endogenous secretory receptor for advanced glycation end products (esRAGE) is a risk factor for prevalent vertebral fractures independent of bone mineral density in patients with type 2 diabetes. Diabetes Care 2009, 32, 2263–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, A.V.; Garnero, P.; Hillier, T.A.; Sellmeyer, D.E.; Strotmeyer, E.S.; Feingold, K.R.; Resnick, H.E.; Tylavsky, F.A.; Black, D.M.; Cummings, S.R.; et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Nyomba, B.L.; Verhaeghe, J.; Thomasset, M.; Lissens, W.; Bouillon, R. Bone mineral homeostasis in spontaneously diabetic BB rats. I. Abnormal vitamin D metabolism and impaired active intestinal calcium absorption. Endocrinology 1989, 124, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Balint, E.; Szabo, P.; Marshall, C.F.; Sprague, S.M. Glucose-induced inhibition of in vitro bone mineralization. Bone 2001, 28, 21–28. [Google Scholar] [CrossRef]
- Starup-Linde, J.; Lykkeboe, S.; Gregersen, S.; Hauge, E.M.; Langdahl, B.L.; Handberg, A.; Vestergaard, P. Differences in biochemical bone markers by diabetes type and the impact of glucose. Bone 2016, 83, 149–155. [Google Scholar] [CrossRef]
- Krakauer, J.C.; McKenna, M.J.; Buderer, N.F.; Rao, D.S.; Whitehouse, F.W.; Parfitt, A.M. Bone loss and bone turnover in diabetes. Diabetes 1995, 44, 775–782. [Google Scholar] [CrossRef]
- Suzuki, A.; Minamide, M.; Iwaya, C.; Ogata, K.; Iwata, J. Role of Metabolism in Bone Development and Homeostasis. Int. J. Mol. Sci. 2020, 21, 8992. [Google Scholar] [CrossRef]
- Gong, F.; Gao, L.; Ma, L.; Li, G.; Yang, J. Uncarboxylated osteocalcin alleviates the inhibitory effect of high glucose on osteogenic differentiation of mouse bone marrow-derived mesenchymal stem cells by regulating TP63. BMC Mol. Cell Biol. 2021, 22, 24. [Google Scholar] [CrossRef]
- Wang, N.; Xu, P.; Wu, R.; Wang, X.; Wang, Y.; Shou, D.; Zhang, Y. Timosaponin BII improved osteoporosis caused by hyperglycemia through promoting autophagy of osteoblasts via suppressing the mTOR/NFkappaB signaling pathway. Free Radic. Biol. Med. 2021, 171. [Google Scholar] [CrossRef]
- Murray, C.E.; Coleman, C.M. Impact of Diabetes Mellitus on Bone Health. Int. J. Mol. Sci. 2019, 20, 4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahwa, H.; Khan, M.T.; Sharan, K. Hyperglycemia impairs osteoblast cell migration and chemotaxis due to a decrease in mitochondrial biogenesis. Mol. Cell. Biochem. 2020, 469, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Chuang, P.Y.; Yang, T.Y.; Huang, T.W.; Chang, S.F. Hyperglycemia inhibits osteoblastogenesis of rat bone marrow stromal cells via activation of the Notch2 signaling pathway. Int. J. Med. Sci. 2019, 16, 696–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, K.; Hao, P.; Xu, S.; Liu, S.; Zhou, W.; Yue, X.; Rausch-Fan, X.; Liu, Z. Alpha-Lipoic Acid Alleviates High-Glucose Suppressed Osteogenic Differentiation of MC3T3-E1 Cells via Antioxidant Effect and PI3K/Akt Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 1897–1906. [Google Scholar] [CrossRef]
- Wu, M.; Ai, W.; Chen, L.; Zhao, S.; Liu, E. Bradykinin receptors and EphB2/EphrinB2 pathway in response to high glucose-induced osteoblast dysfunction and hyperglycemia-induced bone deterioration in mice. Int. J. Mol. Med. 2016, 37, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Midura, R.J.; Vasanji, A.; Wang, A.J.; Hascall, V.C. Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. J. Biol. Chem. 2014, 289, 11410–11420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseley, K.F.; Doyle, M.E.; Jan De Beur, S.M. Diabetic serum from older women increases adipogenic differentiation in mesenchymal stem cells. Endocr. Res. 2018, 43, 155–165. [Google Scholar] [CrossRef]
- Garcia-Hernandez, A.; Arzate, H.; Gil-Chavarria, I.; Rojo, R.; Moreno-Fierros, L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 2012, 50, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Lechleitner, M.; Koch, T.; Herold, M.; Dzien, A.; Hoppichler, F. Tumour necrosis factor-alpha plasma level in patients with type 1 diabetes mellitus and its association with glycaemic control and cardiovascular risk factors. J. Intern. Med. 2000, 248, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Camargo, W.A.; de Vries, R.; van Luijk, J.; Hoekstra, J.W.; Bronkhorst, E.M.; Jansen, J.A.; van den Beucken, J. Diabetes Mellitus and Bone Regeneration: A Systematic Review and Meta-Analysis of Animal Studies. Tissue Eng. Part B Rev. 2017, 23, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J. Clin. Pathol. 2008, 61, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Osteoclasts: What do they do and how do they do it? Am. J. Pathol. 2007, 170, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Blair, J.M.; Zheng, Y.; Dunstan, C.R. RANK ligand. Int. J. Biochem. Cell Biol. 2007, 39, 1077–1081. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-alpha Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Kang, M.K.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Oh, S.Y.; Na, W.; Kang, Y.H. Coumarin Ameliorates Impaired Bone Turnover by Inhibiting the Formation of Advanced Glycation End Products in Diabetic Osteoblasts and Osteoclasts. Biomolecules 2020, 10, 1052. [Google Scholar] [CrossRef]
- Graves, D.T.; Ding, Z.; Yang, Y. The impact of diabetes on periodontal diseases. Periodontol. 2000 2020, 82, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.S.; Graves, D.T.; Loureiro, A.P.; Rossa Junior, C.; Abdalla, D.S.; Faulin Tdo, E.; Olsen Camara, N.; Andriankaja, O.M.; Orrico, S.R. Lipid peroxidation is associated with the severity of periodontal disease and local inflammatory markers in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E1353–E1362. [Google Scholar] [CrossRef] [Green Version]
- Zayzafoon, M.; Stell, C.; Irwin, R.; McCabe, L.R. Extracellular glucose influences osteoblast differentiation and c-Jun expression. J. Cell. Biochem. 2000, 79, 301–310. [Google Scholar] [CrossRef]
- Botolin, S.; McCabe, L.R. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J. Cell. Biochem. 2006, 99, 411–424. [Google Scholar] [CrossRef]
- Tanaka, K.; Yamaguchi, T.; Kanazawa, I.; Sugimoto, T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem. Biophys. Res. Commun. 2015, 461, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Elrick, H.; Stimmler, L.; Hlad, C.J., Jr.; Arai, Y. Plasma Insulin Response to Oral and Intravenous Glucose Administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.J. Long-acting glucagon-like peptide 1 receptor agonists: A review of their efficacy and tolerability. Diabetes Care 2011, 34 (Suppl. 2), S279–S284. [Google Scholar] [CrossRef] [Green Version]
- Jones, B. The therapeutic potential of GLP-1 receptor biased agonism. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Aroda, V.R. A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes. Metab. 2018, 20 (Suppl. 1), 22–33. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, C.H.; Demuth, H.U.; Pospisilik, J.A.; Pederson, R. Dipeptidyl peptidase IV inhibitors: How do they work as new antidiabetic agents? Regul. Pept. 2005, 128, 159–165. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes, 2015: A patient-centered approach: Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, M.; Shen, W.R.; Kimura, K.; Kishikawa, A.; Shima, K.; Ogawa, S.; Qi, J.; Ohori, F.; Noguchi, T.; Marahleh, A.; et al. DPP-4 inhibitor impedes lipopolysaccharide-induced osteoclast formation and bone resorption in vivo. Biomed. Pharmacother. 2019, 109, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.R.; Kimura, K.; Ishida, M.; Sugisawa, H.; Kishikawa, A.; Shima, K.; Ogawa, S.; Qi, J.; Kitaura, H. The Glucagon-Like Peptide-1 Receptor Agonist Exendin-4 Inhibits Lipopolysaccharide-Induced Osteoclast Formation and Bone Resorption via Inhibition of TNF-alpha Expression in Macrophages. J. Immunol. Res. 2018, 2018, 5783639. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Kitaura, H.; Shen, W.R.; Ogawa, S.; Ohori, F.; Noguchi, T.; Marahleh, A.; Nara, Y.; Adya, P.; Mizoguchi, I. Effect of a DPP-4 Inhibitor on Orthodontic Tooth Movement and Associated Root Resorption. Biomed. Res. Int. 2020, 2020, 7189084. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.R.; Kitaura, H.; Qi, J.; Ogawa, S.; Ohori, F.; Noguchi, T.; Marahleh, A.; Nara, Y.; Adya, P.; Mizoguchi, I. Local administration of high-dose diabetes medicine exendin-4 inhibits orthodontic tooth movement in mice. Angle Orthod. 2021, 91, 111–118. [Google Scholar] [CrossRef]
- Gutniak, M.; Orskov, C.; Holst, J.J.; Ahren, B.; Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Schreiber, E.; Fogel, H.; Mojsov, S.; Habener, J.F. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992, 15, 270–276. [Google Scholar] [CrossRef]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Habener, J.F. The glucagon-like peptides. Endocr. Rev. 1999, 20, 876–913. [Google Scholar] [CrossRef]
- Creutzfeldt, W.; Nauck, M. Gut hormones and diabetes mellitus. Diabetes Metab. Rev. 1992, 8, 149–177. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [Green Version]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. Circulation and degradation of GIP and GLP-1. Horm. Metab. Res. 2004, 36, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 1), 5–21. [Google Scholar] [CrossRef]
- Aoyama, E.; Watari, I.; Podyma-Inoue, K.A.; Yanagishita, M.; Ono, T. Expression of glucagon-like peptide-1 receptor and glucosedependent insulinotropic polypeptide receptor is regulated by the glucose concentration in mouse osteoblastic MC3T3-E1 cells. Int. J. Mol. Med. 2014, 34, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, R.J.; Zhong, Q.; Phillips, P.; Min, L.; Zhong, L.; Cameron, R.; Mulloy, A.L.; Rasmussen, H.; Qin, F.; Ding, K.H.; et al. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 2000, 141, 1228–1235. [Google Scholar] [CrossRef]
- Pacheco-Pantoja, E.L.; Ranganath, L.R.; Gallagher, J.A.; Wilson, P.J.; Fraser, W.D. Receptors and effects of gut hormones in three osteoblastic cell lines. BMC Physiol. 2011, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama, K.; Yamada, Y.; Yamada, C.; Harada, N.; Kawasaki, Y.; Ogura, M.; Bessho, K.; Li, M.; Amizuka, N.; Sato, M.; et al. Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol. Endocrinol. 2006, 20, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Berlier, J.L.; Kharroubi, I.; Zhang, J.; Dalla Valle, A.; Rigutto, S.; Mathieu, M.; Gangji, V.; Rasschaert, J. Glucose-Dependent Insulinotropic Peptide Prevents Serum Deprivation-Induced Apoptosis in Human Bone Marrow-Derived Mesenchymal Stem Cells and Osteoblastic Cells. Stem Cell Rev. Rep. 2015, 11, 841–851. [Google Scholar] [CrossRef]
- Mansur, S.A.; Mieczkowska, A.; Bouvard, B.; Flatt, P.R.; Chappard, D.; Irwin, N.; Mabilleau, G. Stable Incretin Mimetics Counter Rapid Deterioration of Bone Quality in Type 1 Diabetes Mellitus. J. Cell. Physiol. 2015, 230, 3009–3018. [Google Scholar] [CrossRef]
- Henriksen, D.B.; Alexandersen, P.; Bjarnason, N.H.; Vilsboll, T.; Hartmann, B.; Henriksen, E.E.; Byrjalsen, I.; Krarup, T.; Holst, J.J.; Christiansen, C. Role of gastrointestinal hormones in postprandial reduction of bone resorption. J. Bone Miner. Res. 2003, 18, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Cheng, H.; Hamrick, M.; Zhong, Q.; Ding, K.H.; Correa, D.; Williams, S.; Mulloy, A.; Bollag, W.; Bollag, R.J.; et al. Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone 2005, 37, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Gaudin-Audrain, C.; Irwin, N.; Mansur, S.; Flatt, P.R.; Thorens, B.; Basle, M.; Chappard, D.; Mabilleau, G. Glucose-dependent insulinotropic polypeptide receptor deficiency leads to modifications of trabecular bone volume and quality in mice. Bone 2013, 53, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieczkowska, A.; Irwin, N.; Flatt, P.R.; Chappard, D.; Mabilleau, G. Glucose-dependent insulinotropic polypeptide (GIP) receptor deletion leads to reduced bone strength and quality. Bone 2013, 56, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.; Jeyabalan, J.; Jorgensen, C.S.; Hopkinson, M.; Al-Jazzar, A.; Roux, J.P.; Chavassieux, P.; Orriss, I.R.; Cleasby, M.E.; Chenu, C. Chronic administration of Glucagon-like peptide-1 receptor agonists improves trabecular bone mass and architecture in ovariectomised mice. Bone 2015, 81, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Su, L.; Zhong, X.; Guohong, W.; Xiao, H.; Li, Y.; Xiu, L. Exendin-4 promotes proliferation and differentiation of MC3T3-E1 osteoblasts by MAPKs activation. J. Mol. Endocrinol. 2016, 56, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Nuche-Berenguer, B.; Portal-Nunez, S.; Moreno, P.; Gonzalez, N.; Acitores, A.; Lopez-Herradon, A.; Esbrit, P.; Valverde, I.; Villanueva-Penacarrillo, M.L. Presence of a functional receptor for GLP-1 in osteoblastic cells, independent of the cAMP-linked GLP-1 receptor. J. Cell. Physiol. 2010, 225, 585–592. [Google Scholar] [CrossRef]
- Sanz, C.; Vazquez, P.; Blazquez, C.; Barrio, P.A.; Alvarez Mdel, M.; Blazquez, E. Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E634–E643. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.K.; Bae, M.J.; Kim, J.I.; Kim, J.H.; Choi, S.J.; Kwon, S.K.; An, J.H.; Kim, S.S.; Kim, B.H.; Kim, Y.K.; et al. Expression of Glucagon-Like Peptide 1 Receptor during Osteogenic Differentiation of Adipose-Derived Stem Cells. Endocrinol. Metab. 2014, 29, 567–573. [Google Scholar] [CrossRef]
- Meng, J.; Ma, X.; Wang, N.; Jia, M.; Bi, L.; Wang, Y.; Li, M.; Zhang, H.; Xue, X.; Hou, Z.; et al. Activation of GLP-1 Receptor Promotes Bone Marrow Stromal Cell Osteogenic Differentiation through beta-Catenin. Stem Cell Rep. 2016, 6, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Joo, B.S.; Lee, C.H.; Kim, H.Y.; Ock, J.H.; Lee, Y.S. Effect of Glucagon-like Peptide-1 on the Differentiation of Adipose-derived Stem Cells into Osteoblasts and Adipocytes. J. Menopausal Med. 2015, 21, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Nuche-Berenguer, B.; Moreno, P.; Esbrit, P.; Dapia, S.; Caeiro, J.R.; Cancelas, J.; Haro-Mora, J.J.; Villanueva-Penacarrillo, M.L. Effect of GLP-1 treatment on bone turnover in normal, type 2 diabetic, and insulin-resistant states. Calcif. Tissue Int. 2009, 84, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Yamada, C.; Yamada, Y.; Tsukiyama, K.; Yamada, K.; Udagawa, N.; Takahashi, N.; Tanaka, K.; Drucker, D.J.; Seino, Y.; Inagaki, N. The murine glucagon-like peptide-1 receptor is essential for control of bone resorption. Endocrinology 2008, 149, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Mabilleau, G.; Mieczkowska, A.; Irwin, N.; Flatt, P.R.; Chappard, D. Optimal bone mechanical and material properties require a functional glucagon-like peptide-1 receptor. J. Endocrinol. 2013, 219, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pederson, R.A.; Satkunarajah, M.; McIntosh, C.H.; Scrocchi, L.A.; Flamez, D.; Schuit, F.; Drucker, D.J.; Wheeler, M.B. Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor -/- mice. Diabetes 1998, 47, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Pamir, N.; Lynn, F.C.; Buchan, A.M.; Ehses, J.; Hinke, S.A.; Pospisilik, J.A.; Miyawaki, K.; Yamada, Y.; Seino, Y.; McIntosh, C.H.; et al. Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E931–E939. [Google Scholar] [CrossRef] [Green Version]
- Mieczkowska, A.; Mansur, S.; Bouvard, B.; Flatt, P.R.; Thorens, B.; Irwin, N.; Chappard, D.; Mabilleau, G. Double incretin receptor knock-out (DIRKO) mice present with alterations of trabecular and cortical micromorphology and bone strength. Osteoporos. Int. 2015, 26, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Itokawa, T.; Sridhar, S.; Ding, K.H.; Xie, D.; Kang, B.; Bollag, W.B.; Bollag, R.J.; Hamrick, M.; Insogna, K.; et al. Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E543–E548. [Google Scholar] [CrossRef]
- Nissen, A.; Christensen, M.; Knop, F.K.; Vilsboll, T.; Holst, J.J.; Hartmann, B. Glucose-dependent insulinotropic polypeptide inhibits bone resorption in humans. J. Clin. Endocrinol. Metab. 2014, 99, E2325–E2329. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Zhong, Q.; Ding, K.H.; Cheng, H.; Williams, S.; Correa, D.; Bollag, W.B.; Bollag, R.J.; Insogna, K.; Troiano, N.; et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone 2007, 40, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Bollag, R.J.; Zhong, Q.; Ding, K.H.; Phillips, P.; Zhong, L.; Qin, F.; Cranford, J.; Mulloy, A.L.; Cameron, R.; Isales, C.M. Glucose-dependent insulinotropic peptide is an integrative hormone with osteotropic effects. Mol. Cell. Endocrinol. 2001, 177, 35–41. [Google Scholar] [CrossRef]
- Nuche-Berenguer, B.; Lozano, D.; Gutierrez-Rojas, I.; Moreno, P.; Marinoso, M.L.; Esbrit, P.; Villanueva-Penacarrillo, M.L. GLP-1 and exendin-4 can reverse hyperlipidic-related osteopenia. J. Endocrinol. 2011, 209, 203–210. [Google Scholar] [CrossRef]
- Mansur, S.A.; Mieczkowska, A.; Flatt, P.R.; Chappard, D.; Irwin, N.; Mabilleau, G. The GLP-1 Receptor Agonist Exenatide Ameliorates Bone Composition and Tissue Material Properties in High Fat Fed Diabetic Mice. Front. Endocrinol. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Meng, J.; Jia, M.; Bi, L.; Zhou, Y.; Wang, Y.; Hu, J.; He, G.; Luo, X. Exendin-4, a glucagon-like peptide-1 receptor agonist, prevents osteopenia by promoting bone formation and suppressing bone resorption in aged ovariectomized rats. J. Bone Miner. Res. 2013, 28, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Bjerre Knudsen, L.; Madsen, L.W.; Andersen, S.; Almholt, K.; de Boer, A.S.; Drucker, D.J.; Gotfredsen, C.; Egerod, F.L.; Hegelund, A.C.; Jacobsen, H.; et al. Glucagon-like Peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010, 151, 1473–1486. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, L.L.; Baron, A.D. Pharmacology of exenatide (synthetic exendin-4) for the treatment of type 2 diabetes. Curr. Opin. Investig. Drugs 2003, 4, 401–405. [Google Scholar]
- Ma, J.; Shi, M.; Zhang, X.; Liu, X.; Chen, J.; Zhang, R.; Wang, X.; Zhang, H. GLP1R agonists ameliorate peripheral nerve dysfunction and inflammation via p38 MAPK/NFkappaB signaling pathways in streptozotocininduced diabetic rats. Int. J. Mol. Med. 2018, 41, 2977–2985. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Xu, H.; Hu, F.; Wu, J.; Kong, X.; Sun, X. Exendin-4, a GLP-1 receptor agonist regulates retinal capillary tone and restores microvascular patency after ischaemia-reperfusion injury. Br. J. Pharmacol. 2020, 177, 3389–3402. [Google Scholar] [CrossRef]
- Fuchigami, A.; Shigiyama, F.; Kitazawa, T.; Okada, Y.; Ichijo, T.; Higa, M.; Hiyoshi, T.; Inoue, I.; Iso, K.; Yoshii, H.; et al. Efficacy of dapagliflozin versus sitagliptin on cardiometabolic risk factors in Japanese patients with type 2 diabetes: A prospective, randomized study (DIVERSITY-CVR). Cardiovasc. Diabetol. 2020, 19, 1. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, A.; Oyama, J.I.; Nishikido, T.; Node, K. GLP-1 analog liraglutide-induced cardiac dysfunction due to energetic starvation in heart failure with non-diabetic dilated cardiomyopathy. Cardiovasc. Diabetol. 2019, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Y.; Tan, S.; Lin, Y.; Liao, S.; Zhang, B.; Chen, X.; Wang, J.; Deng, Z.; Zeng, Q.; Zhang, L.; et al. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J. Neuroinflamm. 2019, 16, 242. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.S.S.; Tencerova, M.; Frolich, J.; Kassem, M.; Frost, M. Effects of gastric inhibitory polypeptide, glucagon-like peptide-1 and glucagon-like peptide-1 receptor agonists on Bone Cell Metabolism. Basic Clin. Pharmacol. Toxicol. 2018, 122, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, S.; Wang, N.; Xue, P.; Li, Y. Liraglutide, a glucagon-like peptide-1 receptor agonist, suppresses osteoclastogenesis through the inhibition of NF-kappaB and MAPK pathways via GLP-1R. Biomed. Pharmacother. 2020, 130, 110523. [Google Scholar] [CrossRef] [PubMed]
- Bunck, M.C.; Eliasson, B.; Corner, A.; Heine, R.J.; Shaginian, R.M.; Taskinen, M.R.; Yki-Jarvinen, H.; Smith, U.; Diamant, M. Exenatide treatment did not affect bone mineral density despite body weight reduction in patients with type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Mabilleau, G.; Mieczkowska, A.; Chappard, D. Use of glucagon-like peptide-1 receptor agonists and bone fractures: A meta-analysis of randomized clinical trials. J. Diabetes 2014, 6, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Sheng, H.; Zhang, M.; Bu, L.; Yang, P.; Li, L.; Li, F.; Sheng, C.; Han, Y.; Qu, S.; et al. Risk of bone fractures associated with glucagon-like peptide-1 receptor agonists’ treatment: A meta-analysis of randomized controlled trials. Endocrine 2015, 48, 107–115. [Google Scholar] [CrossRef]
- Mabilleau, G. Use of GLP-1 mimetic in type 2 diabetes mellitus: Is it the end of fragility fractures? Endocrine 2015, 48, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Lu, N.; Sun, H.; Yu, J.; Wang, X.; Liu, D.; Zhao, L.; Sun, L.; Zhao, H.; Tao, B.; Liu, J. Glucagon-like peptide-1 receptor agonist Liraglutide has anabolic bone effects in ovariectomized rats without diabetes. PLoS ONE 2015, 10, e0132744. [Google Scholar] [CrossRef]
- Zhao, C.; Liang, J.; Yang, Y.; Yu, M.; Qu, X. The Impact of Glucagon-Like Peptide-1 on Bone Metabolism and Its Possible Mechanisms. Front. Endocrinol. 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Sedky, A.A. Improvement of cognitive function, glucose and lipid homeostasis and serum osteocalcin levels by liraglutide in diabetic rats. Fundam. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Sawada, N.; Adachi, K.; Nakamura, N.; Miyabe, M.; Ito, M.; Kobayashi, S.; Miyajima, S.I.; Suzuki, Y.; Kikuchi, T.; Mizutani, M.; et al. Glucagon-Like Peptide-1 Receptor Agonist Liraglutide Ameliorates the Development of Periodontitis. J. Diabetes Res. 2020, 2020, 8843310. [Google Scholar] [CrossRef]
- Sun, H.X.; Lu, N.; Liu, D.M.; Zhao, L.; Sun, L.H.; Zhao, H.Y.; Liu, J.M.; Tao, B. The bone-preserving effects of exendin-4 in ovariectomized rats. Endocrine 2016, 51, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, S.; Xue, P.; Li, Y. Liraglutide, a glucagon-like peptide-1 receptor agonist, facilitates osteogenic proliferation and differentiation in MC3T3-E1 cells through phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), extracellular signal-related kinase (ERK)1/2, and cAMP/protein kinase A (PKA) signaling pathways involving beta-catenin. Exp. Cell Res. 2017, 360, 281–291. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, S.K.; Jo, K.J.; Song, D.Y.; Lim, D.M.; Park, K.Y.; Bonewald, L.F.; Kim, B.J. Exendin-4 increases bone mineral density in type 2 diabetic OLETF rats potentially through the down-regulation of SOST/sclerostin in osteocytes. Life Sci. 2013, 92, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.P.; Zheng, G.; Lee, V.W.; Ouyang, L.; Chang, D.H.; Mahajan, D.; Coombs, J.; Wang, Y.M.; Alexander, S.I.; et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007, 72, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Yoshizawa-Smith, S.; Glowacki, A.; Maltos, K.; Pacheco, C.; Shehabeldin, M.; Mulkeen, M.; Myers, N.; Chong, R.; Verdelis, K.; et al. Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models. J. Dent. Res. 2019, 98, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.K.; Chen, Q.; Chen, S.; Huang, B.; Ren, B.G.; Shi, S.S. GLP-1R Agonist Liraglutide Attenuates Inflammatory Reaction and Neuronal Apoptosis and Reduces Early Brain Injury After Subarachnoid Hemorrhage in Rats. Inflammation 2021, 44, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Gohin, S.; Roux, J.P.; Fisher, A.; Cleasby, M.E.; Mabilleau, G.; Chenu, C. Exenatide Improves Bone Quality in a Murine Model of Genetically Inherited Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zheng, J.; Zheng, T.; Wang, P. Exendin-4 regulates Wnt and NF-kappaB signaling in lipopolysaccharide-induced human periodontal ligament stem cells to promote osteogenic differentiation. Int. Immunopharmacol. 2019, 75, 105801. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Jeon, H.H.; Alshabab, A.; Lee, Y.J.; Chung, C.H.; Graves, D.T. RANKL deletion in periodontal ligament and bone lining cells blocks orthodontic tooth movement. Int. J. Oral Sci. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrovola, J.B.; Spyropoulos, M.N.; Makou, M.; Perrea, D. Root resorption and the OPG/RANKL/RANK system: A mini review. J. Oral Sci. 2008, 50, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Yoshimatsu, M.; Fujimura, Y.; Eguchi, T.; Kohara, H.; Yamaguchi, A.; Yoshida, N. An anti-c-Fms antibody inhibits orthodontic tooth movement. J. Dent. Res. 2008, 87, 396–400. [Google Scholar] [CrossRef]
- Ogawa, S.; Kitaura, H.; Kishikawa, A.; Qi, J.; Shen, W.R.; Ohori, F.; Noguchi, T.; Marahleh, A.; Nara, Y.; Ochi, Y.; et al. TNF-alpha is responsible for the contribution of stromal cells to osteoclast and odontoclast formation during orthodontic tooth movement. PLoS ONE 2019, 14, e0223989. [Google Scholar] [CrossRef]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Dorecka, M.; Siemianowicz, K.; Francuz, T.; Garczorz, W.; Chyra, A.; Klych, A.; Romaniuk, W. Exendin-4 and GLP-1 decreases induced expression of ICAM-1, VCAM-1 and RAGE in human retinal pigment epithelial cells. Pharmacol. Rep. 2013, 65, 884–890. [Google Scholar] [CrossRef]
- Garczorz, W.; Gallego-Colon, E.; Kosowska, A.; Klych-Ratuszny, A.; Wozniak, M.; Marcol, W.; Niesner, K.J.; Francuz, T. Exenatide exhibits anti-inflammatory properties and modulates endothelial response to tumor necrosis factor alpha-mediated activation. Cardiovasc. Ther. 2018, 36. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Chen, Z.W.; Gao, Y.H.; Wang, X.X.; Ma, J.Y.; Chang, S.F.; Qian, J.Y.; Ge, J.B. Exenatide Reduces Tumor Necrosis Factor-alpha-induced Apoptosis in Cardiomyocytes by Alleviating Mitochondrial Dysfunction. Chin. Med. J. 2015, 128, 3211–3218. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jun, H.S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediat. Inflamm. 2016, 2016, 3094642. [Google Scholar] [CrossRef] [Green Version]
- Weltman, B.; Vig, K.W.; Fields, H.W.; Shanker, S.; Kaizar, E.E. Root resorption associated with orthodontic tooth movement: A systematic review. Am. J. Orthod. Dentofac. Orthop. 2010, 137, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Gao, J.; Jia, M.; Ma, X.; Lei, Z.; Da, F.; Yan, F.; Zhang, H.; Zhou, Y.; Li, M.; et al. Exendin-4 Induces Bone Marrow Stromal Cells Migration Through Bone Marrow-Derived Macrophages Polarization via PKA-STAT3 Signaling Pathway. Cell. Physiol. Biochem. 2017, 44, 1696–1714. [Google Scholar] [CrossRef] [Green Version]
- Neumiller, J.J. Differential chemistry (structure), mechanism of action, and pharmacology of GLP-1 receptor agonists and DPP-4 inhibitors. J. Am. Pharm. Assoc. 2009, 49 (Suppl. 1), S16–S29. [Google Scholar] [CrossRef]
- Bunck, M.C.; Poelma, M.; Eekhoff, E.M.; Schweizer, A.; Heine, R.J.; Nijpels, G.; Foley, J.E.; Diamant, M. Effects of vildagliptin on postprandial markers of bone resorption and calcium homeostasis in recently diagnosed, well-controlled type 2 diabetes patients. J. Diabetes 2012, 4, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Dicembrini, I.; Antenore, A.; Mannucci, E. Dipeptidyl peptidase-4 inhibitors and bone fractures: A meta-analysis of randomized clinical trials. Diabetes Care 2011, 34, 2474–2476. [Google Scholar] [CrossRef] [Green Version]
- Driessen, J.H.; van Onzenoort, H.A.; Henry, R.M.; Lalmohamed, A.; van den Bergh, J.P.; Neef, C.; Leufkens, H.G.; de Vries, F. Use of dipeptidyl peptidase-4 inhibitors for type 2 diabetes mellitus and risk of fracture. Bone 2014, 68, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.H.; Chang, K.C.; Li, C.Y.; Ou, H.T. Dipeptidyl peptidase-4 inhibitor use is associated with decreased risk of fracture in patients with type 2 diabetes: A population-based cohort study. Br. J. Clin. Pharmacol. 2018, 84, 2029–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagnani, A.; Gonnelli, S. Antidiabetic therapy effects on bone metabolism and fracture risk. Diabetes Obes. Metab. 2013, 15, 784–791. [Google Scholar] [CrossRef]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Gamble, J.M.; Donnan, J.R.; Chibrikov, E.; Twells, L.K.; Midodzi, W.K.; Majumdar, S.R. The risk of fragility fractures in new users of dipeptidyl peptidase-4 inhibitors compared to sulfonylureas and other anti-diabetic drugs: A cohort study. Diabetes Res. Clin. Pract. 2018, 136, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Glorie, L.; Behets, G.J.; Baerts, L.; De Meester, I.; D’Haese, P.C.; Verhulst, A. DPP IV inhibitor treatment attenuates bone loss and improves mechanical bone strength in male diabetic rats. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E447–E455. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: Preclinical biology and mechanisms of action. Diabetes Care 2007, 30, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, J.; Fujii, H.; Ohnuma, K.; Sato, K.; Ito, Y.; Kaji, M.; Sakamoto, E.; Koganei, M.; Sasaki, H.; Nagashima, Y.; et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 2011, 60, 1246–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, S.; Kostev, K.; Jacob, L. Use of dipeptidyl peptidase-4 inhibitors and risk of bone fracture in patients with type 2 diabetes in Germany-A retrospective analysis of real-world data. Osteoporos. Int. 2017, 28, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, C.; Lee, Y.K.; Ha, Y.C.; Jang, S.; Shin, C.S. Risk of fractures and diabetes medications: A nationwide cohort study. Osteoporos. Int. 2016, 27, 2709–2715. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, C.; Liang, J.; Yu, M.; Qu, X. Effect of Dipeptidyl Peptidase-4 Inhibitors on Bone Metabolism and the Possible Underlying Mechanisms. Front. Pharmacol. 2017, 8, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroller-Schon, S.; Knorr, M.; Hausding, M.; Oelze, M.; Schuff, A.; Schell, R.; Sudowe, S.; Scholz, A.; Daub, S.; Karbach, S.; et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc. Res. 2012, 96, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Hausding, M.; Kroller-Schon, S.; Mader, M.; Mikhed, Y.; Stamm, P.; Zinssius, E.; Pfeffer, A.; Welschof, P.; Agdauletova, S.; et al. Gliptin and GLP-1 analog treatment improves survival and vascular inflammation/dysfunction in animals with lipopolysaccharide-induced endotoxemia. Basic Res. Cardiol. 2015, 110, 6. [Google Scholar] [CrossRef]
- Ervinna, N.; Mita, T.; Yasunari, E.; Azuma, K.; Tanaka, R.; Fujimura, S.; Sukmawati, D.; Nomiyama, T.; Kanazawa, A.; Kawamori, R.; et al. Anagliptin, a DPP-4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo E-deficient mice. Endocrinology 2013, 154, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Kitaura, H.; Zhou, P.; Kim, H.J.; Novack, D.V.; Ross, F.P.; Teitelbaum, S.L. M-CSF mediates TNF-induced inflammatory osteolysis. J. Clin. Investig. 2005, 115, 3418–3427. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Inagaki, M.; Tsuji, M.; Gocho, T.; Handa, K.; Hasegawa, H.; Yura, A.; Kawakami, T.; Ohsawa, I.; Goto, Y.; et al. Linagliptin Has Wide-Ranging Anti-Inflammatory Points of Action in Human Umbilical Vein Endothelial Cells. Jpn. Clin. Med. 2016, 7, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Yamadera, S.; Nakamura, Y.; Inagaki, M.; Kenmotsu, S.; Nohara, T.; Sato, N.; Oguchi, T.; Tsuji, M.; Ohsawa, I.; Gotoh, H.; et al. Linagliptin inhibits lipopolysaccharide-induced inflammation in human U937 monocytes. Inflamm. Regen. 2018, 38, 13. [Google Scholar] [CrossRef]
- Jo, C.H.; Kim, S.; Park, J.S.; Kim, G.H. Anti-Inflammatory Action of Sitagliptin and Linagliptin in Doxorubicin Nephropathy. Kidney Blood Press. Res. 2018, 43, 987–999. [Google Scholar] [CrossRef]
- Oshiro, T.; Shiotani, A.; Shibasaki, Y.; Sasaki, T. Osteoclast induction in periodontal tissue during experimental movement of incisors in osteoprotegerin-deficient mice. Anat. Rec. Off. Publ. Am. Assoc. Anat. 2002, 266, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, H.; Chiba, M.; Takahashi, I.; Haruyama, N.; Nishimura, M.; Mitani, H. Local OPG gene transfer to periodontal tissue inhibits orthodontic tooth movement. J. Dent. Res. 2004, 83, 920–925. [Google Scholar] [CrossRef]
- Yamaguchi, M. RANK/RANKL/OPG during orthodontic tooth movement. Orthod. Craniofac. Res. 2009, 12, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.M.; Wang, A.; Parhar, K.S.; Johnston, M.J.; Van Rooijen, N.; Beck, P.L.; McKay, D.M. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 2010, 138, 1395–1405. [Google Scholar] [CrossRef]
- He, D.; Kou, X.; Luo, Q.; Yang, R.; Liu, D.; Wang, X.; Song, Y.; Cao, H.; Zeng, M.; Gan, Y.; et al. Enhanced M1/M2 macrophage ratio promotes orthodontic root resorption. J. Dent. Res. 2015, 94, 129–139. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yuan, G.; Cheng, F.; Zhang, J.; Guo, X. Mast Cell and M1 Macrophage Infiltration and Local Pro-Inflammatory Factors Were Attenuated with Incretin-Based Therapies in Obesity-Related Glomerulopathy. Metab. Syndr. Relat. Disord. 2017, 15, 344–353. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitaura, H.; Ogawa, S.; Ohori, F.; Noguchi, T.; Marahleh, A.; Nara, Y.; Pramusita, A.; Kinjo, R.; Ma, J.; Kanou, K.; et al. Effects of Incretin-Related Diabetes Drugs on Bone Formation and Bone Resorption. Int. J. Mol. Sci. 2021, 22, 6578. https://doi.org/10.3390/ijms22126578
Kitaura H, Ogawa S, Ohori F, Noguchi T, Marahleh A, Nara Y, Pramusita A, Kinjo R, Ma J, Kanou K, et al. Effects of Incretin-Related Diabetes Drugs on Bone Formation and Bone Resorption. International Journal of Molecular Sciences. 2021; 22(12):6578. https://doi.org/10.3390/ijms22126578
Chicago/Turabian StyleKitaura, Hideki, Saika Ogawa, Fumitoshi Ohori, Takahiro Noguchi, Aseel Marahleh, Yasuhiko Nara, Adya Pramusita, Ria Kinjo, Jinghan Ma, Kayoko Kanou, and et al. 2021. "Effects of Incretin-Related Diabetes Drugs on Bone Formation and Bone Resorption" International Journal of Molecular Sciences 22, no. 12: 6578. https://doi.org/10.3390/ijms22126578