
CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension

Abstract
1. Introduction
2. Results
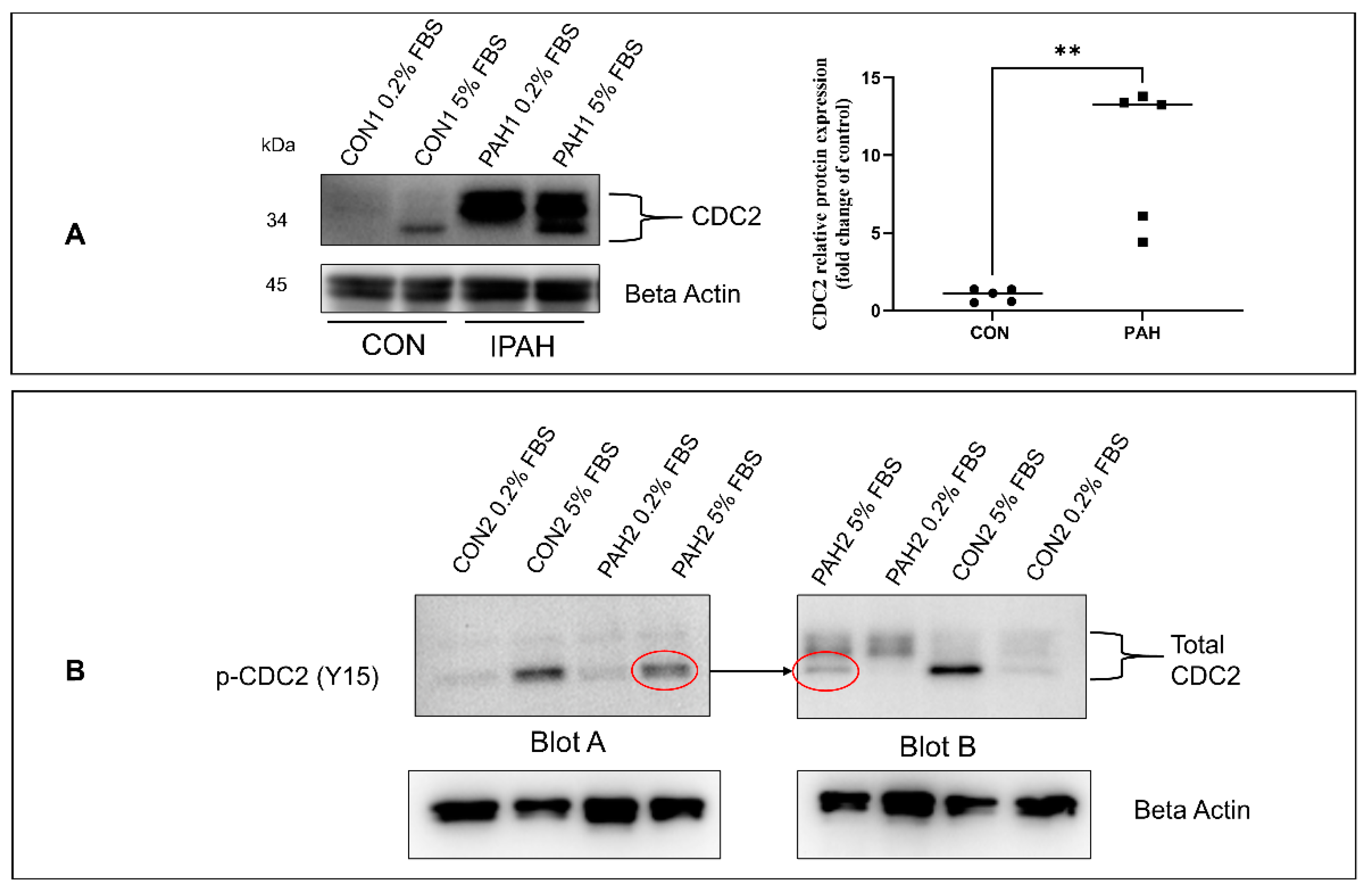
2.1. Expression of CDC2 in HPASMC
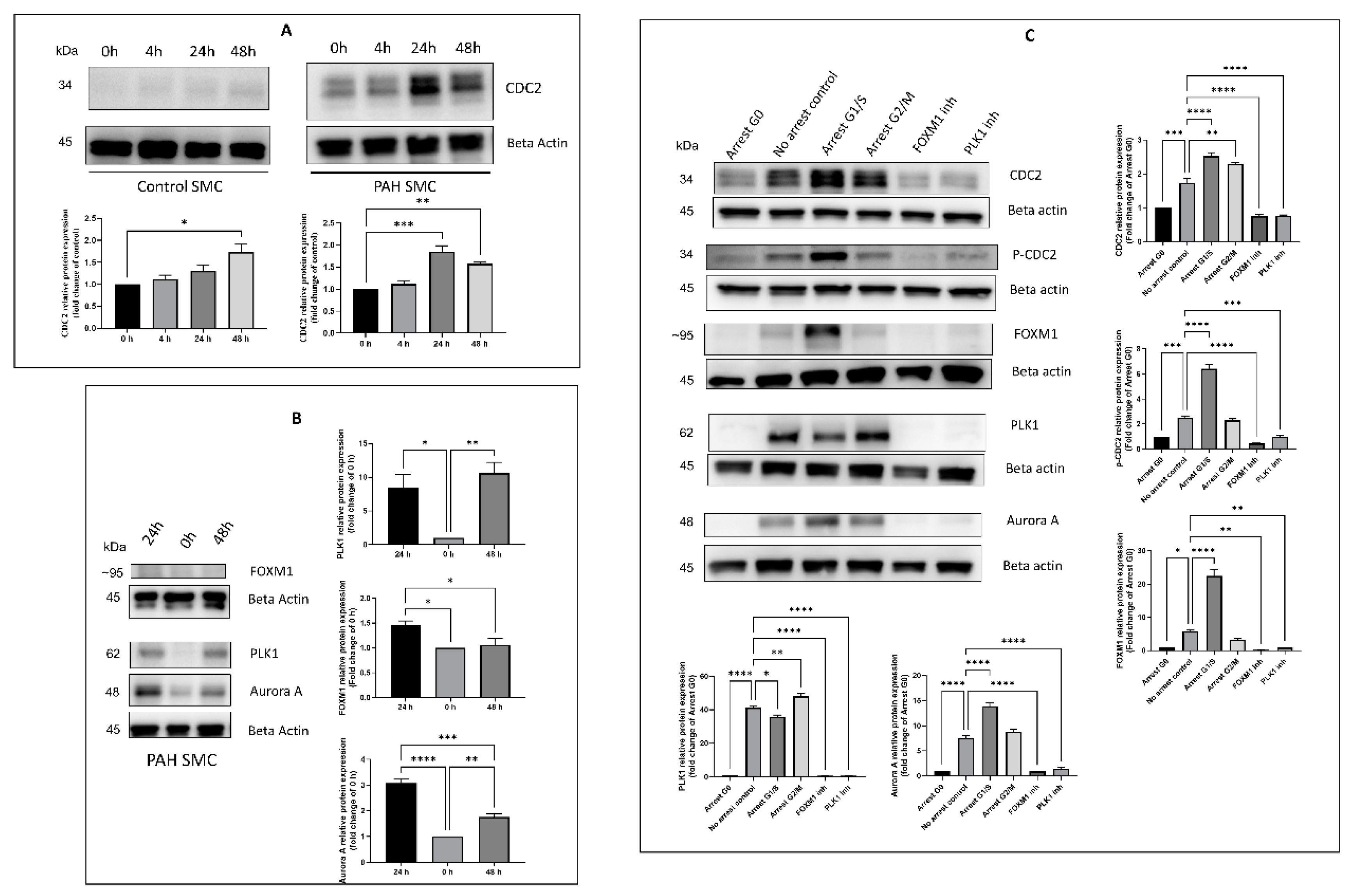
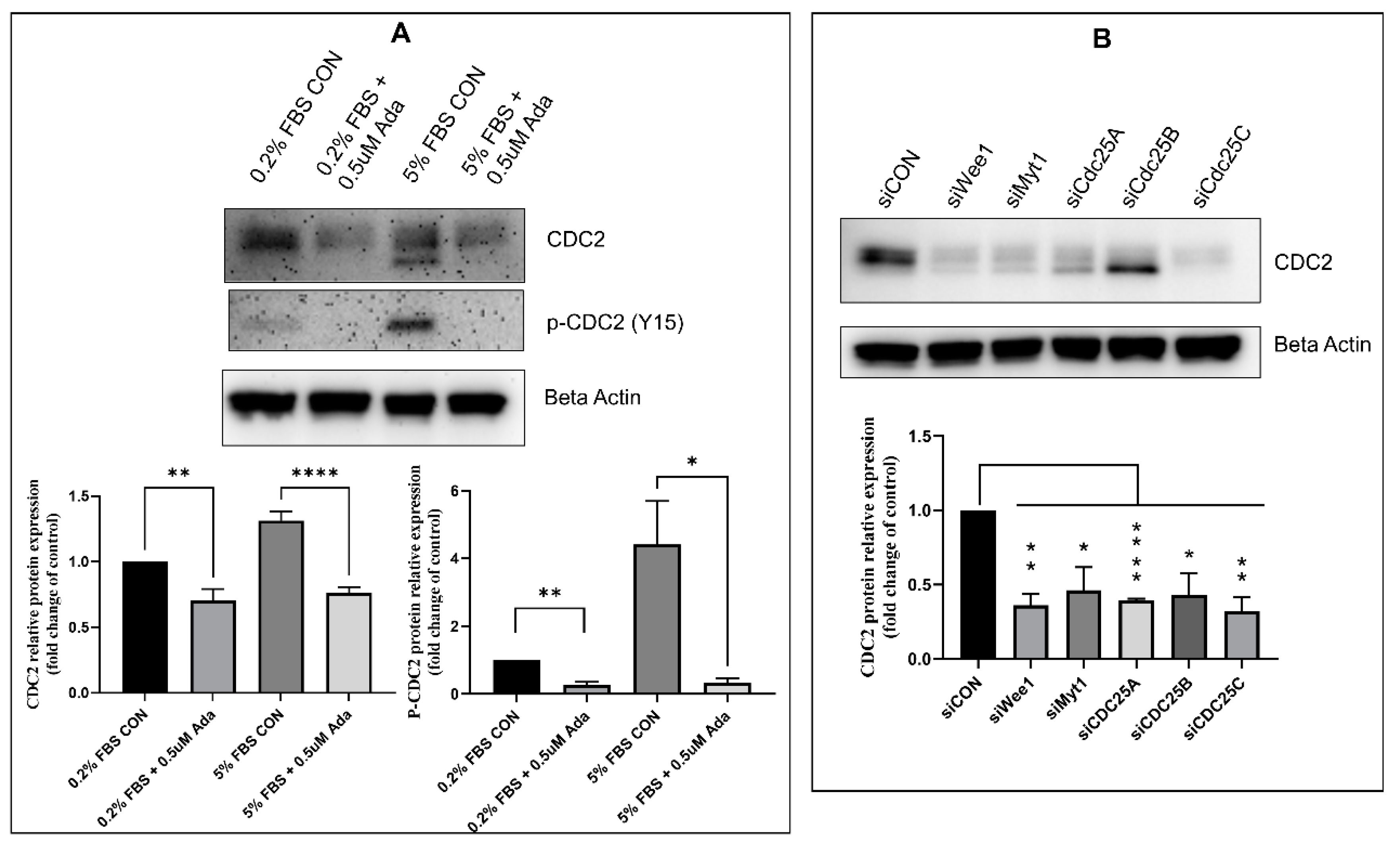
2.2. Effect of Timing, Cell Cycle Arrest, and FOXM1/PLK1 Inhibition on Expression of CDC2
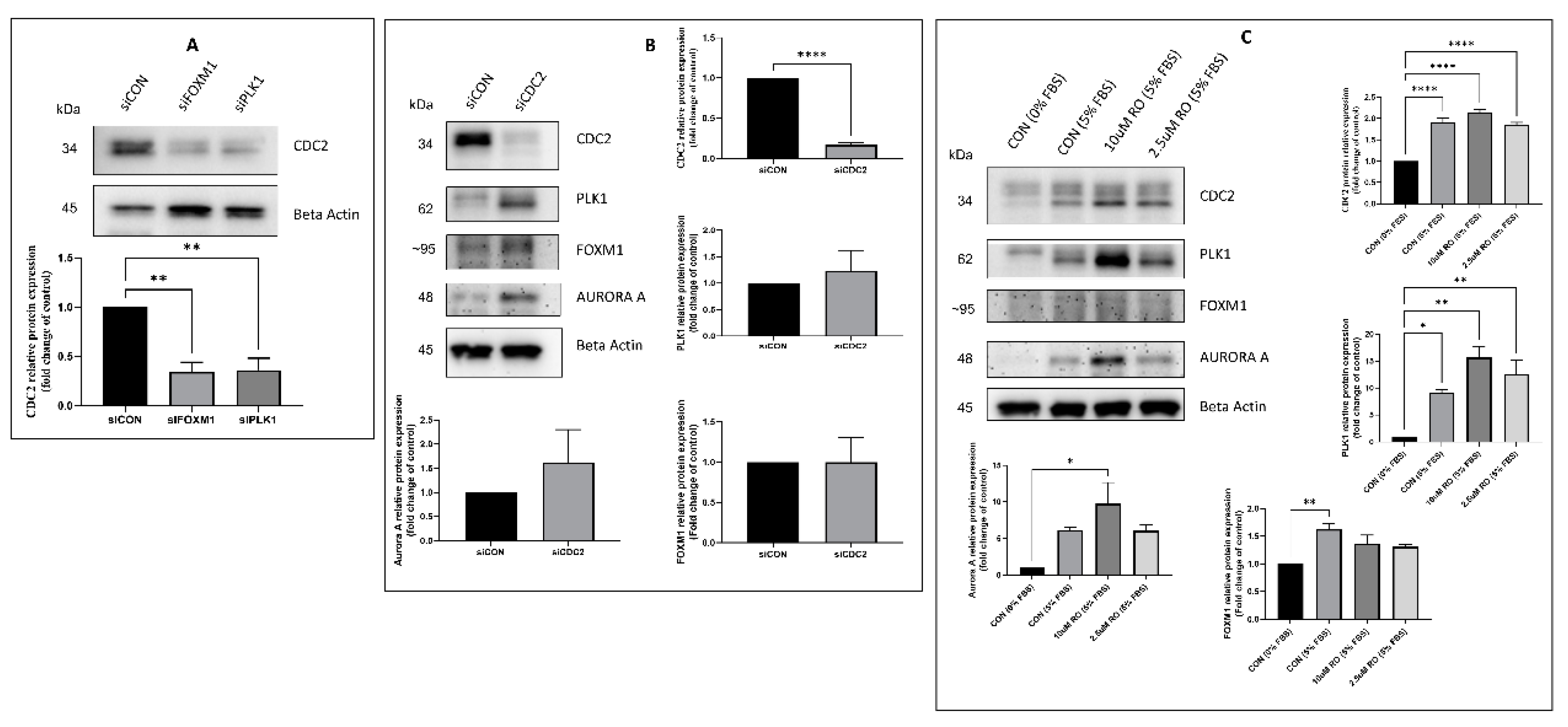
2.3. Effectors of CDC2 Expression
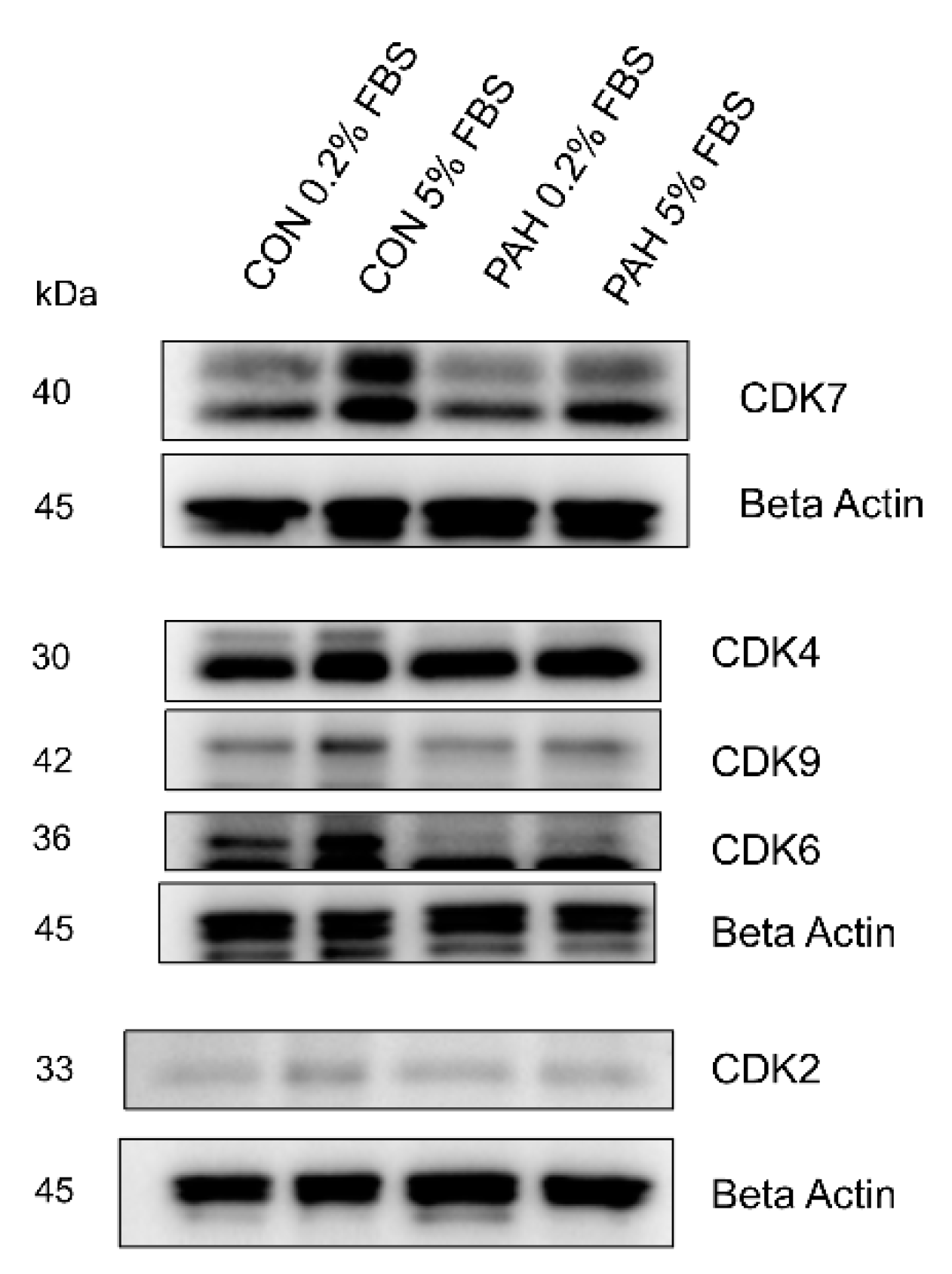
2.4. Expression of Other CDKs
2.5. Regulators of CDC2
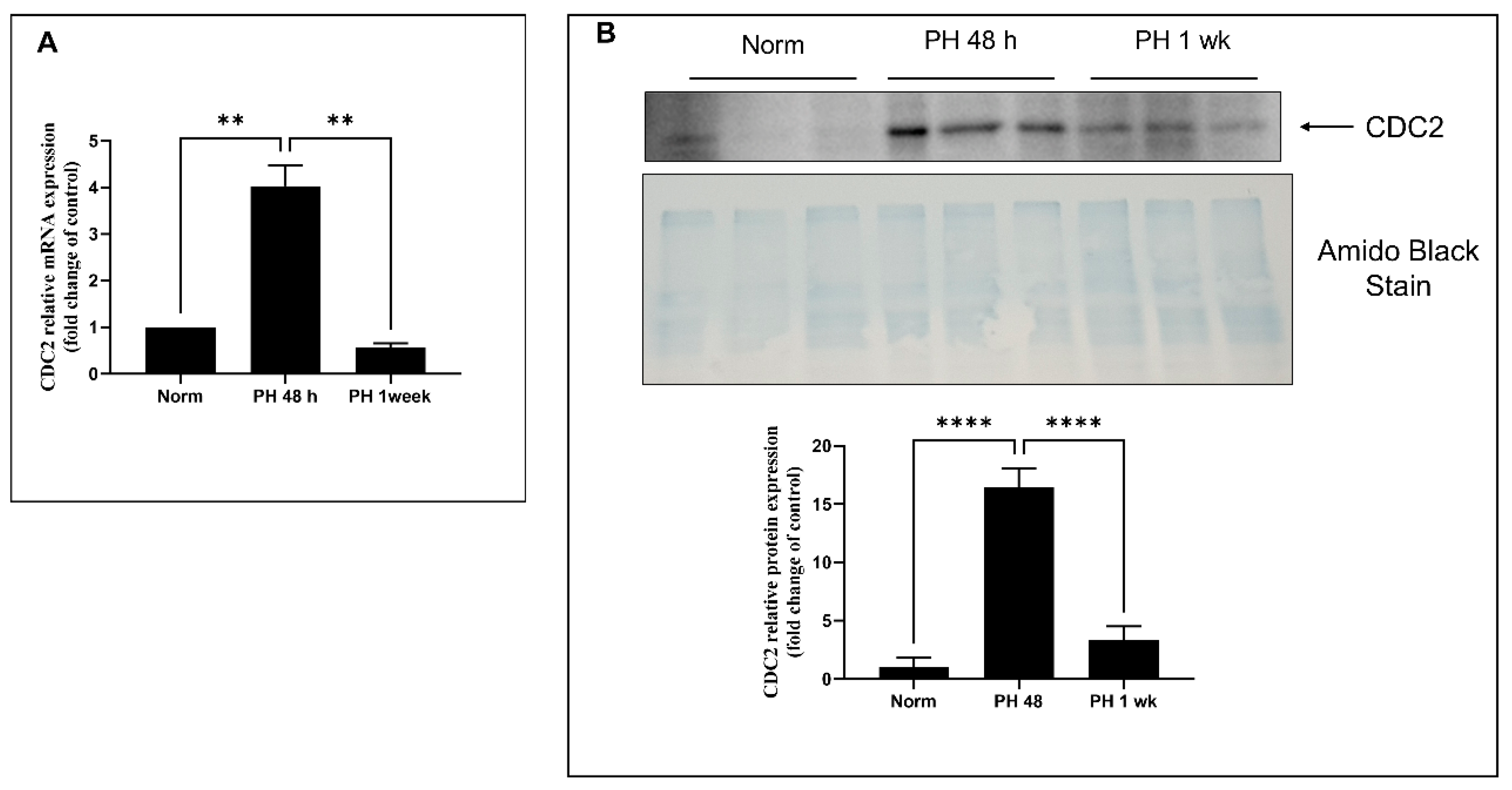
2.6. Expression of CDC2 in Pulmonary Arteries from Sugen/Hypoxia-Treated Rats
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Transfection with siRNA
4.4. Real-Time qPCR
4.5. Western Blot
4.6. Animal Experiments
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef]
- Kudryashova, T.V.; Goncharov, D.A.; Pena, A.; Kelly, N.; Vanderpool, R.; Baust, J.; Kobir, A.; Shufesky, W.; Mora, A.L.; Morelli, A.E.; et al. HIPPO-Integrin-linked Kinase Cross-Talk Controls Self-Sustaining Proliferation and Survival in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 866–877. [Google Scholar] [CrossRef]
- Kudryashova, T.V.; Shen, Y.; Pena, A.; Cronin, E.; Okorie, E.; Goncharov, D.A.; Goncharova, E.A. Inhibitory Antibodies against Activin A and TGF-beta Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2957. [Google Scholar] [CrossRef]
- Wilson, J.L.; Wang, L.; Zhang, Z.; Hill, N.S.; Polgar, P. Participation of PLK1 and FOXM1 in the hyperplastic proliferation of pulmonary artery smooth muscle cells in pulmonary arterial hypertension. PLoS ONE 2019, 14, e0221728. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Yu, J.; Taylor, L.; Polgar, P. Hyperplastic Growth of Pulmonary Artery Smooth Muscle Cells from Subjects with Pulmonary Arterial Hypertension Is Activated through JNK and p38 MAPK. PLoS ONE 2015, 10, e0123662. [Google Scholar] [CrossRef]
- Yu, J.; Wilson, J.; Taylor, L.; Polgar, P. DNA microarray and signal transduction analysis in pulmonary artery smooth muscle cells from heritable and idiopathic pulmonary arterial hypertension subjects. J. Cell. Biochem. 2015, 116, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Cell Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef]
- Perros, F.; Sentenac, P.; Boulate, D.; Manaud, G.; Kotsimbos, T.; Lecerf, F.; Lamrani, L.; Fadel, E.; Mercier, O.; Londono-Vallejo, A.; et al. Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous. Int. J. Mol. Sci. 2019, 20, 3575. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Rupasinghe, C.; Usheva, A.; Warburton, R.; Kaplan, C.; Taylor, L.; Hill, N.; Mierke, D.F.; Polgar, P. Modulating the dysregulated migration of pulmonary arterial hypertensive smooth muscle cells with motif mimicking cell permeable peptides. Curr. Top. Pept. Protein Res. 2015, 16, 1–17. [Google Scholar] [PubMed]
- Tu, L.; De Man, F.S.; Girerd, B.; Huertas, A.; Chaumais, M.C.; Lecerf, F.; Francois, C.; Perros, F.; Dorfmuller, P.; Fadel, E.; et al. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am. J. Respir. Crit. Care Med. 2012, 186, 666–676. [Google Scholar] [CrossRef]
- Paulin, R.; Meloche, J.; Courboulin, A.; Lambert, C.; Haromy, A.; Courchesne, A.; Bonnet, P.; Provencher, S.; Michelakis, E.D.; Bonnet, S. Targeting cell motility in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 531–544. [Google Scholar] [CrossRef]
- Bourgeois, A.; Lambert, C.; Habbout, K.; Ranchoux, B.; Paquet-Marceau, S.; Trinh, I.; Breuils-Bonnet, S.; Paradis, R.; Nadeau, V.; Paulin, R.; et al. FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J. Mol. Med. 2018, 96, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA Damage and Pulmonary Hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, A.; Bonnet, S.; Breuils-Bonnet, S.; Habbout, K.; Paradis, R.; Tremblay, E.; Lampron, M.C.; Orcholski, M.E.; Potus, F.; Bertero, T.; et al. Inhibition of CHK 1 (Checkpoint Kinase 1) Elicits Therapeutic Effects in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Kuhr, F.K.; Smith, K.A.; Song, M.Y.; Levitan, I.; Yuan, J.X. New mechanisms of pulmonary arterial hypertension: Role of Ca(2)(+) signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1546–H1562. [Google Scholar] [CrossRef]
- Yu, J.; Taylor, L.; Wilson, J.; Comhair, S.; Erzurum, S.; Polgar, P. Altered expression and signal transduction of endothelin-1 receptors in heritable and idiopathic pulmonary arterial hypertension. J. Cell. Physiol. 2013, 228, 322–329. [Google Scholar] [CrossRef]
- Yuan, J.X.; Aldinger, A.M.; Juhaszova, M.; Wang, J.; Conte, J.V., Jr.; Gaine, S.P.; Orens, J.B.; Rubin, L.J. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998, 98, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Montani, D.; Dorfmuller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef]
- Wu, K.; Tang, H.; Lin, R.; Carr, S.G.; Wang, Z.; Babicheva, A.; Ayon, R.J.; Jain, P.P.; Xiong, M.; Rodriguez, M.; et al. Endothelial platelet-derived growth factor-mediated activation of smooth muscle platelet-derived growth factor receptors in pulmonary arterial hypertension. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802. [Google Scholar] [CrossRef]
- Chen, X.; Muller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers 2017, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.; Sun, Z.; Guo, Q.; Shi, H.; Jia, Y. The clinical and prognostic value of polo-like kinase 1 in lung squamous cell carcinoma patients: Immunohistochemical analysis. Biosci. Rep. 2017. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Humbert, M.; Hoeper, M.M. Severe pulmonary arterial hypertension: A forme fruste of cancer? Am. J. Respir. Crit. Care Med. 2008, 178, 551–552. [Google Scholar] [CrossRef]
- Rai, P.R.; Cool, C.D.; King, J.A.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef]
- Dai, J.; Zhou, Q.; Tang, H.; Chen, T.; Li, J.; Raychaudhuri, P.; Yuan, J.X.; Zhou, G. Smooth muscle cell-specific FoxM1 controls hypoxia-induced pulmonary hypertension. Cell. Signal. 2018, 51, 119–129. [Google Scholar] [CrossRef]
- Dibb, M.; Han, N.; Choudhury, J.; Hayes, S.; Valentine, H.; West, C.; Ang, Y.S.; Sharrocks, A.D. The FOXM1-PLK1 axis is commonly upregulated in oesophageal adenocarcinoma. Br. J. Cancer 2012, 107, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef]
- Qi, F.; Chen, Q.; Chen, H.; Yan, H.; Chen, B.; Xiang, X.; Liang, C.; Yi, Q.; Zhang, M.; Cheng, H.; et al. WAC Promotes Polo-like Kinase 1 Activation for Timely Mitotic Entry. Cell Rep. 2018, 24, 546–556. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, C.; Wu, J.; Elsayed, Z.; Fu, Z. Polo-like kinase 1-mediated phosphorylation of Forkhead box protein M1b antagonizes its SUMOylation and thereby facilitates its mitotic function. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Chen, Y.J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef]
- Wang, I.C.; Zhang, Y.; Snyder, J.; Sutherland, M.J.; Burhans, M.S.; Shannon, J.M.; Park, H.J.; Whitsett, J.A.; Kalinichenko, V.V. Increased expression of FoxM1 transcription factor in respiratory epithelium inhibits lung sacculation and causes Clara cell hyperplasia. Dev. Biol. 2010, 347, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Andres, V. Control of vascular cell proliferation and migration by cyclin-dependent kinase signalling: New perspectives and therapeutic potential. Cardiovasc. Res. 2004, 63, 11–21. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Norbury, C.; Blow, J.; Nurse, P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991, 10, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, S.; Sundermann, L.; Labbe, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650 e637. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.R.; Dunphy, W.G. Cdc2 regulatory factors. Curr. Opin. Cell Biol. 1994, 6, 877–882. [Google Scholar] [CrossRef]
- Liu, F.; Stanton, J.J.; Wu, Z.; Piwnica-Worms, H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol. 1997, 17, 571–583. [Google Scholar] [CrossRef]
- McGowan, C.H.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [CrossRef]
- Gabrielli, B.G.; Clark, J.M.; McCormack, A.K.; Ellem, K.A. Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J. Biol. Chem. 1997, 272, 28607–28614. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Rodriguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: Feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Gorlick, R.; Kolb, E.A.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Billups, C.A.; Kurmasheva, R.T.; et al. Initial testing (stage 1) of the Polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 158–164. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef]
- Bi, S.; Wei, Q.; Zhao, Z.; Chen, L.; Wang, C.; Xie, S. Wee1 Inhibitor AZD1775 Effectively Inhibits the Malignant Phenotypes of Esophageal Squamous Cell Carcinoma In Vitro and In Vivo. Front. Pharmacol. 2019, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.P.; Zhu, Y.L.; Ratner, E.S. Targeting Cyclin-Dependent Kinases for Treatment of Gynecologic Cancers. Front. Oncol. 2018, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Ni, H.; Ding, C.; Zhang, X.; Zhang, Z. FoxM1 overexpression promotes cell proliferation and migration and inhibits apoptosis in hypopharyngeal squamous cell carcinoma resulting in poor clinical prognosis. Int. J. Oncol. 2017, 51, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.E.; Myatt, S.S.; Krol, J.; Hartman, J.; Peck, B.; McGovern, U.B.; Wang, J.; Guest, S.K.; Filipovic, A.; Gojis, O.; et al. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int. J. Oncol. 2009, 35, 57–68. [Google Scholar] [PubMed]
- Zhao, C.; Gong, L.; Li, W.; Chen, L. Overexpression of Plk1 promotes malignant progress in human esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 9–16. [Google Scholar] [CrossRef]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef]
- Prevo, R.; Pirovano, G.; Puliyadi, R.; Herbert, K.J.; Rodriguez-Berriguete, G.; O’Docherty, A.; Greaves, W.; McKenna, W.G.; Higgins, G.S. CDK1 inhibition sensitizes normal cells to DNA damage in a cell cycle dependent manner. Cell Cycle 2018, 17, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.P.; Poon, R.Y.; Ma, H.T. Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol. Cell. Biol. 2011, 31, 1478–1491. [Google Scholar] [CrossRef]
- Watanabe, N.; Arai, H.; Iwasaki, J.; Shiina, M.; Ogata, K.; Hunter, T.; Osada, H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 11663–11668. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef] [PubMed]
- Wells, N.J.; Watanabe, N.; Tokusumi, T.; Jiang, W.; Verdecia, M.A.; Hunter, T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 1999, 112 Pt 19, 3361–3371. [Google Scholar] [CrossRef]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12. [Google Scholar] [CrossRef]
- Lobjois, V.; Froment, C.; Braud, E.; Grimal, F.; Burlet-Schiltz, O.; Ducommun, B.; Bouche, J.P. Study of the docking-dependent PLK1 phosphorylation of the CDC25B phosphatase. Biochem. Biophys. Res. Commun. 2011, 410, 87–90. [Google Scholar] [CrossRef]
- Sullivan, C.; Liu, Y.; Shen, J.; Curtis, A.; Newman, C.; Hock, J.M.; Li, X. Novel interactions between FOXM1 and CDC25A regulate the cell cycle. PLoS ONE 2012, 7, e51277. [Google Scholar] [CrossRef][Green Version]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002, 3, 341–348. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef]
- L’Italien, L.; Tanudji, M.; Russell, L.; Schebye, X.M. Unmasking the redundancy between Cdk1 and Cdk2 at G2 phase in human cancer cell lines. Cell Cycle 2006, 5, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Dominguez-Brauer, C.; Wang, Z.; Asara, J.M.; Costa, R.H.; Tyner, A.L.; Lau, L.F.; Raychaudhuri, P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J. Biol. Chem. 2009, 284, 30695–30707. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.Y.; Tong, T.H.; Cheung, A.M.; Tsang, A.C.; Leung, W.Y.; Yao, K.M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005, 118, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Neubauer, M.C.; Yerabolu, D.; Kojonazarov, B.; Schlueter, B.C.; Neubert, L.; Jonigk, D.; Baal, N.; Ruppert, C.; Dorfmuller, P.; et al. Targeting cyclin-dependent kinases for the treatment of pulmonary arterial hypertension. Nat. Commun. 2019, 10, 2204. [Google Scholar] [CrossRef]
- Ferguson, A.M.; White, L.S.; Donovan, P.J.; Piwnica-Worms, H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol. Cell. Biol. 2005, 25, 2853–2860. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Lighthouse, J.K.; Greif, D.M. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014, 6, 809–817. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef]
- Wilson, J.L.; Warburton, R.; Taylor, L.; Toksoz, D.; Hill, N.; Polgar, P. Unraveling endothelin-1 induced hypercontractility of human pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. PLoS ONE 2018, 13, e0195780. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Frid, M.G.; Graham, B.B.; Tuder, R.M. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc. Res. 2018, 114, 551–564. [Google Scholar] [CrossRef]
- de Raaf, M.A.; Schalij, I.; Gomez-Arroyo, J.; Rol, N.; Happe, C.; de Man, F.S.; Vonk-Noordegraaf, A.; Westerhof, N.; Voelkel, N.F.; Bogaard, H.J. SuHx rat model: Partly reversible pulmonary hypertension and progressive intima obstruction. Eur. Respir. J. 2014, 44, 160–168. [Google Scholar] [CrossRef]
- Sallum, C.O.; Wilson, J.L.; Rupasinghe, C.; Berg, E.; Yu, J.; Green, D.S.; Taylor, L.; Mierke, D.; Polgar, P. Enhancing and limiting endothelin-1 signaling with a cell-penetrating peptide mimicking the third intracellular loop of the ETB receptor. Chem. Biol. Drug Des. 2012, 80, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Rupasinghe, C.; Wilson, J.L.; Taylor, L.; Rahimi, N.; Mierke, D.; Polgar, P. Targeting receptor tyrosine kinases and their downstream signaling with cell-penetrating peptides in human pulmonary artery smooth muscle and endothelial cells. Chem. Biol. Drug Des. 2015, 85, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Taylor, L.; Mierke, D.; Berg, E.; Shia, M.; Fishman, J.; Sallum, C.; Polgar, P. Limiting angiotensin II signaling with a cell-penetrating peptide mimicking the second intracellular loop of the angiotensin II type-I receptor. Chem. Biol. Drug Des. 2010, 76, 70–76. [Google Scholar] [CrossRef]
- Comhair, S.A.; Xu, W.; Mavrakis, L.; Aldred, M.A.; Asosingh, K.; Erzurum, S.C. Human primary lung endothelial cells in culture. Am. J. Respir. Cell Mol. Biol. 2012, 46, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, P.; Courtman, D.; Babaie, S.; Stewart, D.J. Role of apoptosis in pulmonary hypertension: From experimental models to clinical trials. Pharmacol. Ther. 2010, 126, 1–8. [Google Scholar] [CrossRef]
- Krymskaya, V.P.; Snow, J.; Cesarone, G.; Khavin, I.; Goncharov, D.A.; Lim, P.N.; Veasey, S.C.; Ihida-Stansbury, K.; Jones, P.L.; Goncharova, E.A. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011, 25, 1922–1933. [Google Scholar] [CrossRef]
- Savai, R.; Al-Tamari, H.M.; Sedding, D.; Kojonazarov, B.; Muecke, C.; Teske, R.; Capecchi, M.R.; Weissmann, N.; Grimminger, F.; Seeger, W.; et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat. Med. 2014, 20, 1289–1300. [Google Scholar] [CrossRef]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Code ID | Gender | Age | Germline Mutation |
|---|---|---|---|---|
| CON1 | C128 | Male | 36 | None |
| CON2 | A678 | Male | 39 | None |
| CON3 | TRL-CON-4, E352 | Female | 48 | None |
| CON4 | Lot#1189 | Female | 17 | None |
| CON5 | Lot#1487 | Male | 21 | None |
| PAH1 | D355 | Male | 42 | None |
| PAH2 | ST012 F281 | Female | 26 | Smad-8 R294X |
| PAH3 | CCF005 E037 | Female | 47 | None |
| PAH4 | IPAH16 F518 | ? | ? | None |
| PAH5 | CC008 | ? | ? | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal-Ghosh, R.; Xue, D.; Warburton, R.; Hill, N.; Polgar, P.; Wilson, J.L. CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 6943. https://doi.org/10.3390/ijms22136943
Pal-Ghosh R, Xue D, Warburton R, Hill N, Polgar P, Wilson JL. CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2021; 22(13):6943. https://doi.org/10.3390/ijms22136943
Chicago/Turabian StylePal-Ghosh, Ruma, Danfeng Xue, Rod Warburton, Nicholas Hill, Peter Polgar, and Jamie L. Wilson. 2021. "CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 22, no. 13: 6943. https://doi.org/10.3390/ijms22136943
APA StylePal-Ghosh, R., Xue, D., Warburton, R., Hill, N., Polgar, P., & Wilson, J. L. (2021). CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 22(13), 6943. https://doi.org/10.3390/ijms22136943