
Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways

 , ,
, ,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Patients’ Characteristics
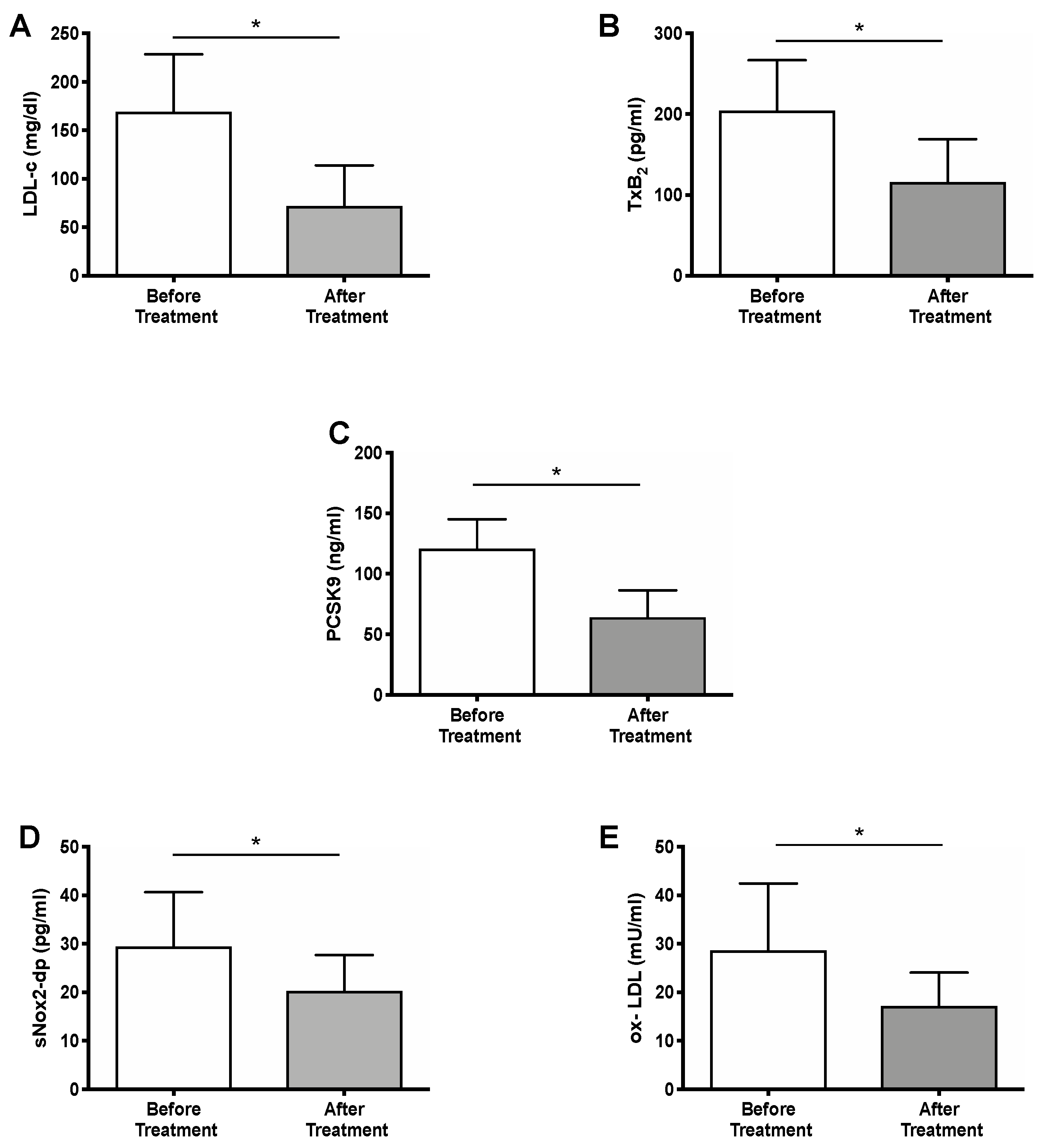
2.2. Effect of PCSK9i Treatment
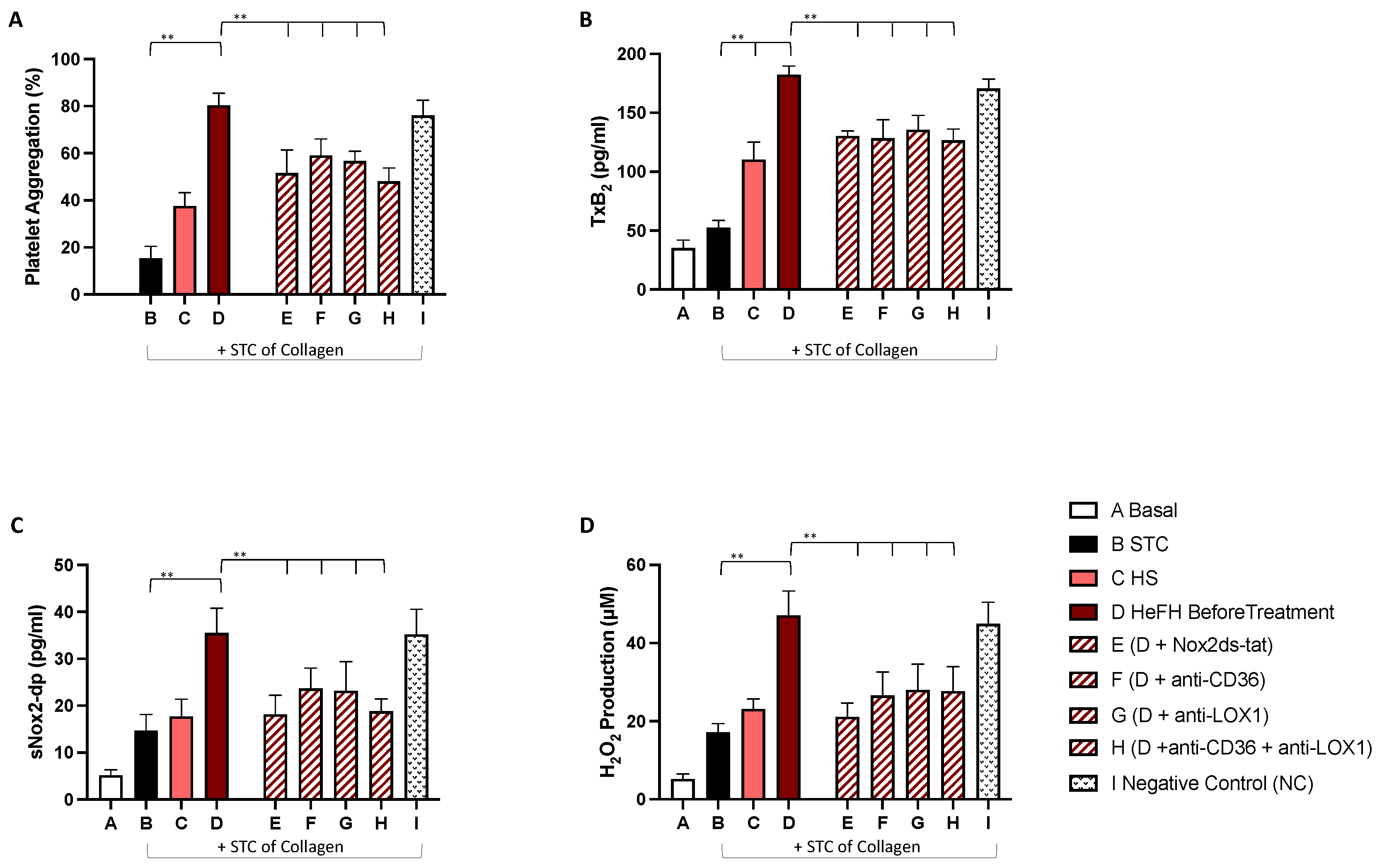
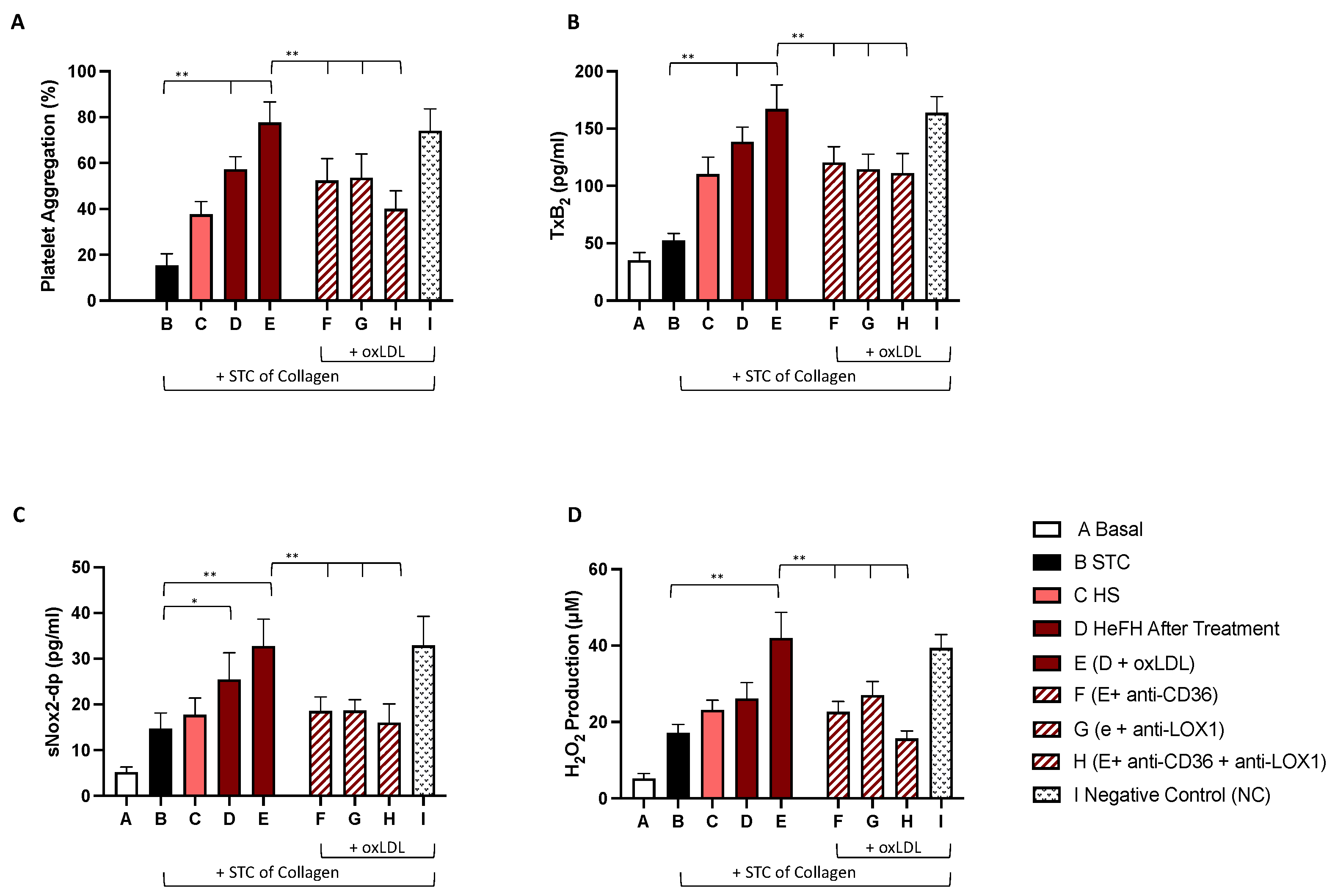
2.3. In Vitro Study
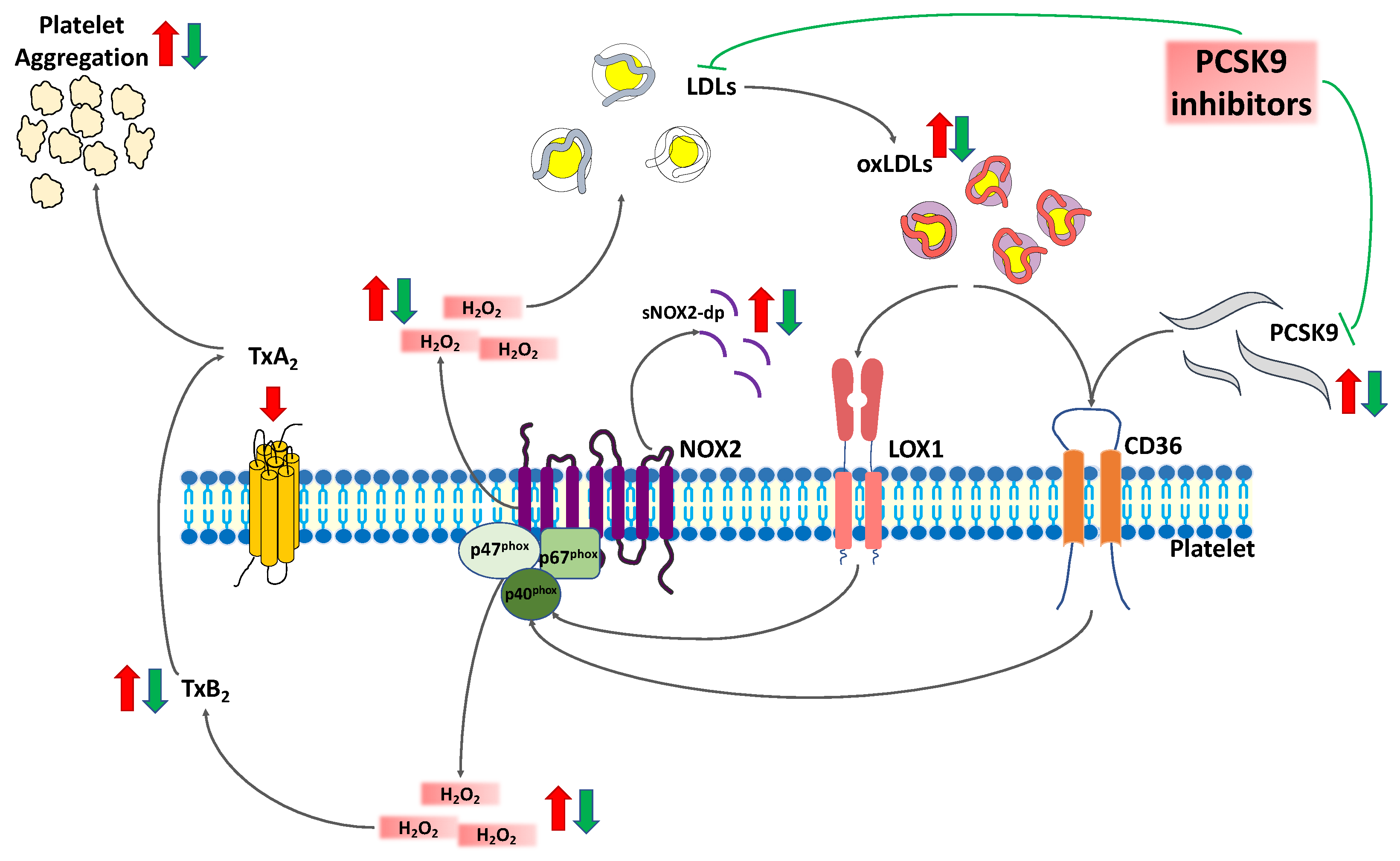
3. Discussion
4. Materials and Methods
- Plasma and platelets sNOX2-dp production
- Plasma Detection of ox-LDL
- Plasma and platelets TxB2 Assay
- Plasma and platelets H2O2 production
- Plasma PCSK9 detection
4.1. In Vitro Study
4.1.1. Platelet Preparation
4.1.2. oxLDL Preparation
4.1.3. Platelet Aggregation
4.2. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hooper, A.J.; Marais, A.D.; Tanyanyiwa, D.M.; Burnett, J.R. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007, 193, 445–448. [Google Scholar] [CrossRef]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenney, J.M.; Koren, M.J.; Kereiakes, D.J.; Hanotin, C.; Ferrand, A.C.; Stein, E.A. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J. Am. Coll. Cardiol. 2012, 59, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.G.; Nedergaard, B.S.; Rogers, W.J.; Fialkow, J.; Neutel, J.M.; Ramstad, D.; Somaratne, R.; Legg, J.C.; Nelson, P.; Scott, R.; et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: The LAPLACE-2 randomized clinical trial. JAMA 2014, 311, 1870–1882. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Ako, J.; Hibi, K.; Tsujita, K.; Hiro, T.; Morino, Y.; Kozuma, K.; Shinke, T.; Otake, H.; Uno, K.; Louie, M.J.; et al. Effect of Alirocumab on Coronary Atheroma Volume in Japanese Patients with Acute Coronary Syndrome—The ODYSSEY J-IVUS Trial. Circ. J. 2019, 83, 2025–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Safarova, M.S.; Satterfield, B.A.; Fan, X.; Austin, E.E.; Ye, Z.; Bastarache, L.; Zheng, N.; Ritchie, M.D.; Borthwick, K.M.; Williams, M.S.; et al. A phenome-wide association study to discover pleiotropic effects of. NPJ Genom. Med. 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, T.D.; Christopoulou, E.C.; Elisaf, M.S. Pleiotropic effects of proprotein convertase subtilisin/kexin type 9 inhibitors? Curr. Opin. Lipidol. 2018, 29, 333–339. [Google Scholar] [CrossRef]
- Paciullo, F.; Momi, S.; Gresele, P. PCSK9 in Haemostasis and Thrombosis: Possible Pleiotropic Effects of PCSK9 Inhibitors in Cardiovascular Prevention. Thromb. Haemost. 2019, 119, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandraffino, G.; Scicali, R.; Rodriguez-Carrio, J.; Savarino, F.; Mamone, F.; Scuruchi, M.; Cinquegrani, M.; Imbalzano, E.; Di Pino, A.; Piro, S.; et al. Arterial stiffness improvement after adding on PCSK9 inhibitors or ezetimibe to high-intensity statins in patients with familial hypercholesterolemia: A Two-Lipid Center Real-World Experience. J. Clin. Lipidol. 2020, 14, 231–240. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, W.; Lu, M.; Wang, Y. Association Between Circulating Proprotein Convertase Subtilisin/Kexin Type 9 and Major Adverse Cardiovascular Events, Stroke, and All-Cause Mortality: Systemic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 617249. [Google Scholar] [CrossRef]
- Cammisotto, V.; Pastori, D.; Nocella, C.; Bartimoccia, S.; Castellani, V.; Marchese, C.; Scavalli, A.S.; Ettorre, E.; Viceconte, N.; Violi, F.; et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants 2020, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Nocella, C.; Cammisotto, V.; Bartimoccia, S.; Castellani, V.; Loffredo, L.; Pastori, D.; Pignatelli, P.; Sanguigni, V.; Violi, F.; Carnevale, R. A novel role of MMP2 in regulating platelet NOX2 activation. Free Radic. Biol. Med. 2020, 152, 355–362. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, G.A. Mechanisms of platelet activation: Thromboxane A2 as an amplifying signal for other agonists. Am. J. Cardiol. 1991, 68, 11B–15B. [Google Scholar] [CrossRef]
- Carnevale, D.; Vecchione, C.; Mascio, G.; Esposito, G.; Cifelli, G.; Martinello, K.; Landolfi, A.; Selvetella, G.; Grieco, P.; Damato, A.; et al. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc. Res. 2012, 93, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Tiseo, G.; Cavarretta, E.; Forniti, A.; Nocella, C.; Sciarretta, S.; Spagnolello, O.; Baldini, E.; Peruzzi, M.; Bertazzoni, G.; Menichetti, F.; et al. Interplay between Nox2 Activity and Platelet Activation in Patients with Sepsis and Septic Shock: A Prospective Study. Oxid. Med. Cell. Longev. 2020, 2020, 4165358. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Andreozzi, P.; Carnevale, R.; Bartimoccia, S.; Limaj, S.; Melandri, S.; Brunori, M.; Spallacci, G.; Violi, F.; Pignatelli, P. Does the Coexistence of Chronic Obstructive Pulmonary Disease and Atrial Fibrillation Affect Nox2 Activity and Urinary Isoprostanes Excretion? Antioxid. Redox Signal. 2019, 31, 786–790. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Bartimoccia, S.; Cammisotto, V.; Cocomello, N.; Colantoni, A.; Nocella, C.; Carnevale, R.; Ferro, D.; Angelico, F.; et al. Poor Adherence to Mediterranean Diet and Serum Lipopolysaccharide are Associated with Oxidative Stress in Patients with Non-Alcoholic Fatty Liver Disease. Nutrients 2020, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef]
- Stein, E.A.; Gipe, D.; Bergeron, J.; Gaudet, D.; Weiss, R.; Dufour, R.; Wu, R.; Pordy, R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A phase 2 randomised controlled trial. Lancet 2012, 380, 29–36. [Google Scholar] [CrossRef]
- Pastori, D.; Nocella, C.; Farcomeni, A.; Bartimoccia, S.; Santulli, M.; Vasaturo, F.; Carnevale, R.; Menichelli, D.; Violi, F.; Pignatelli, P.; et al. Relationship of PCSK9 and Urinary Thromboxane Excretion to Cardiovascular Events in Patients With Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1455–1462. [Google Scholar] [CrossRef]
- Afanasieva, O.; Ezhov, M.V.; Klesareva, E.; Razova, O.; Chubykina, U.; Egiazaryan, M.; Sherstyuk, E.; Afanasieva, M.; Utkina, E.; Pokrovsky, S. Effect of Evolocumab on Lipoprotein(a) and PCSK9 in Healthy Individuals with Elevated Lipoprotein(a) Level. J. Cardiovasc. Dev. Dis. 2020, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kanazawa, M.; Kagaya, Y.; Kondo, M.; Sato, K.; Endo, H.; Nozaki, E. Plasma kinetics of mature PCSK9, furin-cleaved PCSK9, and Lp(a) with or without administration of PCSK9 inhibitors in acute myocardial infarction. J. Cardiol. 2020, 76, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Miles, J.; Tavori, H.; Fazio, S. Diagnosing Resistance to a Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor. Ann. Intern. Med. 2018, 168, 376–379. [Google Scholar] [CrossRef]
- Desai, N.R.; Giugliano, R.P.; Wasserman, S.M.; Gibbs, J.P.; Liu, T.; Scott, R.; Sabatine, M.S. Association Between Circulating Baseline Proprotein Convertase Subtilisin Kexin Type 9 Levels and Efficacy of Evolocumab. JAMA Cardiol. 2017, 2, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.aifa.gov.it/web/guest/-/attivazione-web-e-pubblicazione-schede-di-monitoraggio-registro-praluent-cvd-; https://www.aifa.gov.it/-/attivazione-web-e-pubblicazione-schede-di-monitoraggio-registro-repatha-est-; (accessed on 8 August 2020).
- Casula, M.; Olmastroni, E.; Pirillo, A.; Catapano, A.L. Evaluation of the performance of Dutch Lipid Clinic Network score in an Italian FH population: The LIPIGEN study. Atherosclerosis 2018, 277, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Erasmo, L.; Minicocci, I.; Di Costanzo, A.; Pigna, G.; Commodari, D.; Ceci, F.; Montali, A.; Brancato, F.; Stanca, I.; Nicolucci, A.; et al. Clinical Implications of Monogenic Versus Polygenic Hypercholesterolemia: Long-Term Response to Treatment, Coronary Atherosclerosis Burden, and Cardiovascular Events. J. Am. Heart Assoc. 2021, 10, e018932. [Google Scholar] [CrossRef] [PubMed]
- Winocour, P.D. Platelet abnormalities in diabetes mellitus. Diabetes 1992, 41 (Suppl. 2), 26–31. [Google Scholar] [CrossRef]
- Carnevale, R.; Silvestri, R.; Loffredo, L.; Novo, M.; Cammisotto, V.; Castellani, V.; Bartimoccia, S.; Nocella, C.; Violi, F. Oleuropein, a component of extra virgin olive oil, lowers postprandial glycaemia in healthy subjects. Br. J. Clin. Pharmacol. 2018, 84, 1566–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emelyanov, A.; Fedoseev, G.; Abulimity, A.; Rudinski, K.; Fedoulov, A.; Karabanov, A.; Barnes, P.J. Elevated concentrations of exhaled hydrogen peroxide in asthmatic patients. Chest 2001, 120, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Pagano, E.; Csanyi, G.; Pagano, P.J. NADPH oxidase inhibitors: A decade of discovery from Nox2ds to HTS. Cell. Mol. Life Sci. 2012, 69, 2315–2325. [Google Scholar] [CrossRef] [Green Version]
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Jay, A.G.; Chen, A.N.; Paz, M.A.; Hung, J.P.; Hamilton, J.A. CD36 binds oxidized low density lipoprotein (LDL) in a mechanism dependent upon fatty acid binding. J. Biol. Chem. 2015, 290, 4590–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HeFH Patients Before Treatment (n = 80) | |
|---|---|
| Age (years) | 57.7 ± 10.8 |
| Female | 45% |
| BMI (kg/m2) | 26.5 ± 4.2 |
| Smokers (%) | 27.5% |
| Total Cholesterol (mg/dL) | 246.0 ± 66.0 |
| HDL cholesterol (mg/dL) | 50.8 ± 11.3 |
| LDL cholesterol (mg/dL) | 169.2 ± 59.3 |
| Triglycerides (mg/dL) | 121.9 ± 53.5 |
| Hypertension (%) | 52.5% |
| Prior Cardiovascular Events (%) | 51.5% |
| Antiplatelet drugs | 52.5% |
| Diabetes | 0% |
| Statins | 100% |
| Δ TxB2 | Δ ox-LDL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| B | E.S | Beta | p | B | E.S | Beta | p | ||
| Δ PCSK9 | −0.025 | 0.343 | −0.008 | 0.942 | Δ PCSK9 | 0.153 | 0.062 | 0.277 | 0.016 |
| Δ ox-LDL | 1.246 | 0.607 | 0.233 | 0.044 | - | ||||
| Δ LDL-c | 0.387 | 0.140 | 0.316 | 0.007 | Δ LDL-c | 0.044 | 0.027 | 0.191 | 0.113 |
| Δ sNOX2-dp | 0.315 | 0.595 | 0.061 | 0.597 | Δ sNOX2-dp | 0.223 | 0.109 | 0.231 | 0.045 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’Erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193. https://doi.org/10.3390/ijms22137193
Cammisotto V, Baratta F, Castellani V, Bartimoccia S, Nocella C, D’Erasmo L, Cocomello N, Barale C, Scicali R, Di Pino A, et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. International Journal of Molecular Sciences. 2021; 22(13):7193. https://doi.org/10.3390/ijms22137193
Chicago/Turabian StyleCammisotto, Vittoria, Francesco Baratta, Valentina Castellani, Simona Bartimoccia, Cristina Nocella, Laura D’Erasmo, Nicholas Cocomello, Cristina Barale, Roberto Scicali, Antonino Di Pino, and et al. 2021. "Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways" International Journal of Molecular Sciences 22, no. 13: 7193. https://doi.org/10.3390/ijms22137193
APA StyleCammisotto, V., Baratta, F., Castellani, V., Bartimoccia, S., Nocella, C., D’Erasmo, L., Cocomello, N., Barale, C., Scicali, R., Di Pino, A., Piro, S., Del Ben, M., Arca, M., Russo, I., Purrello, F., Carnevale, R., Violi, F., Pastori, D., & Pignatelli, P. (2021). Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. International Journal of Molecular Sciences, 22(13), 7193. https://doi.org/10.3390/ijms22137193