
Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1?

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
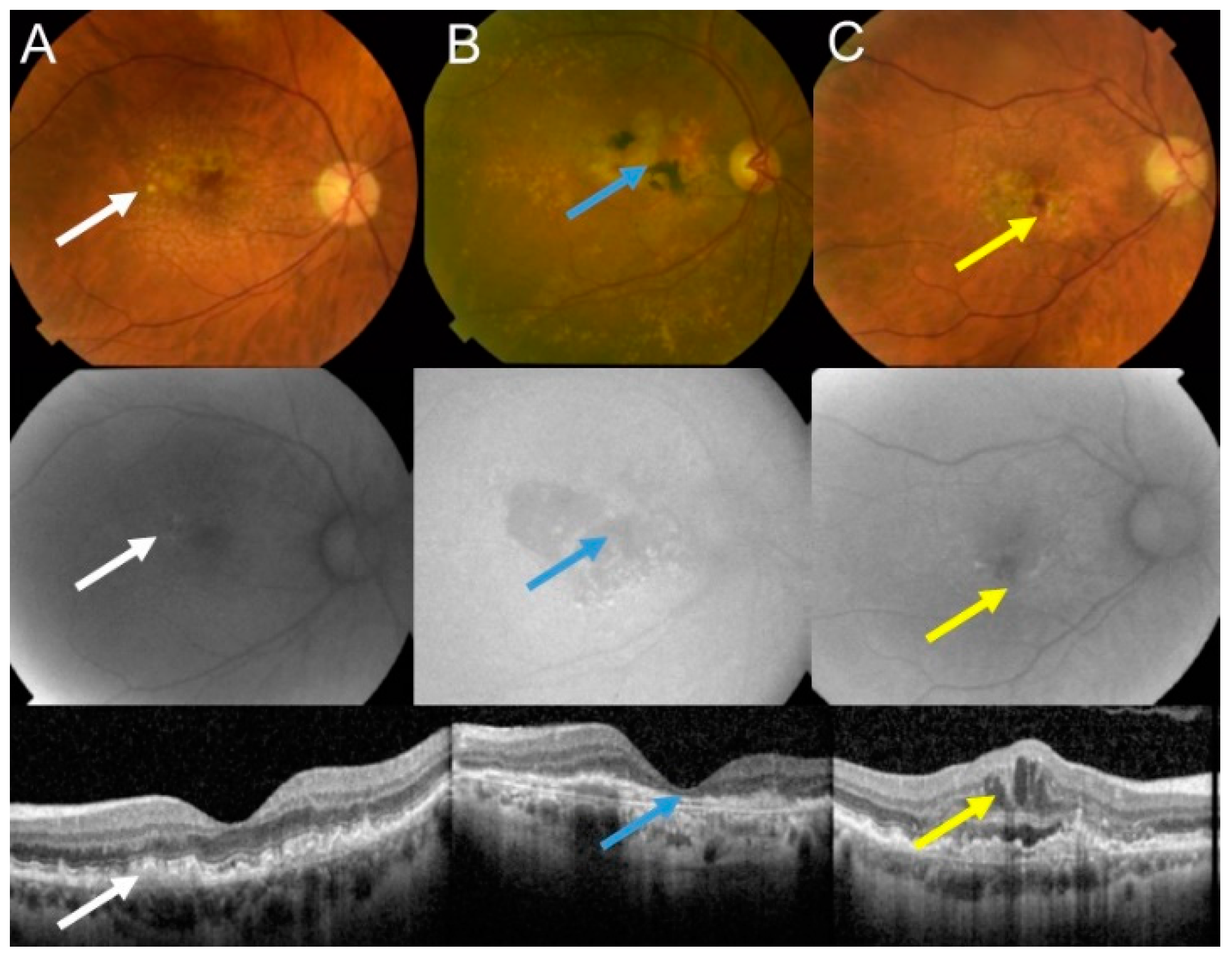
2. Age-Related Macular Degeneration
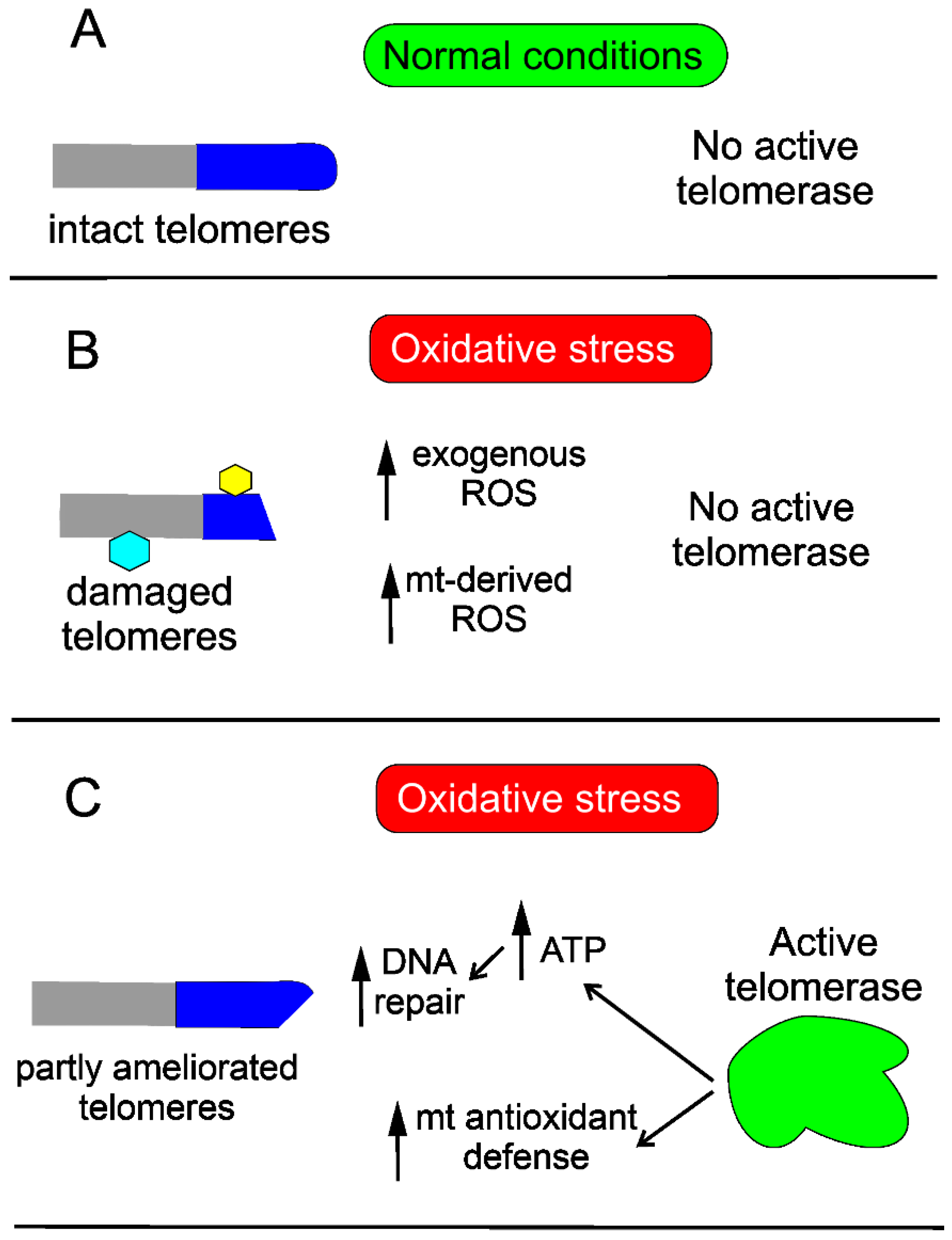
3. Telomeres and Telomerase in AMD
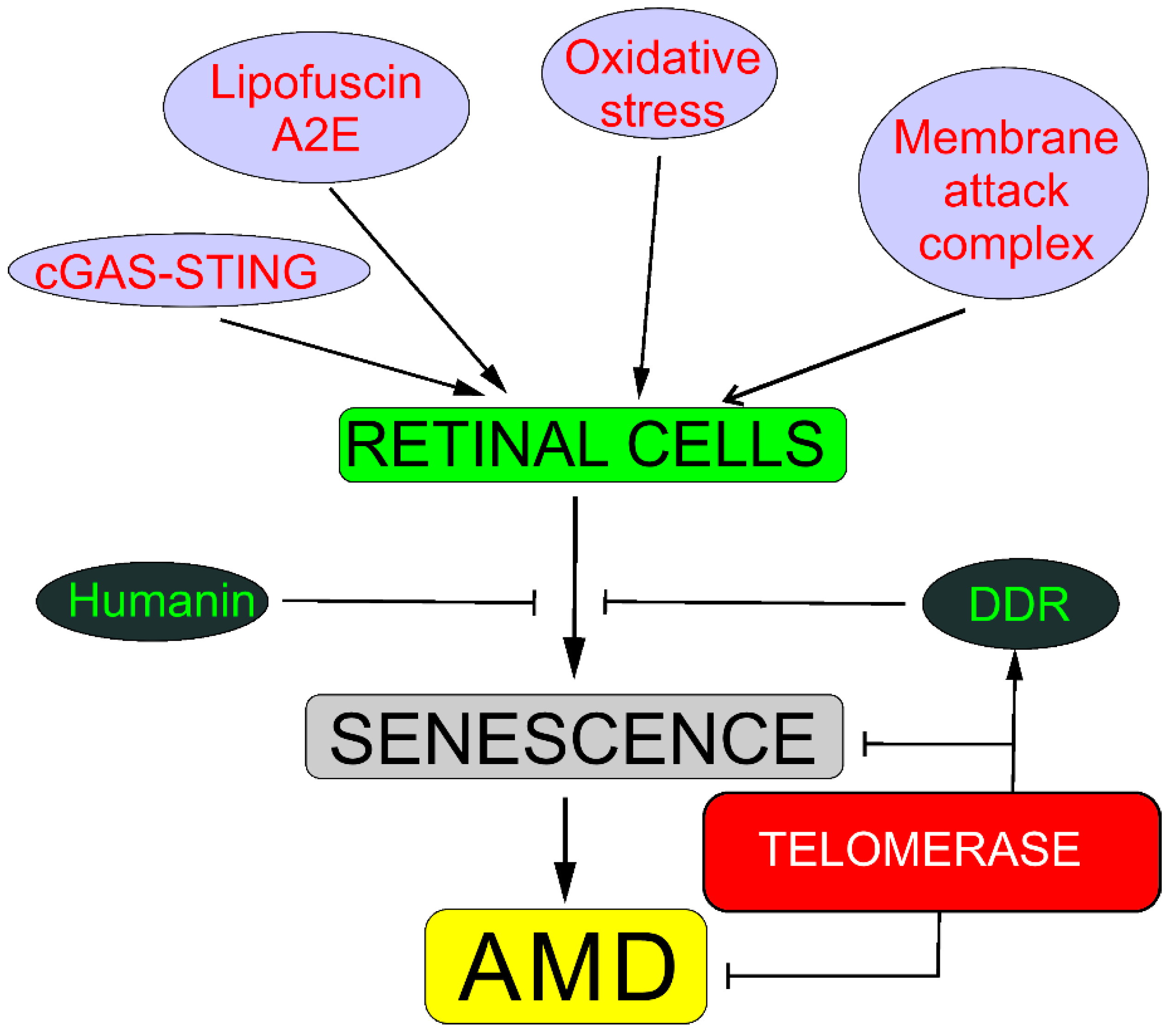
4. Senescence, DNA Damage Response, and Autophagy May Underline the Involvement of Telomerase in AMD
4.1. Senescence
4.2. DNA Damage Response
4.3. Autophagy
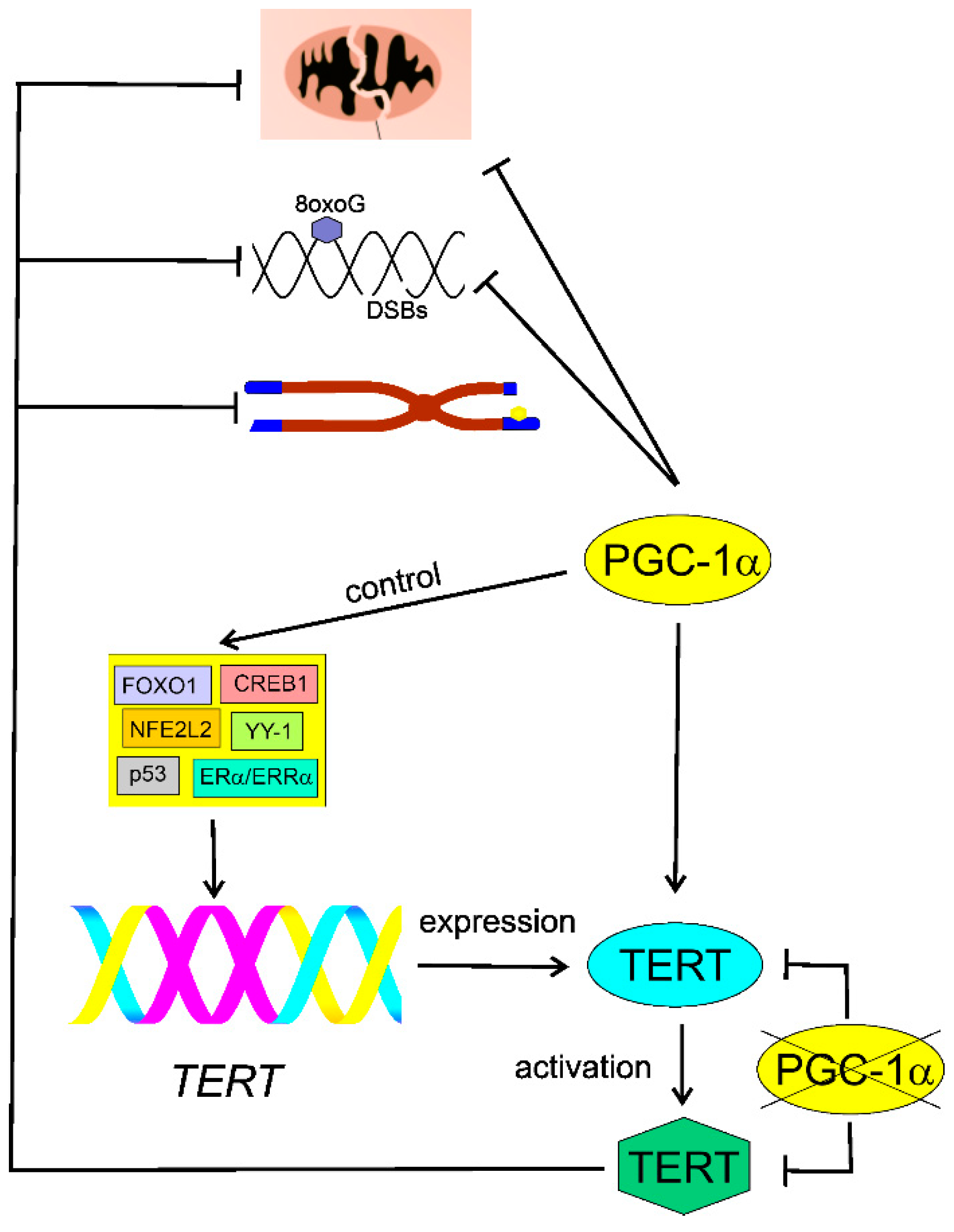
5. PGC-1α May Link Telomerase with AMD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casagrande, S.; Hau, M. Telomere attrition: Metabolic regulation and signalling function? Biol. Lett. 2019, 15, 20180885. [Google Scholar] [CrossRef]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis 2016, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, Amd41–amd47. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.; Mitchell, P.; Freund, K.B.; Sadda, S.; Holz, F.G.; Brittain, C.; Henry, E.C.; Ferrara, D. The Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 369–390. [Google Scholar] [CrossRef]
- Bellezza, I. Oxidative Stress in Age-Related Macular Degeneration: Nrf2 as Therapeutic Target. Front. Pharm. 2018, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Cousins, S.W.; Espinosa-Heidmann, D.G.; Alexandridou, A.; Sall, J.; Dubovy, S.; Csaky, K. The role of aging, high fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp. Eye. Res. 2002, 75, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, T.J.; Lorés-Motta, L.; Hoyng, C.B.; Lechanteur, Y.T.E.; den Hollander, A.I. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol Opt. 2020, 40, 140–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.M.; Groot, A.J.; van der Groep, P.; Sersansie, R.; Vooijs, M.; van Diest, P.J.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Active HIF-1 in the normal human retina. J. Histochem. Cytochem. 2010, 58, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Mockett, R.J.; Orr, W.C. Mechanisms of aging: An appraisal of the oxidative stress hypothesis. Free. Radic. Biol. Med. 2002, 33, 575–586. [Google Scholar] [CrossRef]
- Vatner, S.F.; Zhang, J.; Oydanich, M.; Berkman, T.; Naftalovich, R.; Vatner, D.E. Healthful Aging Mediated by Inhibition of Oxidative Stress. Ageing Res. Rev. 2020, 101194. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye. Res. 2020, 100858. [Google Scholar] [CrossRef]
- Campbell, J.P.; Zhang, M.; Hwang, T.S.; Bailey, S.T.; Wilson, D.J.; Jia, Y.; Huang, D. Detailed Vascular Anatomy of the Human Retina by Projection-Resolved Optical Coherence Tomography Angiography. Sci. Rep. 2017, 7, 42201. [Google Scholar] [CrossRef] [Green Version]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.; Ma, J.X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. physiology. Heart Circ. Physiol. 2016, 311, H738–749. [Google Scholar] [CrossRef] [Green Version]
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharm. 2019, 10, 1363. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hartnett, M.E. Regulation of signaling events involved in the pathophysiology of neovascular AMD. Mol. Vis. 2016, 22, 189–202. [Google Scholar]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; André, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell. Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef]
- de Carlo, T.E.; Romano, A.; Waheed, N.K.; Duker, J.S. A review of optical coherence tomography angiography (OCTA). Int. J. Retin. Vitr. 2015, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, A.J.; Duker, J.S.; Reichel, E. Geographic atrophy: Where we are now and where we are going. Curr. Opin. Ophthalmol. 2021, 32, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Busquets, M.A.; Sabbagh, O. Current status of home monitoring technology for age-related macular degeneration. Curr. Opin. Ophthalmol. 2021, 32, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, F1000 Faculty Rev-1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, W.R.; Meeker, A.K.; Rizzo, A.; Rajpara, S.; Rosenthal, I.M.; Flores Bellver, M.; Aparicio Domingo, S.; Zhong, X.; Barber, J.R.; Joshu, C.E.; et al. A unique telomere DNA expansion phenotype in human retinal rod photoreceptors associated with aging and disease. Brain Pathol. 2019, 29, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dow, C.T.; Harley, C.B. Evaluation of an oral telomerase activator for early age-related macular degeneration—a pilot study. Clin. Ophthalmol. 2016, 10, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.K.; Kim, B.H.; Han, Y.S.; Park, I.K. The effect of telomerase expression on the escape from M2 crisis in virus-transformed human retinal pigment epithelial cells. Exp. Mol. Med. 2002, 34, 107–113. [Google Scholar] [CrossRef]
- Rastmanesh, R. Potential of melatonin to treat or prevent age-related macular degeneration through stimulation of telomerase activity. Med. Hypotheses 2011, 76, 79–85. [Google Scholar] [CrossRef]
- Rowe-Rendleman, C.; Glickman, R.D. Possible therapy for age-related macular degeneration using human telomerase. Brain Res. Bull. 2004, 62, 549–553. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Teo, S.H.; Jackson, S.P. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004, 18, 1781–1799. [Google Scholar] [CrossRef] [Green Version]
- Doksani, Y. The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes 2019, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Nagpal, N.; Agarwal, S. Telomerase RNA processing: Implications for human health and disease. Stem Cells 2020. [Google Scholar] [CrossRef]
- Podlevsky, J.D.; Bley, C.J.; Omana, R.V.; Qi, X.; Chen, J.J. The telomerase database. Nucleic Acids Res. 2008, 36, D339–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sung, Y.H.; Cheong, C.; Choi, Y.S.; Jeon, H.K.; Sun, W.; Hahn, W.C.; Ishikawa, F.; Lee, H.W. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene 2008, 27, 3754–3760. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Barszczewska, G.; Gralewska, P.; Kaarniranta, K. Oxidative stress induces mitochondrial dysfunction and autophagy in ARPE-19 cells. Acta Ophthalmol. 2019, 97. [Google Scholar] [CrossRef]
- Drigeard Desgarnier, M.C.; Zinflou, C.; Mallet, J.D.; Gendron, S.P.; Méthot, S.J.; Rochette, P.J. Telomere Length Measurement in Different Ocular Structures: A Potential Implication in Corneal Endothelium Pathogenesis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5547–5555. [Google Scholar] [CrossRef] [Green Version]
- Immonen, I.; Seitsonen, S.; Saionmaa, O.; Fyhrquist, F. Leucocyte telomere length in age-related macular degeneration. Acta Ophthalmol 2013, 91, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Tsukakoshi, K.; Ikebukuro, K. G-quadruplex: Flexible conformational changes by cations, pH, crowding and its applications to biosensing. Biosens. Bioelectron. 2021, 178, 113030. [Google Scholar] [CrossRef]
- Shafirovich, V.; Geacintov, N.E. Excision of Oxidatively Generated Guanine Lesions by Competitive DNA Repair Pathways. Int. J. Mol. Sci. 2021, 22, 2698. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Lee, J.; Lee, H. Alternative paths to telomere elongation. Semin. Cell Dev. Biol. 2021, 113, 88–96. [Google Scholar] [CrossRef]
- Banevicius, M.; Gedvilaite, G.; Vilkeviciute, A.; Kriauciuniene, L.; Zemaitiene, R.; Liutkeviciene, R. Association of relative leukocyte telomere length and genetic variants in telomere-related genes (TERT, TERT-CLPTM1, TRF1, TNKS2, TRF2) with atrophic age-related macular degeneration. Ophthalmic Genet. 2021, 42, 189–194. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Rambhatla, L.; Chiu, C.P.; Glickman, R.D.; Rowe-Rendleman, C. In vitro differentiation capacity of telomerase immortalized human RPE cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1622–1630. [Google Scholar]
- Lau, B.W.; Tsao, G.S.; So, K.F.; Yip, H.K. Expression of telomerase reverse transcriptase in adult goldfish retina. J. Mol. Neurosci. 2007, 32, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.W.; Wong, A.O.; Tsao, G.S.; So, K.F.; Yip, H.K. Molecular cloning and characterization of the zebrafish (Danio rerio) telomerase catalytic subunit (telomerase reverse transcriptase, TERT). J. Mol. Neurosci. 2008, 34, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Min, X.J.; Zhou, Q.J.; Liu, T.; Yin, H.M.; Dong, X.G.; Xie, L.X. Expression of mouse telomerase reverse transcription in a mouse model of oxygen-induced retinopathy. Zhonghua Yan Ke Za Zhi 2009, 45, 199–205. (In Chinese) [Google Scholar] [PubMed]
- Wright, W.E.; Shay, J.W. Telomere positional effects and the regulation of cellular senescence. Trends Genet. 1992, 8, 193–197. [Google Scholar] [CrossRef]
- Harley, C.B.; Liu, W.; Flom, P.L.; Raffaele, J.M. A natural product telomerase activator as part of a health maintenance program: Metabolic and cardiovascular response. Rejuvenation Res. 2013, 16, 386–395. [Google Scholar] [CrossRef]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between Autophagy and the Ubiquitin-Proteasome System and Its Role in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Haferkamp, S.; Tran, S.L.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging 2009, 1, 542–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, H.; Handa, J.T.; Aotaki-Keen, A.; Sherwood, S.W.; West, M.D.; Hjelmeland, L.M. Beta-galactosidase histochemistry and telomere loss in senescent retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1999, 40, 197–202. [Google Scholar]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowski, M.R. RPE cell senescence: A key contributor to age-related macular degeneration. Med. hypotheses 2012, 78, 505–510. [Google Scholar] [CrossRef]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxidative Med. Cell. Longev. 2017, 2017, 5293258. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Van Houten, B.; Jin, G.F.; Conklin, C.A.; Godley, B.F. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 1999, 68, 765–772. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef] [Green Version]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [Green Version]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishnav, R.A.; Singh, I.N.; Miller, D.M.; Hall, E.D. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. J. Neurotrauma 2010, 27, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Pawlowska, E.; Sobczuk, A.; Szczepanska, J.; Kaarniranta, K. The Aging Stress Response and Its Implication for AMD Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8840. [Google Scholar] [CrossRef]
- Honda, S.; Hjelmeland, L.M.; Handa, J.T. Oxidative stress--induced single-strand breaks in chromosomal telomeres of human retinal pigment epithelial cells in vitro. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2139–2144. [Google Scholar]
- Honda, S.; Weigel, A.; Hjelmeland, L.M.; Handa, J.T. Induction of telomere shortening and replicative senescence by cryopreservation. Biochem. Biophys. Res. Commun. 2001, 282, 493–498. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Jia, P.; Her, C.; Chai, W. DNA excision repair at telomeres. DNA repair 2015, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, A.; Schmid, B.; Akintola, O.; Saretzki, G. Telomerase Does Not Improve DNA Repair in Mitochondria upon Stress but Increases MnSOD Protein under Serum-Free Conditions. Int. J. Mol. Sci. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Gupta, A.; Wang, H.; Scherthan, H.; Dhar, S.; Gandhi, V.; Iliakis, G.; Shay, J.W.; Young, C.S.; Pandita, T.K. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 2003, 22, 131–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassour, J.; Radford, R.; Correia, A.; Fusté, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A., Jr.; Grant, M.B.; Boulton, M.E. Autophagy in the retina: A potential role in age-related macular degeneration. Adv. Exp. Med. Biol. 2012, 723, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy participates in, well, just about everything. Cell Death Differ. 2020, 27, 831–832. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxid. 2020, 9, 761. [Google Scholar] [CrossRef]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, L.; Gao, J.; Wen, L. Pro-Death or Pro-Survival: Contrasting Paradigms on Nanomaterial-Induced Autophagy and Exploitations for Cancer Therapy. Acc. Chem. Res. 2019, 52, 3164–3176. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Geli, V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front. cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Fan, X.; Lawson, W.E.; Paueksakon, P.; Harris, R.C. Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney Int. 2015, 88, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Yang, L.; Wang, T.; Zhang, J.; Li, T.; Ren, Y.; Wang, M.; Chen, X.; Lv, Y.; Wu, R. Irisin Improves Autophagy of Aged Hepatocytes via Increasing Telomerase Activity in Liver Injury. Oxidative Med. Cell Longev. 2020, 2020, 6946037. [Google Scholar] [CrossRef]
- Ali, M.; Devkota, S.; Roh, J.I.; Lee, J.; Lee, H.W. Telomerase reverse transcriptase induces basal and amino acid starvation-induced autophagy through mTORC1. Biochem. Biophys. Res. Commun. 2016, 478, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara-Romeo, I.; Martinez, P.; Saraswati, S.; Whittemore, K.; Graña-Castro, O.; Thelma Poluha, L.; Serrano, R.; Hernandez-Encinas, E.; Blanco-Aparicio, C.; Maria Flores, J.; et al. The mTOR pathway is necessary for survival of mice with short telomeres. Nat. Commun. 2020, 11, 1168. [Google Scholar] [CrossRef] [Green Version]
- Green, P.D.; Sharma, N.K.; Santos, J.H. Telomerase Impinges on the Cellular Response to Oxidative Stress Through Mitochondrial ROS-Mediated Regulation of Autophagy. Int. J. Mol. Sci. 2019, 20, 1509. [Google Scholar] [CrossRef] [Green Version]
- Hughes, W.E.; Chabowski, D.S.; Ait-Aissa, K.; Fetterman, J.L.; Hockenberry, J.; Beyer, A.M.; Gutterman, D.D. Critical Interaction between Telomerase and Autophagy in Mediating Flow-Induced Human Arteriolar Vasodilation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 446–457. [Google Scholar] [CrossRef]
- Roh, J.I.; Kim, Y.; Oh, J.; Kim, Y.; Lee, J.; Lee, J.; Chun, K.H.; Lee, H.W. Hexokinase 2 is a molecular bridge linking telomerase and autophagy. PLoS ONE 2018, 13, e0193182. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.H.; Chung, K.C. Human telomerase reverse transcriptase positively regulates mitophagy by inhibiting the processing and cytoplasmic release of mitochondrial PINK1. Cell Death Dis. 2020, 11, 425. [Google Scholar] [CrossRef]
- Ding, X.; Nie, Z.; She, Z.; Bai, X.; Yang, Q.; Wang, F.; Wang, F.; Geng, X. The Regulation of ROS- and BECN1-Mediated Autophagy by Human Telomerase Reverse Transcriptase in Glioblastoma. Oxidative Med. Cell. Longev. 2021, 2021, 6636510. [Google Scholar] [CrossRef]
- Supruniuk, E.; Mikłosz, A.; Chabowski, A. The Implication of PGC-1α on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Diao, J.; Zhao, H.; You, P.; You, H.; Wu, H.; Shou, X.; Cheng, G. Rosmarinic acid ameliorated cardiac dysfunction and mitochondrial injury in diabetic cardiomyopathy mice via activation of the SIRT1/PGC-1α pathway. Biochem. Biophys. Res. Commun. 2021, 546, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Zheng, A.; Zhang, H.; Cao, H. Down-regulation of betatrophin enhances insulin sensitivity in type 2 diabetes mellitus through activation of the GSK-3β/PGC-1α signaling pathway. J. Endocrinol. Investig. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moon, J.; Park, C.H.; Lee, J.; Cheng, H.; Floyd, Z.E.; Chang, J.S. NT-PGC-1α deficiency attenuates high-fat diet-induced obesity by modulating food intake, fecal fat excretion and intestinal fat absorption. Sci. Rep. 2021, 11, 1323. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Li, P.; Yin, X.; Yan, Z. PGC-1α promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 2011, 178, 1738–1748. [Google Scholar] [CrossRef]
- Guo, A.; Li, K.; Xiao, Q. Fibroblast growth factor 19 alleviates palmitic acid-induced mitochondrial dysfunction and oxidative stress via the AMPK/PGC-1α pathway in skeletal muscle. Biochem. Biophys. Res. Commun. 2020, 526, 1069–1076. [Google Scholar] [CrossRef]
- Tormos, A.M.; Pérez-Garrido, S.; Taléns-Visconti, R.; Nebreda, Á.R.; Sastre, J. Long term p38-a deficiency up-regulates antioxidant enzymes through compensatory NF-κB activation. Free. Radic. Biol Med. 2014, 75 (Suppl. S1), S52. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Jin, Y.; Yang, Q.; Meng, Q.; Liu, Q.; Dai, Y.; Cai, L.; Liu, Z.; Liu, K.; et al. Activating the PGC-1α/TERT Pathway by Catalpol Ameliorates Atherosclerosis via Modulating ROS Production, DNA Damage, and Telomere Function: Implications on Mitochondria and Telomere Link. Oxidative Med. Cell. Longev. 2018, 2018, 2876350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Patrushev, N.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. PGC-1α Modulates Telomere Function and DNA Damage in Protecting against Aging-Related Chronic Diseases. Cell Rep. 2015, 12, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Agurell, E.; Beevers, C.; Brendler-Schwaab, S.; Burlinson, B.; Clay, P.; Collins, A.; Smith, A.; Speit, G.; Thybaud, V.; et al. Recommendations for conducting the in vivo alkaline Comet assay. 4th International Comet Assay Workshop. Mutagenesis 2003, 18, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tice, R. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. JaCVAM-organized international validation study of the in vivo rat alkaline comet assay. Preface. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 786–788, 1. [Google Scholar] [CrossRef]
- Patten, I.S.; Arany, Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol. Metab. 2012, 23, 90–97. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Telomerase Reverse Transcriptase and Peroxisome Proliferator-Activated Receptor γ Co-Activator-1α Cooperate to Protect Cells from DNA Damage and Mitochondrial Dysfunction in Vascular Senescence. Rejuvenation Res. 2015, 18, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega Á, L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Satish, S.; Philipose, H.; Rosales, M.A.B.; Saint-Geniez, M. Pharmaceutical Induction of PGC-1α Promotes Retinal Pigment Epithelial Cell Metabolism and Protects against Oxidative Damage. Oxidative Med. Cell. Longev. 2018, 2018, 9248640. [Google Scholar] [CrossRef] [Green Version]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasiak, J.; Koskela, A.; Pawlowska, E.; Liukkonen, M.; Ruuth, J.; Toropainen, E.; Hyttinen, J.M.T.; Viiri, J.; Eriksson, J.E.; Xu, H.; et al. Epithelial-Mesenchymal Transition and Senescence in the Retinal Pigment Epithelium of NFE2L2/PGC-1α Double Knock-Out Mice. Int. J. Mol. Sci. 2021, 22, 1684. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra197. [Google Scholar] [CrossRef] [Green Version]
- Vaiserman, A.; Krasnienkov, D. Telomere Length as a Marker of Biological Age: State-of-the-Art, Open Issues, and Future Perspectives. Front. Genet. 2020, 11, 630186. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Zhang, W.J.; Bird, K.E.; McMillen, T.S.; LeBoeuf, R.C.; Hagen, T.M.; Frei, B. Dietary alpha-lipoic acid supplementation inhibits atherosclerotic lesion development in apolipoprotein E-deficient and apolipoprotein E/low-density lipoprotein receptor-deficient mice. Circulation 2008, 117, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, D.S.; Wei, F.F.; Cox, B.; Plusquin, M.; Thijs, L.; Winckelmans, E.; Zhang, Z.Y.; Nawrot, T.S.; Staessen, J.A. Retinal microcirculation and leukocyte telomere length in the general population. Sci. Rep. 2018, 8, 7095. [Google Scholar] [CrossRef]
- Blasiak, J.; Watala, C.; Tuuminen, R.; Kivinen, N.; Koskela, A.; Uusitalo-Jarvinen, H.; Tuulonen, A.; Winiarczyk, M.; Mackiewicz, J.; Zmorzynski, S.; et al. Expression of VEGFA-regulating miRNAs and mortality in wet AMD. J. Cell. Mol. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T. The age-related macular degeneration as a vascular disease/part of systemic vasculopathy: Contributions to its pathogenesis. Orv. Hetil. 2015, 156, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, M.R. Senescent retinal pigment epithelial cells are more sensitive to vascular endothelial growth factor: Implications for “wet” age-related macular degeneration. J. Ocul. Pharmacol. Ther. 2015, 31, 87–92. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasiak, J.; Szczepanska, J.; Fila, M.; Pawlowska, E.; Kaarniranta, K. Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1? Int. J. Mol. Sci. 2021, 22, 7194. https://doi.org/10.3390/ijms22137194
Blasiak J, Szczepanska J, Fila M, Pawlowska E, Kaarniranta K. Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1? International Journal of Molecular Sciences. 2021; 22(13):7194. https://doi.org/10.3390/ijms22137194
Chicago/Turabian StyleBlasiak, Janusz, Joanna Szczepanska, Michal Fila, Elzbieta Pawlowska, and Kai Kaarniranta. 2021. "Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1?" International Journal of Molecular Sciences 22, no. 13: 7194. https://doi.org/10.3390/ijms22137194
APA StyleBlasiak, J., Szczepanska, J., Fila, M., Pawlowska, E., & Kaarniranta, K. (2021). Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1? International Journal of Molecular Sciences, 22(13), 7194. https://doi.org/10.3390/ijms22137194