
Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. Results
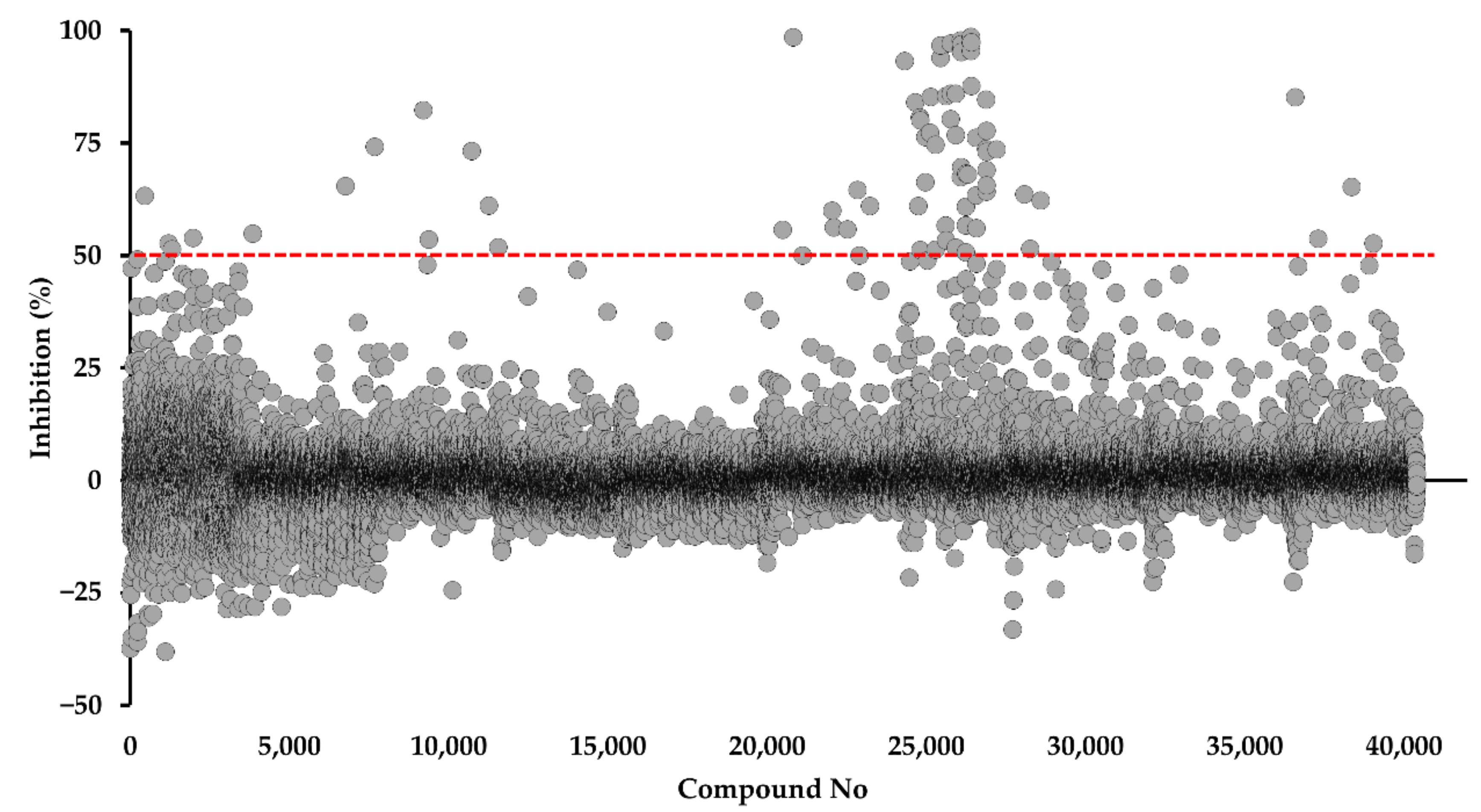
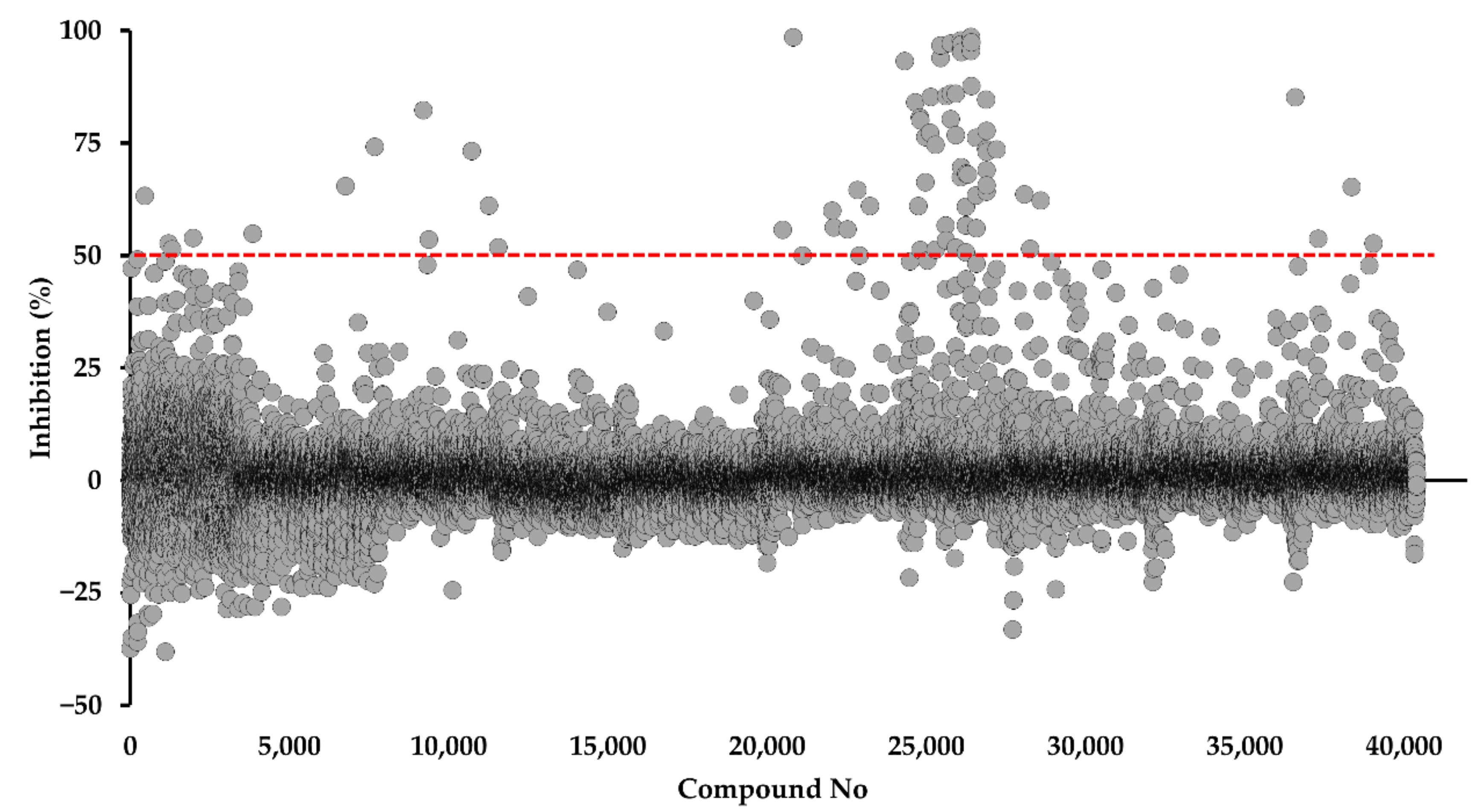
2.1. Identification of DPBI and DBIP Derivatives as PfDHODH Inhibitors
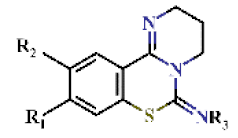
2.2. Structure–Activity Relationship of DPBI Derivatives
2.3. Structure–Activity Relationship of DBIP Derivatives
2.4. Counter-Assays Against HsDHODH and Mammalian Complex I, II, and III Activities
2.5. Antimalarial Activity of PfDHODH Inhibitors from the Kyoto University Chemical Library
2.6. Cytotoxicity Assay of Mammalian Cells
2.7. Confirmation Assay against the Transgenic Parasites
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Recombinant PfDHODH
4.2. Screening of the Kyoto University chemical Library
4.3. HsDHODH Assay
4.4. Mammalian Complex I, II, and III Activity Assays
4.5. In vitro Antimalarial Assay
4.6. Cytotoxicity Assays of Mammalian Cells
4.7. Generation of Transgenic Parasite
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNICEF. Malaria in Africa. Available online: https://data.unicef.org/topic/child-health/malaria/ (accessed on 13 November 2020).
- World Health Organization (WHO). World Malaria Report 2020; World Health Organization (WHO): Geneva, Switzerland, 2020. [Google Scholar]
- Maude, R.J.; Woodrow, C.J.; White, L.J. Artemisinin antimalarials: Preserving the “magic bullet”. Drug Dev. Res. 2010, 71, 12–19. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization (WHO): Geneva, Switzerland, 2015. [Google Scholar]
- Ashley, E.A.; Phyo, A.P. Drugs in development for malaria. Drugs 2018, 78, 861–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; van Huijsduijnen, R.H.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef] [Green Version]
- Wells, T.N.; van Huijsduijnen, R.H.; van Voorhis, W.C. Malaria medicines: A glass half full? Nat. Rev. Drug Discov. 2015, 14, 424–442. [Google Scholar] [CrossRef]
- Medicine for Malaria Venture. MMV Annual Report. 2019. Available online: https://www.mmv.org/newsroom/publications/mmv-annual-report-2019 (accessed on 25 May 2021).
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Belete, T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Devel. Ther. 2020, 14, 3875–3889. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Krungkrai, S.R.; Krungkrai, J. Insights into the pyrimidine biosynthetic pathway of human malaria parasite Plasmodium falciparum as chemotherapeutic target. Asian Pac. J. Trop. Med. 2016, 9, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.E. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets 2007, 8, 31–47. [Google Scholar] [CrossRef] [Green Version]
- Björnberg, O.; Jordan, D.B.; Palfey, B.A.; Jensen, K.F. Dihydrooxonate is a substrate of dihydroorotate dehydrogenase (DHOD) providing evidence for involvement of cysteine and serine residues in base catalysis. Arch. Biochem. Biophys. 2001, 391, 286–294. [Google Scholar] [CrossRef]
- Marcinkeviciene, J.; Tinney, L.M.; Wang, K.H.; Rogers, M.J.; Copeland, R.A. Dihydroorotate dehydrogenase B of Enterococcus faecalis. Characterization and insights into chemical mechanism. Biochemistry 1999, 38, 13129–13137. [Google Scholar] [CrossRef]
- Malmquist, N.A.; Gujjar, R.; Rathod, P.K.; Phillips, M.A. Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry 2008, 47, 2466–2475. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F.S.; Andersen, P.S.; Jensen, K.F. The B form of dihydroorotate dehydrogenase from Lactococcus lactis consists of two different subunits, encoded by the pyrDb and pyrK genes, and contains FMN, FAD, and [FeS] redox centers. J. Biol. Chem. 1996, 271, 29359–29365. [Google Scholar] [CrossRef] [Green Version]
- Rowland, P.; Nørager, S.; Jensen, K.F.; Larsen, S. Structure of dihydroorotate dehydrogenase B: Electron transfer between two flavin groups bridged by an iron-sulphur cluster. Structure 2000, 8, 1227–1238. [Google Scholar] [CrossRef]
- Sarewicz, M.; Osyczka, A. Electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signaling. Physiol. Rev. 2015, 95, 219–243. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.R.M.; Titus, B.R.; Kuenzi, M.R.; Rodriguez-Perez, F.; Brunsch, A.D.L.; Schroll, M.M.; Owen, M.C.; Cronk, J.D.; Anders, K.R.; Shepherd, J.N. Investigation of candidate genes involved in the rhodoquinone biosynthetic pathway in Rhodospirillum rubrum. PLoS ONE 2019, 14, e0217281. [Google Scholar] [CrossRef] [Green Version]
- Schattenkirchner, M. The use of leflunomide in the treatment of rheumatoid arthritis: An experimental and clinical review. Immunopharmacology 2000, 47, 291–298. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Use of Dihydroorotate dehydrogenase inhibitors for treatment of autoimmune diseases and cancer. ACS Med. Chem. Lett. 2020, 11, 2072–2074. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Li, S.; Yang, G.; Luo, Y.; Qi, T.; Liang, Y.; Yang, T.; Zhang, L.; Wang, R.; Zhu, L.; et al. Design, synthesis, molecular modeling, and biological evaluation of acrylamide derivatives as potent inhibitors of human dihydroorotate dehydrogenase for the treatment of rheumatoid arthritis. Acta Pharm. Sin. B 2021, 11, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Marcinkeviciene, J.; Haque, T.S.; Kopcho, L.M.; Jiang, W.; Wang, K.; Ecret, L.D.; Sizemore, C.; Amsler, K.A.; Foster, L.; et al. Helicobacter pylori-selective antibacterials based on inhibition of pyrimidine biosynthesis. J. Biol. Chem. 2000, 275, 33373–33378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohishi, T.; Inaoka, D.K.; Kita, K.; Kawada, M. Dihydroorotate dehydrogenase as a target for the development of novel Helicobacter pylori-specific antimicrobials. Chem. Pharm. Bull. 2018, 66, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, W.; Löffler, M. Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem. Pharmacol. 1998, 56, 1259–1264. [Google Scholar] [CrossRef]
- Chen, S.F.; Perrella, F.W.; Behrens, D.L.; Papp, L.M. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer Res. 1992, 52, 3521–3527. [Google Scholar] [PubMed]
- Sato, D.; Hartuti, E.D.; Inaoka, D.K.; Sakura, T.; Amalia, E.; Nagahama, M.; Yoshioka, Y.; Tsuji, N.; Nozaki, T.; Kita, K.; et al. Structural and biochemical features of Eimeria tenella dihydroorotate dehydrogenase, a potential drug target. Genes 2020, 11, 1468. [Google Scholar] [CrossRef]
- Phillips, M.A.; White, K.L.; Kokkonda, S.; Deng, X.; White, J.; El Mazouni, F.; Marsh, K.; Tomchick, D.R.; Manjalanagara, K.; Rudra, K.R.; et al. A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect. Dis. 2016, 2, 945–957. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.-C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel Plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Medicine for Malaria Venture. MMV-Supported Projects. Available online: https://www.mmv.org/research-development/mmv-supported-projects (accessed on 10 May 2021).
- Duffey, M.; Blasco, B.; Burrows, J.N.; Wells, T.N.C.; Fidock, D.A.; Leroy, D. Assessing risks of Plasmodium falciparum resistance to select next-generation antimalarials. Trends Parasitol. 2021, in press. [Google Scholar] [CrossRef]
- Pramisandi, A.; Dobashi, K.; Mori, M.; Nonaka, K.; Matsumoto, A.; Tokiwa, T.; Higo, M.; Kristiningrum; Amalia, E.; Nurkanto, A.; et al. Microbial inhibitors active against Plasmodium falciparum dihydroorotate dehydrogenase derived from an Indonesian soil fungus, Talaromyces pinophilus BioMCC-f.T.3979. J. Gen. Appl. Microbiol. 2020, 66, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Pharmacogenomics/Chemogenomics Drug Discovery Core Lab, Graduate School of Pharmaceutical Sciences, Kyoto University. Compound Library. Available online: https://www.pharm.kyoto-u.ac.jp/pgcg/library.html (accessed on 10 May 2021).
- Favuzza, P.; de Lera Ruiz, M.; Thompson, J.K.; Triglia, T.; Ngo, A.; Steel, R.W.J.; Vavrek, M.; Christensen, J.; Healer, J.; Boyce, C.; et al. Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 2020, 27, 642–658.e612. [Google Scholar] [CrossRef]
- Leshabane, M.; Dziwornu, G.A.; Coertzen, D.; Reader, J.; Moyo, P.; van der Watt, M.; Chisanga, K.; Nsanzubuhoro, C.; Ferger, R.; Erlank, E.; et al. Benzimidazole derivatives are potent against multiple life cycle stages of Plasmodium falciparum malaria parasites. ACS Infect. Dis. 2021, in press. [Google Scholar] [CrossRef]
- Mizuhara, T.; Oishi, S.; Ohno, H.; Shimura, K.; Matsuoka, M.; Fujii, N. Structure-activity relationship study of pyrimido[1,2-c][1,3]benzothiazin-6-imine derivatives for potent anti-HIV agents. Bioorg. Med. Chem. 2012, 20, 6434–6441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, S.; Oishi, S.; Mizuhara, T.; Shimura, K.; Murayama, H.; Ohno, H.; Matsuoka, M.; Fujii, N. Investigations of possible prodrug structures for 2-(2-mercaptophenyl)tetrahydropyrimidines: Reductive conversion from anti-HIV agents with pyrimidobenzothiazine and isothiazolopyrimidine scaffolds. Org. Biomol. Chem. 2015, 13, 4706–4713. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, S.; Mizuhara, T.; Shimura, K.; Murayama, H.; Ohno, H.; Oishi, S.; Matsuoka, M.; Fujii, N. Identification of anti-HIV agents with a novel benzo[4,5]isothiazolo[2,3-a]pyrimidine scaffold. Bioorg. Med. Chem. 2015, 23, 1447–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuhara, T.; Oishi, S.; Ohno, H.; Shimura, K.; Matsuoka, M.; Fujii, N. Design and synthesis of biotin- or alkyne-conjugated photoaffinity probes for studying the target molecules of PD 404182. Bioorg. Med. Chem. 2013, 21, 2079–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamoun-Emanuelli, A.M.; Bobardt, M.; Moncla, B.; Mankowski, M.K.; Ptak, R.G.; Gallay, P.; Chen, Z. Evaluation of PD 404,182 as an anti-HIV and anti-herpes simplex virus microbicide. Antimicrob. Agents Chemother. 2014, 58, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamoun, A.M.; Chockalingam, K.; Bobardt, M.; Simeon, R.; Chang, J.; Gallay, P.; Chen, Z. PD 404,182 is a virocidal small molecule that disrupts hepatitis C virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 2012, 56, 672–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chockalingam, K.; Simeon, R.L.; Rice, C.M.; Chen, Z. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc. Natl. Acad. Sci. USA 2010, 107, 3764–3769. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S.M.; Morrisey, J.M.; Ke, H.; Painter, H.J.; Laroiya, K.; Phillips, M.A.; Rathod, P.K.; Mather, M.W.; Vaidya, A.B. Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol. Biochem. Parasitol. 2011, 177, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef]
- Baldwin, J.; Michnoff, C.H.; Malmquist, N.A.; White, J.; Roth, M.G.; Rathod, P.K.; Phillips, M.A. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2005, 280, 21847–21853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, Y.; Wu, Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol. Screen. 2007, 12, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Shun, T.Y.; Lazo, J.S.; Sharlow, E.R.; Johnston, P.A. Identifying actives from HTS data sets: Practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J. Biomol. Screen. 2011, 16, 1–14. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Inaoka, D.K.; Shiba, T.; Saimoto, H.; Sakura, T.; Amalia, E.; Kido, Y.; Sakai, C.; Nakamura, M.; Moore, A.L.; et al. Selective cytotoxicity of dihydroorotate dehydrogenase inhibitors to human cancer cells under hypoxia and nutrient-deprived conditions. Front. Pharmacol. 2018, 9, 997. [Google Scholar] [CrossRef]
- Kita, K.; Takamiya, S.; Furushima, R.; Ma, Y.-C.; Suzuki, H.; Ozawa, T.; Oya, H. Electron-transfer complexes of Ascaris suum muscle mitochondria. III. Composition and fumarate reductase activity of complex II. Biochim. Biophy. Acta Bioenerg. 1988, 935, 130–140. [Google Scholar] [CrossRef]
- Takamiya, S.; Furushima, R.; Oya, H. Electron-transfer complexes of Ascaris suum muscle mitochondria. II. Succinate-coenzyme Q reductase (complex II) associated with substrate-reducible cytochrome b-558. Biochim. Biophys. Acta 1986, 848, 99–107. [Google Scholar] [CrossRef]
- Matsubayashi, M.; Inaoka, D.K.; Komatsuya, K.; Hatta, T.; Kawahara, F.; Sakamoto, K.; Hikosaka, K.; Yamagishi, J.; Sasai, K.; Shiba, T.; et al. Novel characteristics of mitochondrial electron transport chain from Eimeria tenella. Genes 2019, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamiya, S.; Furushima, R.; Oya, H. Electron transfer complexes of Ascaris suum muscle mitochondria: I. Characterization of NADH-cytochrome c reductase (complex I-III), with special reference to cytochrome localization. Mol. Biochem. Parasitol. 1984, 13, 121–134. [Google Scholar] [CrossRef]
- Miyadera, H.; Amino, H.; Hiraishi, A.; Taka, H.; Murayama, K.; Miyoshi, H.; Sakamoto, K.; Ishii, N.; Hekimi, S.; Kita, K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J. Biol. Chem. 2001, 276, 7713–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartuti, E.D.; Inaoka, D.K.; Komatsuya, K.; Miyazaki, Y.; Miller, R.J.; Xinying, W.; Sadikin, M.; Prabandari, E.E.; Waluyo, D.; Kuroda, M.; et al. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Miyazaki, Y.; Inaoka, D.K.; Hartuti, E.D.; Watanabe, Y.I.; Shiba, T.; Harada, S.; Saimoto, H.; Burrows, J.N.; Benito, F.J.G.; et al. Identification of Plasmodium falciparum mitochondrial malate: Quinone oxidoreductase inhibitors from the Pathogen Box. Genes 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitsch, K.; Driskill, C.; Wellems, T. Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res. 2001, 29, 850–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Substituents | IC50 (µM) | EC50 (µM) | Growth at 10 µM (%) | |||||
| R1 | R2 | R3 | DHODH | Mammalian Mitochondrial | Pf3D7 | Pf3D7-yDHODH | |||
| Pf | Hs | CI–III | CII–III | ||||||
| 1 |  | H | H | 0.65 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.43 ± 0.18 | −0.62 ± 0.07 |
| 2 | 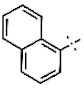 | H | H | 0.85 ± 0.05 | >4.5 | 13.9 ± 2.14 | >22.7 | >10 | nd |
| 3 | 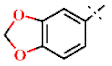 | H | H | 1.18 ± 0.04 | >4.5 | >22.7 | >22.7 | 1.16 ± 0.02 | −0.23 ± 0.01 |
| 4 |  | H | H | 0.97 ± 0.08 | >4.5 | >22.7 | >22.7 | 1.07 ± 0.05 | 24.0 ± 5.76 |
| 5 | 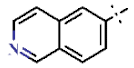 | H | H | 0.96 ± 0.09 | >4.5 | >22.7 | >22.7 | 1.13 ± 0.05 | 26.0 ± 8.27 |
| 6 | 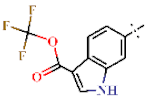 | H | H | 0.70 ± 0.04 | >4.5 | >22.7 | >22.7 | 0.37 ± 0.05 | 104.4 ± 0.11 |
| 7 | 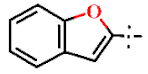 | H | H | 1.30 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.34 ± 0.01 | 36.2 ± 1.01 |
| 8 | 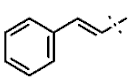 | H | H | 1.07 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.63 ± 0.25 | 0.12 ± 0.21 |
| 9 | H | 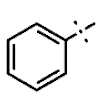 | H | 0.76 ± 0.08 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 10 |  | H | (CH3)3 | 3.84 ± 0.09 | >4.5 | >22.7 | >22.7 | 2.77 ± 0.28 | −0.48 ± 0.07 |
| 11 |  | H | 6.42 ± 0.09 | >4.5 | >22.7 | >22.7 | >10 | nd | |
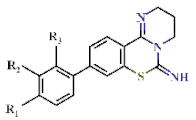 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Substituents | IC50 (µM) | EC50 (µM) | Growth at 10 µM (%) | |||||
| R1 | R2 | R3 | DHODH | Mammalian Mitochondrial | Pf3D7 | Pf3D7- yDHODH | |||
| Pf | Hs | CI-III | CII-III | ||||||
| 12 | H | H | H | 0.68 ± 0.05 | >4.5 | >22.7 | >22.7 | 1.15 ± 0.07 | −0.22 ± 0.12 |
| 13 | 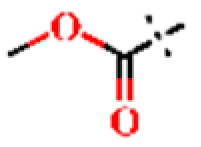 | H | H | 1.22 ± 0.05 | >4.5 | >22.7 | >22.7 | 1.06 ± 0.01 | 70.5 ± 0.73 |
| 14 | 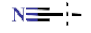 | H | H | 3.89 ± 0.23 | >4.5 | >22.7 | >22.7 | 1.21 ± 0.04 | 105 ± 0.47 |
| 15 |  | H | H | 2.31 ± 0.11 | >4.5 | >22.7 | >22.7 | 1.00 ± 0.05 | −0.38 ± 0.17 |
| 16 | 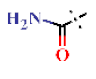 | H | H | 1.82 ± 0.08 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 17 | 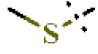 | H | H | 0.96 ± 0.07 | >4.5 | >22.7 | >22.7 | 1.62 ± 0.56 | −0.50 ± 0.01 |
| 18 |  | H | H | 1.43 ± 0.06 | >4.5 | >22.7 | >22.7 | 0.37 ± 0.13 | 2.54 ± 0.42 |
| 19 | 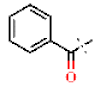 | H | H | 1.07 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.51 ± 0.14 | 68.6 ± 0.43 |
| 20 |  | H | H | 1.21 ± 0.06 | >4.5 | >22.7 | >22.7 | 0.38 ± 0.08 | 0.45 ± 0.74 |
| 21 | H |  | H | 3.68 ± 0.31 | >4.5 | >22.7 | >22.7 | 0.85 ± 0.12 | 103 ± 0.67 |
| 22 | H |  | H | 1.56 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.90 ± 0.44 | −0.06 ± 0.24 |
| 23 | H | 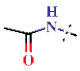 | H | 1.85 ± 0.19 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 24 | H |  | H | 1.94 ± 0.07 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 25 | H | C2H5 | H | 1.52 ± 0.08 | >4.5 | >22.7 | >22.7 | 1.77 ± 0.83 | 0.01 ± 0.15 |
| 26 | H | 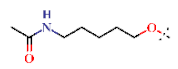 | H | 3.49 ± 0.21 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 27 | H | H |  | 3.30 ± 0.10 | >4.5 | >22.7 | >22.7 | 0.39 ± 0.05 | 0.25 ± 0.04 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | Substituents | IC50 (µM) | EC50 (µM) | Growth at 10 µM (%) | ||||
| R1 | R2 | DHODH | Mammalian Mitochondrial | Pf3D7 | Pf3D7- yDHODH | |||
| Pf | Hs | CI–III | CII–III | |||||
| 28 | 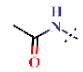 | H | 18.5 ± 0.40 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 29 | 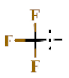 | H | 9.71 ± 0.29 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 30 | 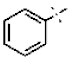 | H | 4.26 ± 0.12 | >4.5 | >22.7 | >22.7 | 9.67 ± 5.09 | 63.6 ± 5.87 |
| 31 |  | H | 1.10 ± 0.08 | >4.5 | >22.7 | >22.7 | 0.33 ± 0.01 | 0 ± 0.07 |
| 32 | 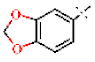 | H | 1.29 ± 0.09 | >4.5 | >22.7 | >22.7 | 1.85 ± 0.53 | 0.11 ± 0.03 |
| 33 | 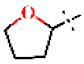 | H | 1.53 ± 0.04 | >4.5 | >22.7 | >22.7 | 3.73 ± 0.88 | 44.1 ± 26.2 |
| 34 |  | H | 0.94 ± 0.05 | >4.5 | >22.7 | >22.7 | 1.64 ± 0.35 | −0.1 ± 0.28 |
| 35 | 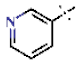 | H | 1.48 ± 0.07 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 36 | H |  | 1.51 ± 0.04 | >4.5 | >22.7 | >22.7 | 4.16 ± 0.09 | 32.5 ± 10.1 |
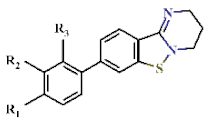 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Substituents | IC50 (µM) | EC50 (µM) | Growth at 10 µM (%) | |||||
| R1 | R2 | R3 | DHODH | Mammalian Mitochondrial | Pf3D7 | Pf3D7- yDHODH | |||
| Pf | Hs | CI–III | CII–III | ||||||
| 37 |  | H | H | 1.15 ± 0.07 | >4.5 | >22.7 | >22.7 | 2.78 ± 0.27 | −0.18 ± 0.05 |
| 38 | 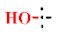 | H | H | 1.81 ± 0.01 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 39 |  | H | H | 1.30 ± 0.04 | >4.5 | >22.7 | >22.7 | 1.14 ± 0.06 | 1.48 ± 0.14 |
| 40 | H | 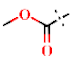 | H | 1.43 ± 0.02 | >4.5 | >22.7 | >22.7 | 1.72 ± 0.48 | −0.09 ± 0.08 |
| 41 | H |  | H | 1.44 ± 0.05 | >4.5 | >22.7 | >22.7 | >10 | nd |
| 42 | H | H |  | 1.18 ± 0.05 | >4.5 | >22.7 | >22.7 | 0.54 ± 0.22 | 0.04 ± 0.15 |
| 43 | H | H |  | 1.15 ± 0.06 | >4.5 | >22.7 | >22.7 | 1.11 ± 0.08 | 31.1 ± 2.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartuti, E.D.; Sakura, T.; Tagod, M.S.O.; Yoshida, E.; Wang, X.; Mochizuki, K.; Acharjee, R.; Matsuo, Y.; Tokumasu, F.; Mori, M.; et al. Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Int. J. Mol. Sci. 2021, 22, 7236. https://doi.org/10.3390/ijms22137236
Hartuti ED, Sakura T, Tagod MSO, Yoshida E, Wang X, Mochizuki K, Acharjee R, Matsuo Y, Tokumasu F, Mori M, et al. Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. International Journal of Molecular Sciences. 2021; 22(13):7236. https://doi.org/10.3390/ijms22137236
Chicago/Turabian StyleHartuti, Endah Dwi, Takaya Sakura, Mohammed S. O. Tagod, Eri Yoshida, Xinying Wang, Kota Mochizuki, Rajib Acharjee, Yuichi Matsuo, Fuyuki Tokumasu, Mihoko Mori, and et al. 2021. "Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase" International Journal of Molecular Sciences 22, no. 13: 7236. https://doi.org/10.3390/ijms22137236
APA StyleHartuti, E. D., Sakura, T., Tagod, M. S. O., Yoshida, E., Wang, X., Mochizuki, K., Acharjee, R., Matsuo, Y., Tokumasu, F., Mori, M., Waluyo, D., Shiomi, K., Nozaki, T., Hamano, S., Shiba, T., Kita, K., & Inaoka, D. K. (2021). Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. International Journal of Molecular Sciences, 22(13), 7236. https://doi.org/10.3390/ijms22137236