
Biochemical Studies of Mitochondrial Malate: Quinone Oxidoreductase from Toxoplasma gondii

 , ,
, ,  , , , , ,
, , , , ,  ,
,
Abstract
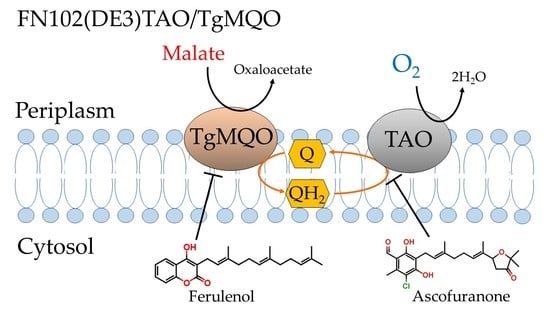
:
1. Introduction
2. Results
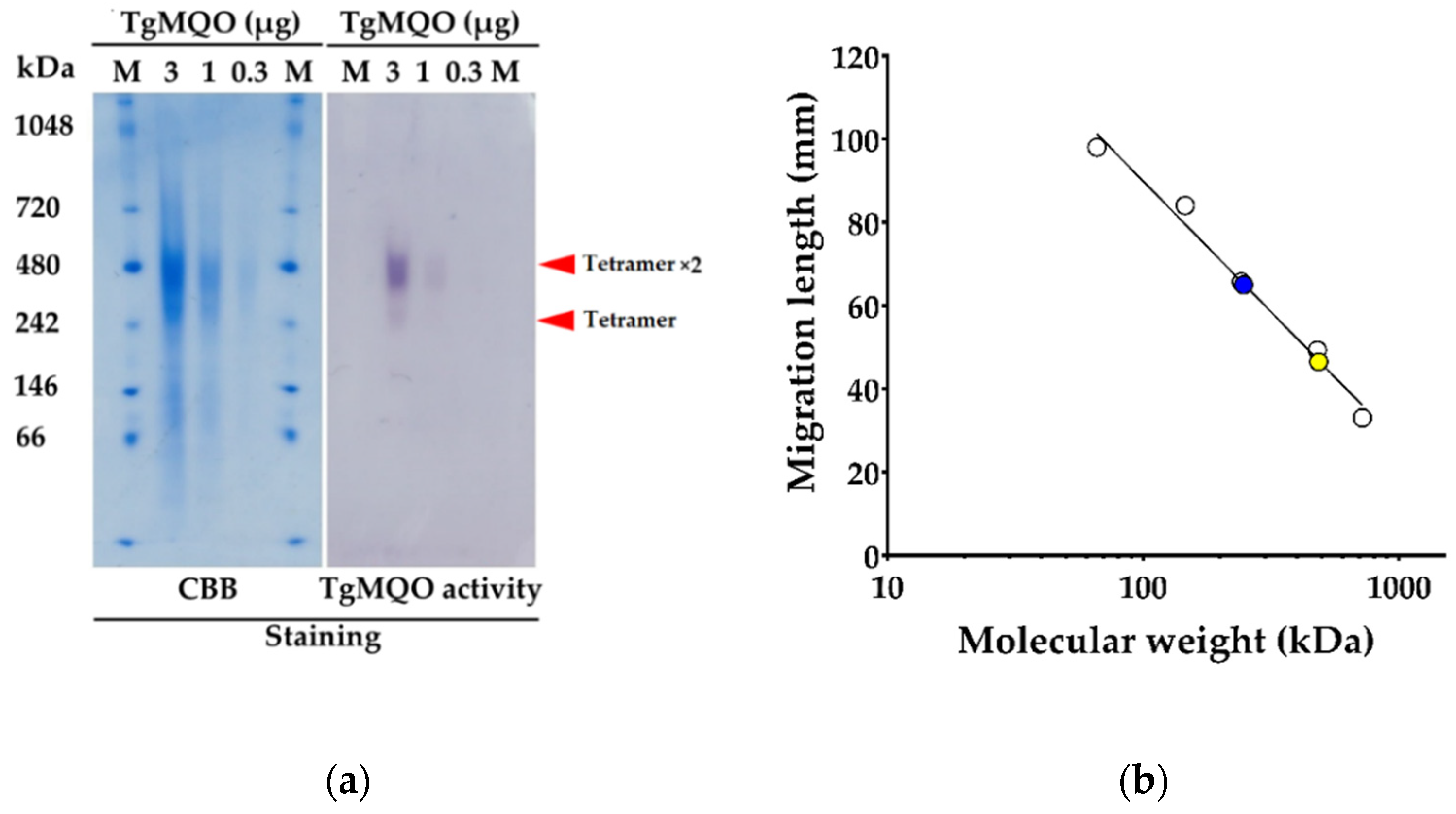
2.1. Functional Expression and Purification of Recombinant TgMQO
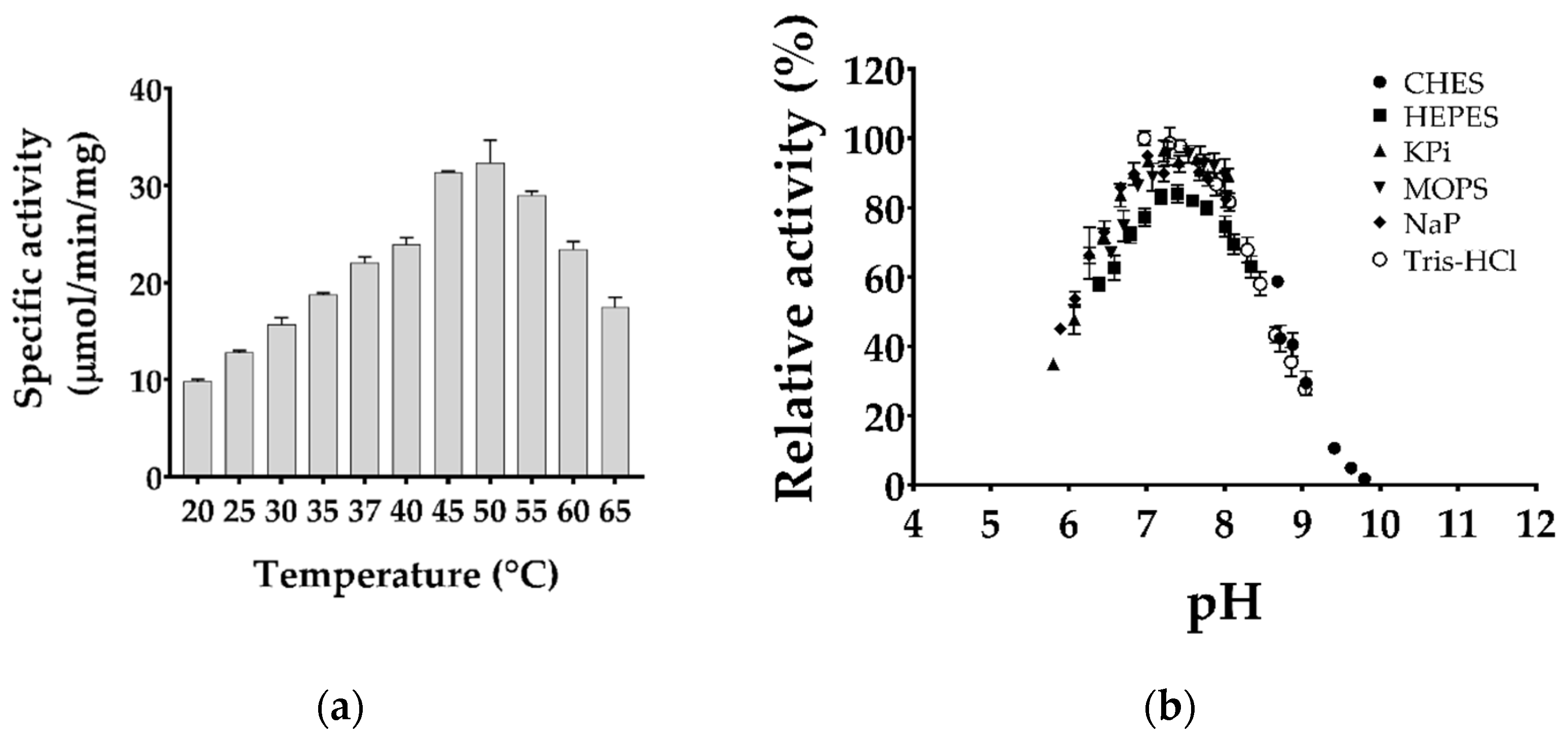
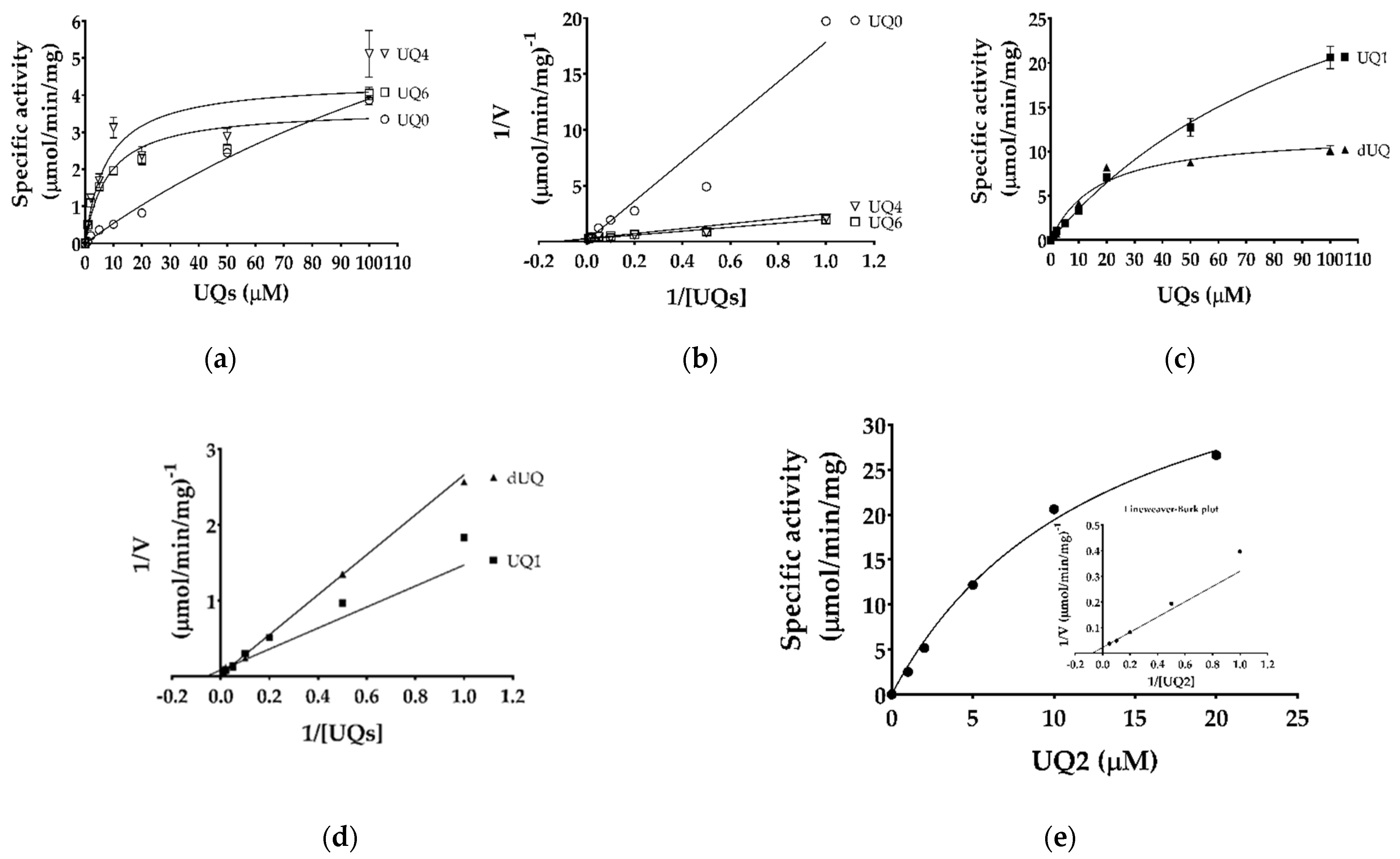
2.2. Biochemical Characterization of Purified TgMQO
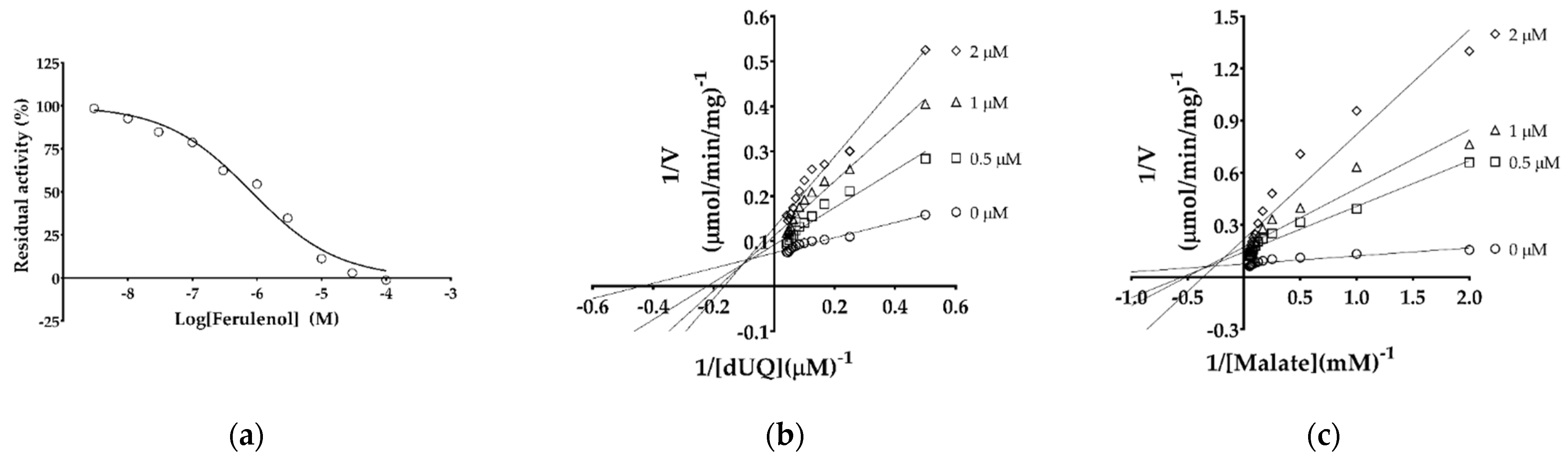
2.3. IC50 and Inhibition Mechanism of Ferulenol with TgMQO
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Reagents for Recombinant TgMQO Expression
4.2. Overexpression of Active TgMQO in the Bacterial Membrane
4.3. Purification of TgMQO from the Membrane Fraction
4.4. Protein Quantification and Electrophoresis
4.5. Optimization of TgMQO Assay Conditions
4.6. Determination of Enzyme Kinetic Parameters and Reaction Mechanism
4.7. TgMQO Inhibition with Ferulenol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demar, M.; Ajzenberg, D.; Maubon, D.; Djossou, F.; Panchoe, D.; Punwasi, W.; Valery, N.; Peneau, C.; Daigre, J.-L.; Aznar, C.; et al. Fatal outbreak of human toxoplasmosis along the Maroni River: Epidemiological, clinical, and parasitological aspects. Clin. Infect. Dis. 2007, 45, e88–e95. [Google Scholar] [CrossRef]
- Dubey, J.P. Toxoplasmosis of Animals and Humans, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 1–336. [Google Scholar]
- Galal, L.; Schares, G.; Stragier, C.; Vignoles, P.; Brouat, C.; Cuny, T.; Dubois, C.; Rohart, T.; Glodas, C.; Dardé, M.L.; et al. Diversity of Toxoplasma gondii strains shaped by commensal communities of small mammals. Int. J. Parasitol. 2019, 49, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Hejlícek, K.; Literák, I.; Nezval, J. Toxoplasmosis in wild mammals from the Czech Republic. J. Wildl. Dis. 1997, 33, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Flegr, J.; Prandota, J.; Sovičková, M.; Israili, Z.H. Toxoplasmosis—A Global Threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 2014, 9, e90203. [Google Scholar] [CrossRef]
- Dubremetz, J.F.; Lebrun, M. Virulence factors of Toxoplasma gondii. Microbes Infect. 2012, 14, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Blume, M.; Seeber, F. Metabolic interactions between Toxoplasma gondii and its host. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef]
- Soête, M.; Camus, D.; Dubremetz, J.F. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp. Parasitol. 1994, 78, 361–370. [Google Scholar] [CrossRef]
- Weilhammer, D.R.; Iavarone, A.T.; Villegas, E.N.; Brooks, G.A.; Sinai, A.P.; Sha, W.C. Host metabolism regulates growth and differentiation of Toxoplasma gondii. Int. J. Parasitol. 2012, 42, 947–959. [Google Scholar] [CrossRef]
- Tomavo, S.; Boothroyd, J.C. Interconnection between organellar functions, development and drug resistance in the protozoan parasite. Toxoplasma gondii. Int. J. Parasitol. 1995, 25, 1293–1299. [Google Scholar] [CrossRef]
- Boothroyd, J.C.; Grigg, M.E. Population biology of Toxoplasma gondii and its relevance to human infection: Do different strains cause different disease? Curr. Opin. Microbiol. 2002, 5, 438–442. [Google Scholar] [CrossRef]
- Ajzenberg, D.; Yera, H.; Marty, P.; Paris, L.; Dalle, F.; Menotti, J.; Aubert, D.; Franck, J.; Bessières, M.H.; Quinio, D.; et al. Genotype of 88 Toxoplasma gondii isolates associated with toxoplasmosis in immunocompromised patients and correlation with clinical findings. J. Infect. Dis. 2009, 199, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Dardé, M.L.; Villena, I.; Pinon, J.M.; Beguinot, I. Severe toxoplasmosis caused by a Toxoplasma gondii strain with a new isoenzyme type acquired in French Guyana. J. Clin. Microbiol. 1998, 36, 324. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.A.; Amouei, A.; Sharif, M.; Sarvi, S.; Galal, L.; Javidnia, J.; Pagheh, A.S.; Gholami, S.; Mizani, A.; Daryani, A. Human toxoplasmosis: A systematic review for genetic diversity of Toxoplasma gondii in clinical samples. Epidemiol. Infect. 2018, 147, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Kruszon-Moran, D.; Wilson, M.; McQuillan, G.; Navin, T.; McAuley, J.B. Toxoplasma gondii Infection in the United States: Seroprevalence and Risk Factors. Am. J. Epidemiol. 2001, 154, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.D.; Christian, D.A.; Hunter, C.A. Immune response and immunopathology during toxoplasmosis. Semin. Immunopathol. 2012, 34, 793–813. [Google Scholar] [CrossRef]
- Ferguson, D.J.P.; Bowker, C.; Jeffery, K.J.M.; Chamberlain, P.; Squier, W. Congenital toxoplasmosis: Continued parasite proliferation in the fetal brain despite maternal immunological control in other tissues. Clin. Infect. Dis. 2013, 56, 204–208. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Konstantinovic, N.; Guegan, H.; Stäjner, T.; Belaz, S.; Robert-Gangneux, F. Treatment of toxoplasmosis: Current options and future perspectives. Food Waterborne Parasitol. 2019, 15, e00036. [Google Scholar] [CrossRef]
- Sinai, A.P.; Watts, E.A.; Dhara, A.; Murphy, R.D.; Gentry, M.S.; Patwardhan, A. Reexamining chronic Toxoplasma gondii infection: Surprising activity for a “dormant” parasite. Curr. Clin. Microbiol. Rep. 2016, 3, 175–185. [Google Scholar] [CrossRef]
- Buxton, D.; Innes, E.A. A commercial vaccine for ovine toxoplasmosis. Parasitology 1995, 110, S11–S16. [Google Scholar] [CrossRef]
- Lyons, R.E.; McLeod, R.; Roberts, C.W. Toxoplasma gondii tachyzoite-bradyzoite interconversion. Trends Parasitol. 2002, 18, 198–201. [Google Scholar] [CrossRef]
- Hartuti, E.D.; Inaoka, D.K.; Komatsuya, K.; Miyazaki, Y.; Miller, R.J.; Xinying, W.; Sadikin, M.; Prabandari, E.E.; Waluyo, D.; Kuroda, M.; et al. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 191–200. [Google Scholar] [CrossRef]
- MacRae, J.I.; Sheiner, L.; Nahid, A.; Tonkin, C.; Striepen, B.; McConville, M.J. Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 2012, 12, 682–692. [Google Scholar] [CrossRef]
- Polonais, V.; Soldati-Favre, D. Versatility in the acquisition of energy and carbon sources by the Apicomplexa. Biol. Cell 2010, 102, 435–445. [Google Scholar] [CrossRef]
- Shukla, A.; Olszewski, K.L.; Llinás, M.; Rommereim, L.M.; Fox, B.A.; Bzik, D.J.; Xia, D.; Wastling, J.; Beiting, D.; Roos, D.S.; et al. Glycolysis is important for optimal asexual growth and formation of mature tissue cysts by Toxoplasma gondii. Int. J. Parasitol. 2018, 48, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Fleige, T.; Pfaff, N.; Gross, U.; Bohne, W. Localisation of gluconeogenesis and tricarboxylic acid (TCA)-cycle enzymes and first functional analysis of the TCA cycle in Toxoplasma gondii. Int. J. Parasitol. 2008, 38, 1121–1132. [Google Scholar] [CrossRef]
- Hikosaka, K.; Komatsuya, K.; Suzuki, S.; Kita, K. Mitochondria of malaria parasites as a drug target. In An Overview of Tropical Diseases; IntechOpen: London, UK, 2015. [Google Scholar]
- Kawahara, K.; Mogi, T.; Tanaka, T.Q.; Hata, M.; Miyoshi, H.; Kita, K. Mitochondrial dehydrogenases in the aerobic respiratory chain of the rodent malaria parasite Plasmodium yoelii yoelii. J. Biochem. 2009, 145, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Mather, M.W.; Henry, K.W.; Vaidya, A.B. Mitochondrial drug targets in apicomplexan parasites. Curr. Drug. Targets 2007, 8, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Mi-Ichi, F.; Miyadera, H.; Kobayashi, T.; Takamiya, S.; Waki, S.; Iwata, S.; Shibata, S.; Kita, K. Parasite mitochondria as a target of chemotherapy: Inhibitory effect of licochalcone A on the Plasmodium falciparum respiratory chain. Ann. N. Y. Acad. Sci. 2005, 1056, 46–54. [Google Scholar] [CrossRef]
- Fueyo González, F.J.; Ebiloma, G.U.; Izquierdo García, C.; Bruggeman, V.; Sánchez Villamañán, J.M.; Donachie, A.; Balogun, E.O.; Inaoka, D.K.; Shiba, T.; Harada, S.; et al. Conjugates of 2,4-dihydroxybenzoate and salicylhydroxamate and lipocations display potent antiparasite effects by efficiently targeting the Trypanosoma brucei and Trypanosoma congolense mitochondrion. J. Med. Chem. 2017, 60, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Kido, Y.; Sakamoto, K.; Inaoka, D.K.; Tsuge, C.; Tatsumi, R.; Takahashi, G.; Balogun, E.O.; Nara, T.; Aoki, T.; et al. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 201218386. [Google Scholar] [CrossRef]
- Yamasaki, S.; Shoji, M.; Kayanuma, M.; Sladek, V.; Inaoka, D.K.; Matsuo, Y.; Shiba, T.; Young, L.; Moore, A.L.; Kita, K.; et al. Weak O2 binding and strong H2O2 binding at the non-heme diiron center of trypanosome alternative oxidase. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148356. [Google Scholar] [CrossRef]
- Young, L.; Rosell-Hidalgo, A.; Inaoka, D.K.; Xu, F.; Albury, M.; May, B.; Kita, K.; Moore, A.L. Kinetic and structural characterisation of the ubiquinol-binding site and oxygen reduction by the trypanosomal alternative oxidase. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148247. [Google Scholar] [CrossRef]
- Seidi, A.; Muellner-Wong, L.S.; Rajendran, E.; Tjhin, E.T.; Dagley, L.F.; Aw, V.Y.T.; Faou, P.; Webb, A.I.; Tonkin, C.J.; van Dooren, G.G. Elucidating the mitochondrial proteome of Toxoplasma gondii reveals the presence of a divergent cytochrome c oxidase. eLife 2018, 7, e38131. [Google Scholar] [CrossRef]
- Hikosaka, K.; Kita, K.; Tanabe, K. Diversity of mitochondrial genome structure in the phylum Apicomplexa. Mol. Biochem. Parasitol. 2013, 188, 26–33. [Google Scholar] [CrossRef]
- Doggett, J.S.; Nilsen, A.; Forquer, I.; Wegmann, K.W.; Jones-Brando, L.; Yolken, R.H.; Bordón, C.; Charman, S.A.; Katneni, K.; Schultz, T.; et al. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc. Natl. Acad. Sci. USA 2012, 109, 15936–15941. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Gujjar, R.; Malmquist, N.A.; White, J.; El Mazouni, F.; Baldwin, J.; Rathod, P.K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Seeber, F.; Limenitakis, J.; Soldati-Favre, D. Apicomplexan mitochondrial metabolism: A story of gains, losses and retentions. Trends Parasitol. 2008, 24, 468–478. [Google Scholar] [CrossRef]
- Lin, S.S.; Gross, U.; Bohne, W. Two internal type II NADH dehydrogenases of Toxoplasma gondii are both required for optimal tachyzoite growth. Mol. Microbiol. 2011, 82, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Denton, H.; Roberts, C.W.; Alexander, J.; Thong, K.W.; Coombs, G.H. Enzymes of energy metabolism in the bradyzoites and tachyzoites of Toxoplasma gondii. FEMS Microbiol. Lett. 1996, 137, 103–108. [Google Scholar] [CrossRef]
- Jacot, D.; Waller, R.F.; Soldati-Favre, D.; MacPherson, D.A.; MacRae, J.I. Apicomplexan energy metabolism: Carbon source promiscuity and the quiescence hyperbole. Trends Parasitol. 2016, 32, 56–70. [Google Scholar] [CrossRef]
- Lin, S.S.; Gross, U.; Bohne, W. Type II NADH dehydrogenase inhibitor 1-hydroxy-2-dodecyl-4(1H)quinolone leads to collapse of mitochondrial inner-membrane potential and ATP depletion in Toxoplasma gondii. Eukaryot. Cell 2009, 8, 877–887. [Google Scholar] [CrossRef]
- Bisanz, C.; Bastien, O.; Grando, D.; Jouhet, J.; Maréchal, E.; Cesbron-Delauw, M.F. Toxoplasma gondii acyl-lipid metabolism: De novo synthesis from apicoplast-generated fatty acids versus scavenging of host cell precursors. Biochem. J. 2006, 394 Pt 1, 197–205. [Google Scholar] [CrossRef]
- Shen, W.; Wei, Y.; Dauk, M.; Tan, Y.; Taylor, D.C.; Selvaraj, G.; Zou, J. Involvement of a glycerol-3-phosphate dehydrogenase in modulating the NADH/NAD+ ratio provides evidence of a mitochondrial glycerol-3-phosphate shuttle in Arabidopsis. Plant Cell 2006, 18, 422. [Google Scholar] [CrossRef]
- Hortua Triana, M.A.; Cajiao Herrera, D.; Zimmermann, B.H.; Fox, B.A.; Bzik, D.J. Pyrimidine pathway-dependent and -independent functions of the Toxoplasma gondii mitochondrial dihydroorotate dehydrogenase. Infect. Immun. 2016, 84, 2974–2981. [Google Scholar] [CrossRef] [PubMed]
- Alday, P.H.; Doggett, J.S. Drugs in development for toxoplasmosis: Advances, challenges, and current status. Drug Des. Devel. Ther. 2017, 11, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Rajendran, E.; Zwahlen, S.M.; Faou, P.; van Dooren, G.G. Divergent features of the coenzyme Q:cytochrome c oxidoreductase complex in Toxoplasma gondii parasites. PLoS Pathog. 2021, 17, e1009211. [Google Scholar] [CrossRef] [PubMed]
- Iltzsch, M.H. Pyrimidine salvage pathways in Toxoplasma gondii. J. Eukaryot. Microbiol. 1993, 40, 24–28. [Google Scholar] [CrossRef]
- Fox, B.A.; Bzik, D.J. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature 2002, 415, 926–929. [Google Scholar] [CrossRef]
- Kather, B.; Stingl, K.; van der Rest, M.E.; Altendorf, K.; Molenaar, D. Another unusual type of citric acid cycle enzyme in Helicobacter pylori: The malate:quinone oxidoreductase. J. Bacteriol. 2000, 182, 3204–3209. [Google Scholar] [CrossRef]
- Bulusu, V.; Jayaraman, V.; Balaram, H. Metabolic fate of fumarate, a side product of the purine salvage pathway in the intraerythrocytic stages of Plasmodium falciparum. J. Biol. Chem. 2011, 286, 9236–9245. [Google Scholar] [CrossRef]
- Ke, H.; Lewis, I.A.; Morrisey, J.M.; McLean, K.J.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Jacobs-Lorena, M.; Llinas, M.; Vaidya, A.B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015, 11, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Morrisey, J.M.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Vaidya, A.B. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot. Cell 2011, 10, 1053–1061. [Google Scholar] [CrossRef]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Mogi, T.; Murase, Y.; Mori, M.; Shiomi, K.; Omura, S.; Paranagama, M.P.; Kita, K. Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: Quinone oxidoreductase from the Gram-positive bacterium Mycobacterium smegmatis. J. Biochem. 2009, 146, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, D.; van der Rest, M.E.; Petrović, S. Biochemical and genetic characterization of the membrane-associated malate dehydrogenase (acceptor) from Corynebacterium glutamicum. Eur. J. Biochem. 1998, 254, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, D.; van der Rest, M.E.; Drysch, A.; Yücel, R. Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Corynebacterium glutamicum. J. Bacteriol. 2000, 182, 6884. [Google Scholar] [CrossRef]
- van der Rest, M.E.; Frank, C.; Molenaar, D. Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Escherichia coli. J. Bacteriol. 2000, 182, 6892–6899. [Google Scholar] [CrossRef]
- Niikura, M.; Komatsuya, K.; Inoue, S.-I.; Matsuda, R.; Asahi, H.; Inaoka, D.K.; Kita, K.; Kobayashi, F. Suppression of experimental cerebral malaria by disruption of malate:quinone oxidoreductase. Malar. J. 2017, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Miyazaki, Y.; Inaoka, D.K.; Hartuti, E.D.; Watanabe, Y.-I.; Shiba, T.; Harada, S.; Saimoto, H.; Burrows, J.N.; Benito, F.J.G.; et al. Identification of Plasmodium falciparum mitochondrial malate: Quinone oxidoreductase inhibitors from the pathogen Box. Genes 2019, 10, 471. [Google Scholar] [CrossRef]
- Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D.K.; et al. Complete biosynthetic pathways of ascofuranone and ascochlorin in Acremonium egyptiacum. Proc Natl. Acad. Sci. USA 2019, 116, 8269. [Google Scholar] [CrossRef]
- Hijikawa, Y.; Matsuzaki, M.; Suzuki, S.; Inaoka, D.K.; Tatsumi, R.; Kido, Y.; Kita, K. Re-identification of the ascofuranone-producing fungus Ascochyta viciae as Acremonium sclerotigenum. J. Antibiot. 2017, 70, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1398–1413. [Google Scholar] [CrossRef]
- Forte, E.; Borisov, V.B.; Falabella, M.; Colaço, H.G.; Tinajero-Trejo, M.; Poole, R.K.; Vicente, J.B.; Sarti, P.; Giuffrè, A. The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci. Rep. 2016, 6, 23788. [Google Scholar] [CrossRef]
- Kita, K.; Konishi, K.; Anraku, Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 1984, 259, 3375–3381. [Google Scholar] [CrossRef]
- Nihei, C.; Fukai, Y.; Kawai, K.; Osanai, A.; Yabu, Y.; Suzuki, T.; Ohta, N.; Minagawa, N.; Nagai, K.; Kita, K. Purification of active recombinant trypanosome alternative oxidase. FEBS Lett. 2003, 538, 35–40. [Google Scholar] [CrossRef]
- Ohshima, T.; Tanaka, S. Dye-linked L-malate dehydrogenase from thermophilic Bacillus species DSM 465. Purification and characterization. Eur. J. Biochem. 1993, 214, 37–42. [Google Scholar] [CrossRef]
- Shinagawa, E.; Fujishima, T.; Moonmangmee, D.; Theeragool, G.; Toyama, H.; Matsushita, K.; Adachi, O. Purification and characterization of membrane-bound Malate dehydrogenase from Acetobacter sp. SKU 14. Biosci. Biotechnol. Biochem. 2002, 66, 298–306. [Google Scholar] [CrossRef]
- Oh, Y.-R.; Jang, Y.-A.; Hong, S.H.; Eom, G.T. Purification and characterization of a malate:quinone oxidoreductase from Pseudomonas taetrolens capable of producing valuable lactobionic acid. J. Agric. Food Chem. 2020, 68, 13770–13778. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, Y.; Sone, N.; Kusumoto, T.; Sakamoto, J. Purification and characterization of malate:quinone oxidoreductase from thermophilic Bacillus sp. PS3. J. Bioenerg. Biomembr. 2013, 45, 131–136. [Google Scholar] [CrossRef]
- Chrétien, D.; Bénit, P.; Ha, H.-H.; Keipert, S.; El-Khoury, R.; Chang, Y.-T.; Jastroch, M.; Jacobs, H.T.; Rustin, P.; Rak, M. Mitochondria are physiologically maintained at close to 50 °C. PLoS Biol. 2018, 16, e2003992. [Google Scholar] [CrossRef]
- Macherel, D.; Haraux, F.; Guillou, H.; Bourgeois, O. The conundrum of hot mitochondria. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148348. [Google Scholar] [CrossRef]
- Lindsay, D.S.; Mitschler, R.R.; Toivio-Kinnucan, M.A.; Upton, S.J.; Dubey, J.P.; Blagburn, B.L. Association of host cell mitochondria with developing Toxoplasma gondii tissue cysts. Am. J. Vet. Res. 1993, 54, 1663–1667. [Google Scholar]
- Nelson, M.M.; Jones, A.R.; Carmen, J.C.; Sinai, A.P.; Burchmore, R.; Wastling, J.M. Modulation of the host cell proteome by the intracellular apicomplexan parasite Toxoplasma gondii. Infect. Immun. 2008, 76, 828. [Google Scholar] [CrossRef]
- Pernas, L.; Adomako-Ankomah, Y.; Shastri, A.J.; Ewald, S.E.; Treeck, M.; Boyle, J.P.; Boothroyd, J.C. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLoS Biol. 2014, 12, e1001845. [Google Scholar] [CrossRef] [PubMed]
- Sinai, A.P.; Webster, P.; Joiner, K.A. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: A high affinity interaction. J. Cell Sci. 1997, 110, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Lahouel, M.; Zini, R.; Zellagui, A.; Rhouati, S.; Carrupt, P.-A.; Morin, D. Ferulenol specifically inhibits succinate ubiquinone reductase at the level of the ubiquinone cycle. Biochem. Biophys. Res. Commun. 2007, 355, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Mercalli, E.; Fuzzati, N.; Arnoldi, L.; Stavri, M.; Gibbons, S.; Ballero, M.; Maxia, A. Antimycobacterial coumarins from the sardinian giant fennel (Ferula communis). J. Nat. Prod. 2004, 67, 2108–2110. [Google Scholar] [CrossRef]
- Bell, R.G.; Sadowski, J.A.; Matschiner, J.T. Mechanism of action of warfarin. Warfarin and metabolism of vitamin K1. Biochemistry 1972, 11, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Fasco, M.J.; Principe, L.M.; Walsh, W.A.; Friedman, P.A. Warfarin inhibition of vitamin K 2,3-epoxide reductase in rat liver microsomes. Biochemistry 1983, 22, 5655–5660. [Google Scholar] [CrossRef]
- Sadler, J.E. K is for koagulation. Nature 2004, 427, 493–494. [Google Scholar] [CrossRef]
- Bocca, C.; Gabriel, L.; Bozzo, F.; Miglietta, A. Microtubule-interacting activity and cytotoxicity of the prenylated coumarin ferulenol. Planta Med. 2002, 68, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M. Synthesis and structure–activity relationships of novel warfarin derivatives. Bioorganic Med. Chem. 2007, 15, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Schulman, S.; Dutton, R.J.; Boyd, D.; Beckwith, J.; Rapoport, T.A. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 2010, 463, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-s.; Luemmen, P.; Berry, E. Crystallographic investigation of the ubiquinone binding site of respiratory Complex II and its inhibitors. Biochim. Biophys. Acta Proteins Proteom. 2021, 3, 140679. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.; Hartuti, E.D.; Inaoka, D.K.; Sakura, T.; Amalia, E.; Nagahama, M.; Yoshioka, Y.; Tsuji, N.; Nozaki, T.; Kita, K.; et al. Structural and biochemical features of Eimeria tenella dihydroorotate dehydrogenase, a potential drug target. Genes 2020, 11, 12. [Google Scholar] [CrossRef]
- Balogun, E.O.; Inaoka, D.K.; Shiba, T.; Tsuge, C.; May, B.; Sato, T.; Kido, Y.; Nara, T.; Aoki, T.; Honma, T.; et al. Discovery of trypanocidal coumarins with dual inhibition of both the glycerol kinase and alternative oxidase of Trypanosoma brucei brucei. FASEB J. 2019, 33, 13002–13013. [Google Scholar] [CrossRef]
- Shiba, T.; Inaoka, D.K.; Takahashi, G.; Tsuge, C.; Kido, Y.; Young, L.; Ueda, S.; Balogun, E.O.; Nara, T.; Honma, T.; et al. Insights into the ubiquinol/dioxygen binding and proton relay pathways of the alternative oxidase. Biochim Biophys. Acta Bioenerg. 2019, 1860, 375–382. [Google Scholar] [CrossRef]
- Holdgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Dis. 2018, 17, 115–132. [Google Scholar] [CrossRef]
- Ebiloma, G.U.; Ayuga, T.D.; Balogun, E.O.; Gil, L.A.; Donachie, A.; Kaiser, M.; Herraiz, T.; Inaoka, D.K.; Shiba, T.; Harada, S.; et al. Inhibition of trypanosome alternative oxidase without its N-terminal mitochondrial targeting signal (ΔMTS-TAO) by cationic and non-cationic 4-hydroxybenzoate and 4-alkoxybenzaldehyde derivatives active against T. brucei and T. Congolense. Eur. J. Med. Chem. 2018, 150, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Honma, T.; Lee, N.; Hashimoto, S.; Matsuoka, S.; Kuranaga, T.; Sato, K.; Shiba, T.; et al. The open form inducer approach for structure-based drug design. PLoS ONE 2016, 11, e0167078. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Km (μM) | Vmax (μmol/min/mg) | |
|---|---|---|---|
| Variable | Fixed | ||
| Malate | Ubiquinone-1 | 370 ± 140 | 6.30 ± 0.5 |
| Malate | Ubiquinone-2 | 637 ± 180 | 24.6 ± 0.8 |
| Malate | Decylubiquinone | 466 ± 291 | 9.55 ± 0.6 |
| Ubiquinone-0 | Malate | 225 ± 77 | 12.7 ± 3.2 |
| Ubiquinone-1 | Malate | 116 ± 16 | 44.2 ± 3.8 |
| Ubiquinone-2 1 | Malate | 13.2 1 | 45.1 1 |
| Ubiquinone-4 | Malate | 7.7 ± 4.3 | 4.39 ± 0.7 |
| Ubiquinone-6 | Malate | 8.0 ± 3.3 | 3.64 ± 0.4 |
| Decylubiquinone | Malate | 17 ± 4.7 | 12.1 ± 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharjee, R.; Talaam, K.K.; Hartuti, E.D.; Matsuo, Y.; Sakura, T.; Gloria, B.M.; Hidano, S.; Kido, Y.; Mori, M.; Shiomi, K.; et al. Biochemical Studies of Mitochondrial Malate: Quinone Oxidoreductase from Toxoplasma gondii. Int. J. Mol. Sci. 2021, 22, 7830. https://doi.org/10.3390/ijms22157830
Acharjee R, Talaam KK, Hartuti ED, Matsuo Y, Sakura T, Gloria BM, Hidano S, Kido Y, Mori M, Shiomi K, et al. Biochemical Studies of Mitochondrial Malate: Quinone Oxidoreductase from Toxoplasma gondii. International Journal of Molecular Sciences. 2021; 22(15):7830. https://doi.org/10.3390/ijms22157830
Chicago/Turabian StyleAcharjee, Rajib, Keith K. Talaam, Endah D. Hartuti, Yuichi Matsuo, Takaya Sakura, Bundutidi M. Gloria, Shinya Hidano, Yasutoshi Kido, Mihoko Mori, Kazuro Shiomi, and et al. 2021. "Biochemical Studies of Mitochondrial Malate: Quinone Oxidoreductase from Toxoplasma gondii" International Journal of Molecular Sciences 22, no. 15: 7830. https://doi.org/10.3390/ijms22157830
APA StyleAcharjee, R., Talaam, K. K., Hartuti, E. D., Matsuo, Y., Sakura, T., Gloria, B. M., Hidano, S., Kido, Y., Mori, M., Shiomi, K., Sekijima, M., Nozaki, T., Umeda, K., Nishikawa, Y., Hamano, S., Kita, K., & Inaoka, D. K. (2021). Biochemical Studies of Mitochondrial Malate: Quinone Oxidoreductase from Toxoplasma gondii. International Journal of Molecular Sciences, 22(15), 7830. https://doi.org/10.3390/ijms22157830