Abstract
Copper is an essential trace element and possesses critical roles in various brain functions. A considerable amount of copper accumulates in the synapse and is secreted in neuronal firings in a manner similar to zinc. Synaptic copper and zinc modulate neuronal transmission and contribute to information processing. It has been established that excess zinc secreted during transient global ischemia plays central roles in ischemia-induced neuronal death and the pathogenesis of vascular dementia. We found that a low concentration of copper exacerbates zinc-induced neurotoxicity, and we have demonstrated the involvement of the endoplasmic reticulum (ER) stress pathway, the stress-activated protein kinases/c-Jun amino-terminal kinases (SAPK/JNK) signaling pathway, and copper-induced reactive oxygen species (ROS) production. On the basis of our results and other studies, we discuss the collaborative roles of copper in zinc-induced neurotoxicity in the synapse and the contribution of copper to the pathogenesis of vascular dementia.
1. Introduction
Copper (Cu) is the third most abundant trace element in the brain. Cu is essential for most life forms and plays crucial roles in various biological functions, including electron transport, oxygen transport and aerobic respiration [1]. Despite its significance, excess free Cu is toxic because Cu produces reactive oxygen species (ROS) in the redox activity between Cu2+ and Cu+. Increasing evidence has suggested that the dyshomeostasis of Cu is implicated in the pathogenesis of various neurodegenerative diseases, such as Alzheimer’s disease (AD), prion diseases, Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and amyotrophic lateral sclerosis (ALS) [2,3,4,5].
Here, we focus on the link between Cu and the pathogenesis of a vascular type of senile dementia (VD). VD mainly occurs after stroke or ischemia. During transient global ischemia, the deprivation of glucose and oxygen by the disruption of blood flow and the subsequent neuronal excitation causes a sustainable release of glutamate, which causes neurodegeneration and leads to the pathogenesis of VD [6]. It is widely established that excess zinc (Zn) co-released with glutamate plays central roles in ischemia-induced neurodegeneration and the pathogenesis of VD [7]. Copper ions (Cu2+) and zinc ions (Zn2+) share similar chemical characteristics and bind the same regions of chelators or metal-binding proteins. Therefore, Cu2+ acts competitively with Zn2+ in several biological functions. The intake of excess Zn causes a Cu deficiency and vice versa [8]. However, we found that the co-existence of a low concentration of Cu2+ markedly exacerbated Zn2+-induced neuronal death [9]. We have investigated the molecular pathways implicated in Cu2+-enhanced Zn2+-induced neurotoxicity and demonstrated the involvement of the endoplasmic reticulum (ER) stress pathway and the stress-activated protein kinases/c-Jun amino-terminal kinases (SAPK/JNK) signaling pathway [9,10]. We also demonstrated that Cu2+-induced ROS may be an upstream factor of the ER stress pathway and the SAPK/JNK pathway in the neurodegeneration processes [11,12]. Considering that Cu2+ and Zn2+ co-exist in the synaptic cleft under ischemic conditions, we hypothesize the involvement of Cu2+ as a collaborative partner of Zn2+-induced neurotoxicity and subsequent pathogenesis of VD. We also discuss the roles of Cu-binding, amyloidogenic proteins in the synapse in the regulation of Cu homeostasis.
2. Copper in the Brain
Cu is an essential trace element and abundantly exists in the liver, kidney and brain [1]. Orally digested Cu is absorbed from the gastrointestinal pathway by divalent cation transporter 1 (DMT1) as Cu2+ or by copper transporter 1 (CTR1) as Cu+. Cu binds to ceruloplasmin and is then transported in the blood system. The copper-transporting ATPases (ATP7A and ATP7B) play essential roles in Cu distribution in the organs. Cu deficiency or excess due to the impairment of Cu transporters leads to severe neurodegenerative diseases, such as Menkes disease or Wilson’s disease [13]. In the brain, Cu especially exists in the thalamus, substantia nigra, striatum, and hippocampus. Cu plays essential functions in the brain in the synthesis of neurotransmitters, myelination, and neuroprotection against ROS as a cofactor of various enzymes and functional proteins, including cytochrome C oxidase, lysyl oxidase, uricase, dopamine hydroxylase, tyrosinase and Cu/Zn superoxide dismutase (Cu/Zn SOD). Cu is also involved in iron (Fe) homeostasis because ceruloplasmin is a ferroxidase, which converts Fe2+ to Fe3+.
Increasing evidence suggests that Cu is involved in neurotransmission and information processing in the brain [14,15]. Although the majority of Cu rigidly binds to proteins, a substantial fraction of Cu is in the form of free Cu (Cu2+) or loosely bound to small molecular compounds in synaptic vesicles of neurons and is released into the synaptic clefts during neuronal excitation. Secreted Cu2+ regulates overall brain excitability by binding to several neurotransmitter receptors, including N-methyl-d-aspartate (NMDA)-type glutamate receptors, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)-type glutamate receptors, γ-aminobutyric acid (GABA) receptors, and purinergic receptors. The accumulation of Cu in the brain reportedly altered GABA-mediated neurotransmission and promoted impulsive behaviors [16]. ATP7A plays a pivotal role in the transport of Cu into synaptic vesicles and, therefore, contributes to axon outgrowth and synaptogenesis [17].
It is well established that a considerable amount of Zn also accumulates in the presynaptic vesicles of glutamatergic neurons as free Zn (Zn2+) or in a loosely bound form [18]. Synaptic Zn2+ is released with glutamate during neuronal excitation and binds to various neurotransmitter receptors, including NMDA-type glutamate receptors, AMPA receptors, and GABA receptors. These characteristics of Zn2+ are quite similar to those of Cu2+.
It is possible that—but has yet to be determined whether—secreted Cu2+ and Zn2+ can diffuse across the synaptic cleft, spill over to neighboring synapses, and modulate the activity of those neighboring synapses, as shown in Figure 1. Synaptic Zn2+ plays critical roles in information processing and memory formation [19,20]. It is plausible that differing concentrations of excitatory glutamate and inhibitory Cu2+ and/or Zn2+ in adjacent synapses transmit spatio-temporal information about neuronal firings and facilitate the precise modulation of neuronal activity. This modulation of neuronal activity at adjacent synapses generates contrasting signals that may enable lateral inhibition and serve as the basis for synaptic plasticity [21]. Therefore, it is possible that “Cu signaling” and “Zn signaling” coordinate at the synapse and modulate the neuronal information [22,23]. Indeed, Cu deficiency caused impaired maturation of the hippocampus in immature rats [24].
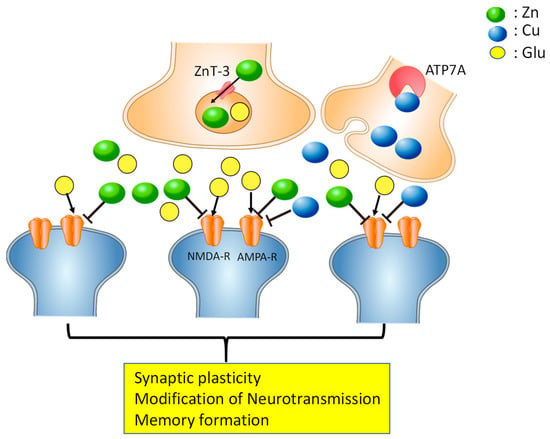
Figure 1.
Copper and zinc in the synapse. Under normal conditions, Cu2+ and Zn2+ are stored in presynaptic vesicles, released with neurotransmitters such as glutamate, and bind to N-methyl-d-aspartate (NMDA)-type glutamate receptors (NMDA-R), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)-type glutamate receptors (AMPA-R), or other receptors. ZnT-3, a Zn transporter, and the copper-transporting ATPase (ATF7A) are involved in the accumulation of Zn and Cu in the synapse, respectively. Cu2+ and Zn2+ may spill over to the neighboring synapses and modulate excitability, and are implicated in the maintenance of synaptic plasticity and memory formation.
Although the concentration of Cu and Zn in the cerebrospinal fluid (CSF) has been reported to be less than 1 μM [25], the concentration in the synaptic cleft may be much higher compared with that in the CSF. The synaptic cleft is a small compartment conceptualized as a cylinder with a radius of 120 nm and a height of 20 nm, and the total volume is estimated to be about 1% of the extracellular space [26]. Thus, it is possible that the concentrations of Cu2+ and Zn2+ are much higher compared with the CSF. Indeed, the concentration of glutamate in the synaptic cleft is estimated to reach the mM range after 1 ms of neuronal depolarization. Although the exact levels of Zn and Cu in the synaptic cleft remain controversial, the Zn concentrations are estimated to be 1–100 μM [27]. Kardos et al. found ~100 μM of Cu released into synaptic clefts by atomic absorption [28]. However, considering the chemical characteristics of Cu2+, it is plausible that synaptic Cu2+ loosely bound to small compounds such as organic acids and ATP. A study using a Cu-sensitive fluorescent probe demonstrated that ~3 μM of Cu is released in the synaptic clefts [29].
3. Vascular Dementia and Zinc
Senile dementia is a serious problem for our rapidly aging society. It is characterized by profound memory loss and the inability to form new memories in older adults, and its prevalence increases with age. The number of patients in Japan, including those with a mild cognitive deficiency, is estimated to be more than 800 million.
Senile dementia is mainly divided into Alzheimer’s disease (AD), vascular dementia (VD), and dementia with Lewy bodies (DLB). Both AD and DLB are characterized by the deposition of abnormally accumulated proteins: β-amyloid protein (AβP) in AD and α-synuclein (α-Syn) in DLB. VD accounts for approximately one-third of senile dementia cases in Japan. VD is a degenerative cerebrovascular disease that is mainly caused by a series of strokes or ischemia [6]. Risk factors for VD include age, male sex, diabetes, and high blood pressure [30]. After transient global ischemia, the interruption of blood flow and its reperfusion cause the deprivation of oxygen and glucose and the production of ROS. Thereafter, abnormal neuronal excitation occurs in large parts of the brain with the excessive release of glutamate into the synaptic clefts. The excess glutamate causes over-stimulation of its receptors, and the successive entry of large quantities of calcium ions (Ca2+) triggers the delayed death of vulnerable neurons, such as pyramidal neurons in the hippocampus, which is an area associated with learning, memory and language. Thus, the development of an infarct and the subsequent cognitive dysfunction characterize the pathogenesis of VD in elderly individuals. An epidemiological study reported the occurrence of dementia symptoms in about 30% of stroke patients within 3 years of the initial stroke [31].
Increasing evidence suggests that Zn has a causative role in neuronal injury after transient global ischemia, ultimately leading to VD [32]. As noted, Zn2+ co-accumulates with glutamate in the presynaptic vesicles and is co-released into the synaptic cleft under ischemic conditions. The concentration of Zn2+ is estimated to be up to 300 µM [33]. Excess Zn2+ reportedly causes the apoptotic death of cultured neurons or neuroblastoma cells (PC12 cells) [34,35]. Koh et al. demonstrated that Zn2+ accumulates in apoptotic neurons in the hippocampus after ischemia [36]. “Zn translocation”, namely the entry of Zn from presynaptic vesicles into the postsynaptic neurons, and the increase in intracellular Zn2+ levels ([Zn2+]i), occurs in vulnerable neurons in the CA1 or CA3 regions of the hippocampus prior to the onset of delayed neuronal death following transient global ischemia, enhancing the appearance of the infarct [37]. There are three routes of Zn2+ entry into the cell: AMPA-type glutamate receptors, NMDA-type glutamate receptors, and the voltage-gated L-type Ca2+ channel (VGLC). Under normal conditions, most hippocampal neurons express AMPA receptors containing the subunit GluR2, which are poorly permeable to Ca2+ and Zn2+ [38]. However, after ischemia, an acute reduction in the expression of the GluR2 subunit occurs, and neurons express a specific type of AMPA receptor that has channels that are directly Ca2+-permeable (Ca-AMPA/kainate channels: Ca-A/K-R). The appearance of Ca-A/K-R channels causes an increased permeability for Ca2+ and Zn2+ and enhances their toxicity. Therefore, the expression of Zn2+-permeable Ca-A/K-R channels and the entry of Ca2+ and/or Zn2+ through these channels mediate delayed neuronal death after ischemia. Furthermore, the administration of calcium ethylenediaminetetraacetic acid (Ca EDTA), a membrane-impermeable Zn chelator, blocks the translocation of Zn, protects hippocampal neurons after transient global ischemia, and reduces infarct volume [39]. Because Ca EDTA attenuates the ischemia-induced downregulation of the GluR2 gene, Zn is also implicated in the transcriptional regulation of Ca-A/K-R channels. These results strongly suggest that Zn plays a key role in delayed neuronal death after transient global ischemia, a process that is potentially involved in the pathogenesis of VD.
4. Copper Enhances Zinc-Induced Neurotoxicity
An understanding of the molecular mechanism underlying Zn2+-induced neurodegeneration will advance the development of treatments for VD. We found that Zn causes the apoptotic death of GT1-7 cells (immortalized hypothalamic neurons) in a dose-dependent and time-dependent manner [40]. GT1-7 cells are much more sensitive to Zn and exhibited much lower viability after Zn exposure compared with other neuronal cells, such as PC-12 cells, primary cultured rat hippocampal neurons, and B-50 neuroblastoma cells [41]. The degenerated GT1-7 cells were terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick-end labeling (TUNEL)-positive and exhibited DNA fragmentation. The GT1-7 cells, which were developed by genetical targeted tumorigenesis of mouse hypothalamic neurons, possess neuronal characteristics, such as the extension of neuritis, secretion of gonadotropin-releasing hormone (GnRH), and expression of neuron-specific proteins or receptors, including microtubule-associated protein 2 (MAP2), tau protein, neurofilaments, synaptophysin, GABA receptors, dopamine receptors, and L-type Ca2+ channels [42]. Notably, the GT1-7 cells are not subject to glutamate toxicity [8] because they either lack or possess low levels of ionotropic glutamate receptors [43]. Because of these properties, we considered the GT1-7 cell line to be an excellent model system for investigating Zn2+-induced neurotoxicity.
First, we examined the effects of treatment with various pharmacological agents prior to Zn2+ treatment of GT1-7 cells and found that neither antagonists nor agonists of excitatory neurotransmitters (D-APV, glutamate and CNQX) nor those of inhibitory neurotransmitters (bicuculline, muscimol, baclofen and GABA) influenced the viability of GT1-7 cells [44,45,46,47]. Thus, it is possible that glutamate receptors and/or GABA receptors are not involved in the Zn-induced neurodegenerative pathways. However, several compounds, including energy substrates (pyruvate, citrate), metal chelators (o-phenanthroline, deferoxamine), dipeptides (carnosine, anserine), and amino acids (histidine), attenuated the Zn2+-induced death of GT1-7 cells.
We examined the effects of other metal ions on Zn-induced neurotoxicity and found that co-exposure with aluminum ions (Al3+), gadolinium ions (Gd3+), or Ca2+ attenuated the Zn2+-induced death of GT1-7 cells [13,48]. However, co-existence with Cu2+ or nickel ions (Ni2+) remarkably exacerbated Zn-induced neurotoxicity [9]. Although cells exposed to Cu2+ (CuCl2) alone (0–80 μM) did not exhibit neurodegenerative changes after exposure for 24 h, 30 μM of Zn2+ caused the loss of 39.0 ± 1.0% of GT1-7 cells (Figure 2). Meanwhile, the co-exposure of 2.5 μM Cu with 30 μM Zn (molar ratio of Cu:Zn = 1:12) resulted in a 76.2 ± 3.3% decrease in cell viability, and 10 μM Cu with 30 μM Zn (molar ratio of Cu:Zn = 1:3) resulted in a 93.8 ± 2.3% decrease in cell viability. Although we applied ZnCl2 or CuCl2 in the culture media, it is plausible that these metals can bind in a labile manner to small compounds such as citrate or ATP. These results were contrary to our expectation that Cu2+ may compete with Zn2+-induced neurotoxicity. Therefore, we focused on the molecular mechanism of Cu2+-enhanced Zn2+-induced neurotoxicity (Cu/Zn neurotoxicity).
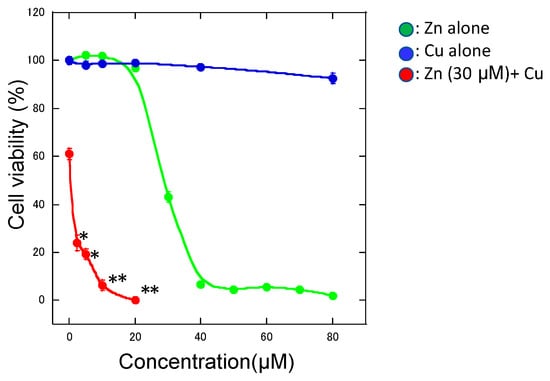
Figure 2.
Cu2+-enhanced Zn2+-induced neurotoxicity. Various concentrations of CuCl2 alone (blue circle), ZnCl2 alone (green circle), and CuCl2 (0–20 μM) with 30 μM ZnCl2 (red circle) were administered to GT1-7 cells in serum-free culture media. After 24 h, cell viability was determined using the CellTiter-Glo® assay. Data are presented as mean ± S.E.M. * p < 0.05, ** p < 0.01 versus 30 μM ZnCl2 alone (Tukey’s test).
5. The Molecular Pathways of Copper-Enhanced Zinc-Induced Neurotoxicity
Examination of the molecular pathways involved in the neurotoxicity induced by co-exposure to Cu/Zn may facilitate the development of drugs for the treatment/prevention of VD. Using DNA microarray analysis and real-time PCR (RT-PCR) techniques, we demonstrated that various genes, including metal-related genes (Zn transporter 1 (ZnT-1), metallothionein 1 (MT1), and metallothionein 2 (MT2)), endoplasmic reticulum (ER)-stress-related genes (CHOP, GADD34), signal-transduction-related genes, and Ca2+-signaling-related genes (Arc), were upregulated after exposure to Zn2+ alone [46,47]. Based on the DNA microarray analysis of GT1-7 cells exposed to Cu2+ and Zn2+, we found that several genes related to ER stress pathways and MAP kinase pathways were especially upregulated in Cu/Zn conditions compared with Zn alone [9,10,11,12]. Herein, we focus on the five neurodegenerative pathways.
5.1. ER Stress Pathway
The ER stress pathway, which impairs ER function and leads to an accumulation of unfolded or misfolded proteins, is implicated in many neurodegenerative diseases, including AD, PD, and cerebral ischemia [49]. ER stress is mediated by three sensors at the ER membrane: PKR-like endoplasmic reticulum eIF2a kinase (PERK), inositol requiring 1 (IRE1), and activating transcription factor 6 (ATF6) [50]. In the PERK branch, activating transcription factor 4 (ATF4) induces C/EBP homologous protein (CHOP), which triggers an intrinsic apoptotic pathway, such as caspase cascades [51], and thereafter CHOP induces GADD34 (protein phosphatase 1 regulatory subunit 15A).
We previously found that Zn2+ induces marked upregulation of endoplasmic reticulum (ER)-stress-related genes, especially CHOP and GADD34 in GT1-7 cells [47,48]. We demonstrated that genes related to the PERK branch (ATF4, CHOP and GADD34) were upregulated during Cu/Zn neurotoxicity using RT-PCR analysis [9]. The induction of CHOP protein and the correlation with cell viability were observed by Western blotting analysis. Although Zn2+ alone induced these ER-stress-related genes, Cu2+ alone did not influence these genes. Therefore, it is possible that the ATF4–CHOP–GADD34 axis is responsible for the apoptotic death observed in Cu/Zn neurotoxicity. We found that the ER stress pathway is also involved in Ni2+-enhanced Zn2+-induced neurotoxicity of GT1-7 cells [52].
5.2. SAPK/JNK Pathway
We also found that genes related to the MAP kinase (MAPK) signaling pathway were upregulated after co-exposure to Cu2+ and Zn2+. The MAPKs are serine/threonine protein kinases that mediate complex signal transduction based on various cellular processes, including proliferation, differentiation, migration, cell death/survival and environmental stress response [53]. There are four subfamilies: the extracellular signal-regulated kinases (ERK1/2), the c-Jun NH2-terminal kinases (JNK 1, 2 and 3), the p38 kinases, and the ERK5 (also known as big MAPK-1, BMK1) subfamilies. Among them, the SAPK/JNK signaling pathway plays an important role in apoptotic cell death, necroptosis, and autophagy [54]. The SAPK/JNK signaling pathway is activated by a variety of environmental stressors, such as oxidative stress, inflammatory cytokines and metals. Upon activation of this pathway by various stressors, MAPK kinase 4 (MKK4) or MKK7 phosphorylates and activates SAPK/JNK. Then, c-Jun and activating transcription factor 2 (ATF2), known major downstream factors of SAPK/JNK, are phosphorylated and activated by SAPK/JNK. Ultimately, phosphorylated forms of c-Jun and ATF2 induce downstream factors related to cell death and mitochondrial injury, leading to cell death.
Using RT-PCR and Western blotting, we found that the phosphorylated (i.e., active) forms of SAPK/JNK were increased by co-treatment of Cu2+ and Zn2+, but not by Zn2+ alone nor by Cu2+ alone [10]. We found that phospho-c-Jun and phospho-ATF2 were also induced by Cu2+ and Zn2+. Moreover, an inhibitor of the SAPK/JNK signaling pathway (SP600125) significantly suppressed the induction of CHOP by co-treatment of Cu2+ and Zn2+ and the activation of the SAPK/JNK signaling pathway and attenuated the neuronal cell death.
5.3. Energy Production Pathway
We have already demonstrated that pyruvate, an energy substrate, attenuated Zn2+-induced neurotoxicity [40]. Shelline and colleagues reported that Zn exposure decreased the levels of nicotinamide adenine dinucleotide (NAD+) and ATP in cultured cortical neurons, and that treatment with pyruvate restored the NAD+ level [55,56]. The administration of pyruvate attenuated neuronal death after ischemia in vivo [57]. We found that pyruvate and citrate attenuated Cu/Zn neurotoxicity [58]. Furthermore, the co-existence of pyruvate and citrate did not influence the intracellular concentrations of Zn2+ and Cu2+ of GT1-7 cells nor the elevations of MTs mRNA. Therefore, it is unlikely that pyruvate and citrate attenuated Cu/Zn neurotoxicity by the chelation to Cu2+ and/or Zn2+. Therefore, it is possible that the mitochondrial energy production pathway is involved in Cu/Zn neurotoxicity as well as Zn2+-induced neurotoxicity.
5.4. Disruption of Ca2+ Homeostasis
The upstream factors that underlie the Cu/Zn-induced ER stress pathway and the SAPK/JNK pathway are a matter of interest. Here, we focus on two possible upstream pathways: Ca2+ homeostasis and ROS production. Both pathways are known to regulate the ER stress pathway and the SAPK/JNK pathway. Notably, disrupted Ca2+ homeostasis is also reportedly involved in Zn2+-induced neuronal death. Kim et al. reported that Zn neurotoxicity in PC-12 cells was attenuated by an L-type Ca2+ channel blocker, nimodipine, and enhanced by the L-type Ca2+ channel activator S(-)-Bay K 8644 [35]. Additionally, Zn neurotoxicity was attenuated by aspirin, which prevents Zn2+ entry through voltage-gated Ca2+ channels (VGLCs) [59].
Using a high-resolution multi-site video imaging system with fura-2 as the cytosolic free calcium reporter fluorescent probe, we previously demonstrated that the exposure of Zn2+ caused an elevation in intracellular concentrations of Ca2+ ([Ca2+]i) in GT1-7 cells after 3–30 min of exposure [41]. The addition of Ca2+, Al3+ and Gd3+ attenuated the Zn2+-induced death of GT1-7 cells. It is widely known that Gd3+ is a blocker of VGLCs [60] and that Al3+ inhibits various types of Ca2+ channels [61]. We found that pretreatment with Al3+ significantly blocked the Zn-induced [Ca2+]i elevations and attenuated the Zn2+-induced neurotoxicity of GT1-7 cells [41]. These results suggest that Ca2+ dyshomeostasis is involved in the mechanism of Zn-induced neurotoxicity. It is plausible that the disruption of Ca2+ homeostasis is also involved in Cu/Zn neurotoxicity.
5.5. ROS Production
It is widely known that oxidative stress is involved in various neurodegenerative diseases. ROS reportedly induce the ER stress pathway [62], the SAPK/JNK pathway [63], and numerous other adverse effects. As previously noted, an inhibitor of SAPK/JNK (SP600125) suppresses Cu2+/Zn2+-dependent increases in CHOP expression, suggesting that the ROS–JNK–CHOP pathway is involved in Cu/Zn neurotoxicity. The ROS–JNK–CHOP pathway is also implicated in other types of cell death, such as TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [64].
Cu is a redox-active metal that exists as oxidized Cu2+ and reduced Cu+, while Zn exists only as Zn2+ and is not directly implicated in the redox pathway. The addition of Cu2+ induced ROS production in GT1-7 cells, while Zn2+ alone did not produce ROS or influence Cu2+-induced ROS production in GT1-7 cells [10]. We examined the involvement of oxidative stress in activation of the SAPK/JNK signaling pathway and found that human serum albumin–thioredoxin fusion protein (HSA-Trx), an antioxidative protein, suppressed activation of the SAPK/JNK signaling pathway, inhibited ROS production, and attenuated Cu/Zn neuronal death of GT1-7 cells [11]. Furthermore, selenomethionine (Se-Met), an endogenous selenium (Se)-containing amino acid, induced glutathione peroxidase and blocked ROS production [12]. Pretreatment with Se-Met significantly suppressed the induction of CHOP and attenuated Cu/Zn neurotoxicity.
6. Hypothetical Scheme Regarding Cu/Zn Neurotoxicity
On the basis of these findings, we established a hypothetical scheme regarding Cu/Zn neurotoxicity and the implication of Cu and/or Zn in the pathogenesis of vascular dementia (Figure 3).

Figure 3.
Hypothetical scheme regarding Cu/Zn neurotoxicity. Under pathological conditions such as transient global ischemia, excess Cu2+ and Zn2+ co-exist in the synaptic cleft. The elevation in [Zn2+]i and [Ca2+]i triggers ER stress pathways, inhibits the energy production pathway in mitochondria, and induces neurodegeneration. The co-existence of Cu2+ with Zn2+ causes the production of ROS, upregulates the ER stress pathway and the SAPK/JNK pathway, and finally exacerbates neuronal death. Cu-binding proteins, including normal cellular prion protein (PrPC), amyloid precursor protein (APP), and α-synuclein (α-Syn), are located in the synapse and regulate the levels of metals such as Cu, Zn and Fe. Additionally, PrPC regulates Zn2+ levels as a ZIP Zn transporter analogue with the ZnT-1 Zn transporter, which is also localized to postsynaptic membranes. PrPC can provide Cu to APP or other Cu-binding proteins in the synapse. APP is mainly localized to the presynaptic membrane. APP binds to Cu and/or Zn and can convert Cu2+ to Cu+. APP can provide Cu+ to CTR1 or other Cu+-binding proteins. APP also regulates Fe2+ efflux from cells via ferroportin. α-Syn is mainly localized to the presynaptic domain and binds Cu, Mn, and Fe. Both PrPC and α-Syn have ferrireductase activity and provide bioavailable Fe2+ to enzymes at the pre- and post-synapse, respectively. Other metal-binding factors such as metallothionein 3 (MT-3) and carnosine (Car) are secreted into synaptic clefts and play critical roles in the maintenance of metal homeostasis. NMDA-R, NMDA-type glutamate receptor; Ca-A/K-R, Ca2+-permeable AMPA/kainate-type glutamate receptor; VGLC, voltage-gated L-type Ca2+ channel; FPN, ferroportin. The colored circles represent Zn, Cu, Fe, Ca and glutamate.
Under normal conditions, Zn2+ and/or Cu2+ are secreted into the synaptic cleft during neuronal excitation in a distinctive manner and modulate neuronal information processing. The co-existence of Zn2+ and Cu2+ in the same synapse does not often occur because the secreted Zn2+ and/or Cu2+ experience rapid re-uptake by Zn transporters or by CTR1.
However, under pathogenetic conditions such as transient global ischemia, long-term neuronal excitation occurs in major parts of the brain and, thereafter, Zn2+ and Cu2+ are released and abundantly co-localize in the same synaptic clefts. Excess Zn2+ induces a reduction in the expression of the GluR2 subunit, and the elevation in [Zn2+]i and [Ca2+]i occurs through Ca-A/K-Rs, NMDA-Rs and VGLCs. Al3+, a known Ca2+ channel blocker, blocks Zn2+-induced elevation of [Ca2+]i and attenuates Zn2+-induced neuronal death. Thereafter, the elevation in [Zn2+]i and [Ca2+]i triggers the PERK branch of the ER stress pathway, induces CHOP, and finally causes neuronal death. The increase in [Zn2+]i also inhibits NAD+ in the mitochondrial energy production pathway and causes the depletion of ATP, which leads to neurodegeneration.
When Cu2+ and Zn2+ co-exist in the same synaptic cleft, Cu2+ produces ROS, which subsequently induces the PERK pathway and the SAPK/JNK pathway. Thereafter, both pathways induce CHOP and ultimately exacerbate Zn2+-induced neuronal death. SP600125, an inhibitor of the SAPK/JNK pathway, inhibits these processes and attenuates Cu/Zn neurotoxicity. Antioxidants such as HSA-Trx or Se-Met inhibit ROS production by Cu2+ and attenuate Cu/Zn neuronal death. In conclusion, synaptic Cu2+ collaborates with Zn2+ in the same synapse and the co-existence of Cu2+ and Zn2+ triggers neuronal death after transient global ischemia, ultimately causing the pathogenesis of VD. This hypothesis can explain various aspects of Cu/Zn neurotoxicity and the involvement of synaptic Cu2+ and Zn2+ in the pathogenesis of VD. Several epidemiological findings suggest that elevated serum Cu is a risk factor of stroke [65,66,67].
Considering the abundance of Cu2+ and Zn2+ in the synapse, regulatory factors of metal homeostasis may exist in the synapse and play critical roles in the pathogenesis of VD. Several Cu-binding proteins are reportedly localized in the synapse [4,68]. Normal cellular prion protein (PrPC) exists at postsynaptic membranes. PrPC binds to Cu at the N-terminal octarepeat domain and contributes to intracellular uptake of Cu [4]. PrPC acts as a ZIP Zn transporter analogue and regulates cellular Zn uptake combined with the AMPA receptor [69]. The ZnT-1 Zn transporter is also localized to postsynaptic membranes binding with glutamate receptors [70]. Therefore, both PrPC and ZnT-1 control Zn2+ levels at the synapse. PrPC also possesses ferrireductase activity to convert Fe3+ and Fe2+ and is involved in cellular uptake of Fe2+ [71]. Amyloid precursor protein (APP) is localized in the presynaptic membranes and possesses Cu- and/or Zn-binding domains. APP can convert Cu2+ to Cu+ and contributes to the cellular uptake of Cu [72]. APP is also implicated in Fe2+ efflux by binding with ferroportin [73]. α-Syn is a Cu-binding protein present in the presynaptic domain. Cu enhanced the oligomerization of α-Syn [74]. α-Syn also possesses ferrireductase activity to convert Fe3+ and Fe2+ [75]. Interestingly, all these proteins are involved in the pathogenesis of neurodegenerative diseases [68]. PrPC and its conformational changes are central to the pathogenesis of prion diseases, including scrapie in sheep, bovine spongiform encephalopathy in cattle, and Creutzfeldt–Jakob disease (CJD) in humans. The accumulation of AβP, which is secreted from APP, is observed in AD brain. The oligomerization and the neurotoxicity of AβP are central in AD. The deposition of α-Syn as Lewy bodies has been characterized in the brains of patients with DLB. These disease-related proteins (amyloidogenic proteins) can act to maintain the homeostasis of Cu, Zn and Fe, while these metals can in turn regulate the expressions and functions of the disease-related proteins. The mRNAs of APP, α-Syn, and PrPC possess an iron-responsive element and, therefore, their expressions are regulated by Fe [76]. Cu regulates the processing of APP [77] and the production and clearance of AβP [78]. Considering that these disease-related proteins co-exist in the synaptic cleft, which is a small compartment filled with Cu2+ and/or Zn2+, it is possible that these proteins can interact with each other in the maintenance of these metals [79,80]. Nibaldo et al. demonstrated that Cu-binding domains of both APP and PrPC prevent neurotoxicity of Cu [81]. AβPs reportedly inhibit the binding of Cu2+ to PrPC [82]. PrPC contributes to AD pathogenesis as a toxic receptor of AβP oligomers [83]. Moreover, recent studies have suggested the involvement of PrPC in ischemia-induced neurotoxicity [84,85].
Metal–metal or metal–protein crosstalk is complex and delicate. Therefore, it is plausible that the disorder of these regulatory proteins and disrupting Cu and/or Zn homeostasis may trigger various neurodegenerative diseases, including AD, prion disease, DLB and VD.
There are other regulatory factors of metal homeostasis in the synapse. Metallothionein-3 (MT-3) and carnosine (β-alanyl histidine) are secreted from glial cells or from neurons to the synaptic cleft [86,87]. Carnosine is an endogenous dipeptide that possesses various neuroprotective functions, including anti-oxidant, anti-crosslinking, and anti-glycation functions [8,88]. Carnosine has the ability to chelate to Zn2+ and/or Cu2+ [88]. The complex of Zn and carnosine (Poraprezinc) is widely used for the treatment of gastric ulcers. We have previously demonstrated that carnosine attenuated Zn2+-induced neuronal death [47]. Administration of carnosine was effective in the animal model of ischemic stroke [89]. Dietary supplementation of carnosine and anserine was revealed to improve the cognitive decline and maintenance of memory in elderly people [90]. Based on our findings regarding carnosine, we obtained a patent about carnosine and related compounds as a possible strategy for the prevention and treatment of VD [91,92].
7. Conclusions
Our findings regarding the role of Cu in Zn-induced neurotoxicity and ischemia-induced neuronal death clarify the role of Cu as a collaborator with Zn in the pathogenesis of vascular-type dementia. The role of Cu in the synapse may enhance our understanding of the pathogenesis of VD and other neurodegenerative diseases. Substances that attenuate Cu/Zn neurotoxicity may lead to strategies for the prevention or treatment of VD. Further research that provides a more detailed analysis of Cu/Zn neurotoxicity and preventive substances is necessary.
Author Contributions
Participated in research design: K.-i.T. and M.K.; Conducted experiments: K.-i.T., M.K. and M.K.-N.; Wrote or contributed to the writing of the manuscript: K.-i.T., M.K. and M.K.-N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (JSPS Kakennhi Grant numbers. JP18K06669 and JP17H03197).
Acknowledgments
This work was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology. We thank Kobayashi, N. for technical assistance.
Conflicts of Interest
The authors declare that there are no conflict of interest.
Abbreviations
| AD | Alzheimer’s disease |
| AβP | Alzheimer’s β-amyloid protein |
| ATF7A | copper-transporting ATPase 7A |
| AMPA | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid |
| CSF | cerebrospinal fluid |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| CJD | Creutzfeldt–Jakob disease |
| CTR1 | copper transporter 1 |
| D-APV | 2-amino-5-phosphonovalerate |
| DLB | dementia with Lewy bodies |
| DMT1 | divalent metal transporter 1 |
| ER | endoplasmic reticulum |
| GABA | γ-aminobutyric acid |
| GADD34 | growth-arrest and DNA-damage-inducible gene 34 |
| [Ca2+]i | intracellular calcium levels |
| NMDA | N-methyl-d-aspartate |
| NAC | non-amyloid component |
| PD | Parkinson’s disease |
| PrP | prion protein |
| ROS | reactive oxygen species |
| SAPK/JNK | stress-activated protein kinases/c-Jun amino-terminal kinases |
| VD | vascular dementia |
| VGCC | voltage-gated Ca2+ channel |
| CSF | cerebrospinal fluid |
| DMT1 | divalent metal transporter-1 |
References
- Scheiber, I.F.; Mercer, J.F.B.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar]
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper dyshomeostasis in neurodegenerative diseases-therapeutic implications. Int. J. Mol. Sci. 2020, 21, 9259. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, K.; Masaldan, S.; Opazo, C.M.; Bush, A.I. Redox active metals in neurodegenerative diseases. J. Biol. Inorg. Chem. 2019, 24, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.-I. Neurometals in the Pathogenesis of Prion Diseases. Int. J. Mol. Sci. 2021, 22, 1267. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bea, F.J.; Aldanondo, G.; Lasa-Fernández, H.; de Munain, A.L.; Vallejo-Illarramendi, A. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev. Mol. Med. 2017, 19, e7. [Google Scholar] [CrossRef]
- Lee, J.M.; Grabb, M.C.; Zipfel, G.Z.; Choi, D.W. Brain tissue responses to ischemia. J. Clin. Investig. 2000, 106, 723–731. [Google Scholar] [CrossRef]
- Pochwat, B.; Nowak, G.; Szewczyk, B. Relationship between Zinc (Zn2+) and glutamate receptors in the processes underlying neurodegeneration. Neural Plast. 2015, 591563. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Sadakane, Y.; Mizuno, K.; Kato-Negishi, M.; Tanaka, K.-I. Carnosine as a possible drug for zinc-induced neurotoxicity and vascular dementia. Int. J. Mol. Sci. 2020, 21, 2570. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Kawahara, M. Copper enhances zinc-induced neurotoxicity and the endoplasmic reticulum stress response in a neuronal model of vascular dementia. Front. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Shimoda, M.; Kasai, M.; Ikeda, M.; Ishima, Y.; Kawahara, M. Involvement of SAPK/JNK signaling pathway in copper enhanced zinc-induced neuronal cell death. Toxicol. Sci. 2019, 169, 293–302. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Shimoda, M.; Chuang, V.T.G.; Nishida, K.; Kawahara, M.; Ishida, T.; Otagiri, M.; Maruyama, T.; Ishima, Y. Thioredoxin-albumin fusion protein prevents copper enhanced zinc-induced neurotoxicity via its antioxidative activity. Int. J. Pharm. 2018, 535, 140–147. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimoda, M.; Okudomi, S.; Kawaraya, S.; Kawahara, M.; Tanaka, K.-I. Seleno-L-methionine suppresses copper-enhanced zinc-induced neuronal cell death via induction of glutathione peroxidase. Metallomics 2020, 12, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, B. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.M.; Greenough, M.; Bush, A.I. Copper: From neurotransmission to neuroproteostasis. Front. Aging Neurosci. 2014, 6, 1–7. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Chang, J.; Kim, J. Loss of divalent metal transporter 1 function promotes brain copper accumulation and increases impulsivity. J. Neurochem. 2016, 138, 918–928. [Google Scholar] [CrossRef]
- Meskini, R.E.; Kelli, J.; Crabtree, L.; Cline, L.B.; Mains, R.E.; Eipper, A.; Ronnett, G.V. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol. Cell. Neurosci. 2007, 34, 409–421. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H. The impact of synaptic Zn2+ dynamics on cognition and its decline. Int. J. Mol. Sci. 2017, 18, 2411. [Google Scholar] [CrossRef]
- Ueno, S.; Tsukamoto, M.; Hirano, T.; Kikuchi, K.; Yamada, M.K.; Nishiyama, N.; Nagano, N.; Matsuki, N.; Ikegaya, Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-d-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 2002, 158, 215–220. [Google Scholar] [CrossRef]
- Huston, J.P.; Wagner, U.; Hasenöhrl, R.U. The tuberomammillary nucleus projections in the control of learning, memory and reinforcement processes: Evidence for an inhibitory role. Behav. Brain Res. 1997, 83, 97–105. [Google Scholar] [CrossRef]
- Kardos, J.; Héja, L.; Simon, A.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T. Roles of zinc signaling in the immune system. J. Immunol. Res. 2016, 6762343. [Google Scholar] [CrossRef] [PubMed]
- Gybina, A.A.; Tkac, I.; Prohaska, J.R. Copper deficiency alters the neurochemical profile of developing rat brain. Nutr. Neurosci. 2009, 12, 114–122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roos, P.M.; Vesterberg, O.; Syversen, T.; Flaten, T.P.; Nordberg, M. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 2013, 151, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef]
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, N.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef]
- Román, G.C. Vascular dementia prevention: A risk factor analysis. Cerebrovasc. Dis. 2005, 20, 91–100. [Google Scholar] [CrossRef]
- De Haan, E.H.; Nys, G.M.; Van Zandvoort, M.J. Cognitive function following stroke and vascular cognitive impairment. Curr. Opin. Neurol. 2006, 19, 559–564. [Google Scholar]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn2+: A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Hernandez, M.D.; Goik, S.A.; Morton, J.D.; McGinty, J.F. Loss of zinc staining from hippocampal mossy fibers during kainic acid induced seizures: A histofluorescence study. Brain Res. 1988, 446, 383–386. [Google Scholar]
- Koh, J.Y.; Choi, D.W. Zinc toxicity of cultured cortical neurons: Involvement of N-methyl-d-asparatate receptors. Neuroscience 1994, 4, 1049–1057. [Google Scholar] [CrossRef]
- Kim, A.H. L-type Ca2+ channel-mediated Zn2+ toxicity and modulation by ZnT-1 in PC12 cells. Brain Res. 2000, 886, 99–107. [Google Scholar] [CrossRef]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Canzoniero, L.M.; Yu, S.P.; Ying, H.S.; Koh, J.Y.; Kerchner, G.A.; Choi, D.W. Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J. Neurosci. 1997, 17, 9554–9564. [Google Scholar]
- Pellegrini-Giampietro, D.E.; Gorter, G.A.; Bennett, M.V.; Zukin, R.S. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar]
- Calderone, A.; Jover, T.; Mashiko, T.; Noh, K.; Tanaka, H.; Bennett, M.V.L.; Zukin, R.S. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J. Neurosci. 2004, 24, 9903–9913. [Google Scholar]
- Kawahara, M.; Kato-Negishi, M.; Kuroda, Y. Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell. Mol. Neurobiol. 2002, 22, 87–93. [Google Scholar] [CrossRef]
- Koyama, H.; Konoha, K.; Sadakane, Y.; Ohkawara, S.; Kawahara, M. Zinc neurotoxicity and the pathogenesis of vascular-type dementia: Involvement of calcium dyshomeostasis and carnosine. J. Clin. Toxicol. 2011, 3. [Google Scholar] [CrossRef]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar] [CrossRef]
- Mahesh, V.B.; Zamorano, P.; De Sevilla, L.; Lewis, D.; Brann, D.W. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1-7 cells). Neuroendocrinology 1999, 69, 397–407. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Hosoda, R.; Kuroda, Y. Characterization of zinc-induced apoptosis of GT1-7 cells. Biomed. Res. Trace Elem. 2002, 13, 280–281. [Google Scholar]
- Kawahara, M.; Konoha, K.; Sadakane, Y. Neurotoxicity of zinc: The involvement of calcium homeostasis and carnosine. Biomed. Res. Trace Elem. 2007, 18, 26–34. [Google Scholar]
- Kawahara, M.; Sadakane, Y.; Koyama, H.; Konoha, K.; Ohkawara, S. D-histidine and L-histidine attenuate zinc-induced neuronal death in GT1-7 cells. Metallomics 2013, 5, 453–460. [Google Scholar] [CrossRef]
- Mizuno, D.; Konoha-Mizuno, D.; Mori, M.; Sadakane, Y.; Koyama, H.; Ohkawara, S.; Kawahara, M. Protective activity of carnosine and anserine against zinc-induced neurotoxicity: A possible treatment for vascular dementia. Metallomics 2015, 7, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Effects of gadolinium and other metal on the neurotoxicity of immortalized hypothalamic neurons induced by zinc. Biomed. Res. Trace Elem. 2004, 15, 275–277. [Google Scholar]
- Koksal, A.R.; Verne, G.N.; Zhou, Q. Endoplasmic reticulum stress in biological processing and disease. J. Investig. Med. 2021, 69, 309–315. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Kasai, M.; Shimoda, M.; Shimizu, A.; Kubota, M.; Kawahara, M. Nickel enhances zinc-induced neuronal cell death by priming the endoplasmic reticulum stress response. Oxid. Med. Cell. Longev. 2019, 2019, 9693726. [Google Scholar] [CrossRef]
- Kyosseva, S.V. Mitogen-activated protein kinase signaling. Int. Rev. Neurobiol. 2004, 59, 201–220. [Google Scholar] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Sheline, C.T.; Behrens, M.M.; Choi, D.W. Zinc-induced cortical neuronal death: Contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 2000, 20, 3139–3154. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci. 2001, 21, RC171. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Shimoda, M.; Kawahara, M. Pyruvic acid prevents Cu2+/Zn2+-induced neurotoxicity by suppressing mitochondrial injury. Biochem. Biophys. Res. Commun. 2018, 495, 1335–1341. [Google Scholar] [CrossRef]
- Kim, E.Y.; Chang, S.Y.; Chung, J.M.; Ryu, B.R.; Joo, C.K.; Moon, H.S.; Kang, K.; Yoon, S.H.; Han, P.L.; Gwag, B.J. Attenuation of Zn2+ neurotoxicity by aspirin: Role of N-type Ca2+ channel and the carboxyl acid group. Neurobiol. Dis. 2001, 8, 774–783. [Google Scholar]
- Gulati, P.; Muthuraman, A.; Jaggi, A.S.; Singh, N. Neuroprotective effect of gadolinium: A stretch-activated calcium channel blocker in mouse model of ischemia-reperfusion injury. Naunyn Schmiedeberg’s Arch. Pharmacol. 2013, 386, 255–264. [Google Scholar] [CrossRef]
- Platt, B.; Büsselberg, D. Combined actions of Pb2+, Zn2+, and Al3+ on voltage-activated calcium channel currents. Cell. Mol. Neurobiol. 1994, 14, 831–840. [Google Scholar] [CrossRef]
- Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular sources of ROS/H2O2 in health and neurodegeneration: Spotlight on endoplasmic reticulum. Cells 2021, 10, 233. [Google Scholar] [CrossRef]
- Dent, P.; Yacoub, A.; Contessa, J.; Caron, R.; Amorino, G.; Valerie, K.; Hagan, M.P.; Grant, S.; Schmidt-Ullrich, R. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res. 2003, 159, 283–300. [Google Scholar] [CrossRef]
- Chang, C.C.; Kuan, C.P.; Lin, J.Y.; Lai, J.Y.; Ho, T.F. Tanshinone IIA facilitates TRAIL sensitization by up-regulating DR5 through the ROS-JNK-CHOP signaling axis in human ovarian carcinoma cell lines. Chem. Res. Toxicol. 2015, 28, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, G.; Fang, J. Association between serum copper and stroke risk factors in adults: Evidence from the National Health and Nutrition Examination Survey, 2011–2016. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef]
- Hu, L.; Bi, C.; Lin, T.; Liu, L.; Song, Y.; Wang, P.; Wang, B.; Fang, C.; Ma, H.; Huang, X.; et al. Association between plasma copper levels and first stroke: A community-based nested case-control study. Nutr. Neurosci. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, W.; Wang, Y.; Wang, T.; Ma, M.; Tian, C. Association between the change of serum copper and ischemic stroke: A systematic review and meta-analysis. J. Mol. Neurosci. 2020, 70, 475–480. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.-I. Amyloids: Regulators of metal homeostasis in the synapse. Molecules 2020, 25, 1441. [Google Scholar] [CrossRef]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Neuronal zinc regulation and the prion protein. Prion 2013, 7, 203–208. [Google Scholar] [CrossRef]
- Mellone, M.; Pelucchi, S.; Alberti, L.; Genazzani, A.A.; Luca, M.D.; Gardoni, F. Zinc transporter-1: A novel NMDA receptor-binding protein at the postsynaptic density. J. Neurochem. 2015, 132, 159–168. [Google Scholar] [CrossRef]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimer’s Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef] [PubMed]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.-M.; Sobott, F. Metal ions shape α-synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Rogers, J.T.; Cahill, C.M. Iron-responsive-like elements and neurodegenerative ferroptosis. Learn. Mem. 2020, 27, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Liu, G.; Zhao, Y.; Shi, Z.; Zheng, Q.; Bu, G.; Xu, H.; Zhang, Y. Role of copper and the copper-related protein CUTA in mediating APP processing and Aβ generation. Neurobiol. Aging 2015, 36, 1310–1315. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Cerpa, W.; Varela-Nallar, L. Copper brain homeostasis: Role of amyloid precursor protein and prion protein. IUBMB Life 2005, 57, 645–650. [Google Scholar] [CrossRef]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef]
- Posadas, Y.; Parra-Ojeda, L.; Perez-Cruz, C.; Quintanar, L. Amyloid β perturbs Cu(II) binding to the prion protein in a site-specific manner: Insights into its potential neurotoxic mechanisms. Inorg. Chem. 2021. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, L.; Yu, W.; Wang, Y.; Chang, W. Cellular prion protein as a receptor of toxic amyloid-β42 oligomers is important for Alzheimer’s disease. Front. Cell. Neurosci. 2019, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Yang, D.; Brenna, S.; Altmeppen, H.C.; Magnus, T. Show me your friends and I tell you who you are: The many facets of prion protein in stroke. Cells 2020, 9, 1609. [Google Scholar] [CrossRef]
- Black, S.A.G.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef]
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, D.S.; Stvolinsky, S.L.; Lopachev, A.V.; Devyatov, A.A.; Lopacheva, O.M.; Kulikova, O.I.; Abaimov, D.A.; Fedorova, T.N. Carnosine as an effective neuroprotector in brain pathology and potential neuromodulator in normal conditions. Amino Acids 2019, 51, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Trombley, P.Q.; Horning, M.S.; Blakemore, L.J. Interactions between carnosine and zinc and copper: Implications for neuromodulation and neuroprotection. Biochemistry 2000, 65, 807–816. [Google Scholar] [PubMed]
- Davis, C.K.; Laud, P.J.; Bahor, Z.; Rajanikant, G.K.; Majid, A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Godos, J.; Castellano, S.; Micek, A.; Murabito, P.; Galvano, F.; Ferri, R.; Grosso, G.; Caraci, F. The Therapeutic Potential of Carnosine/Anserine Supplementation against Cognitive Decline: A Systematic Review with Meta-Analysis. Biomedicines 2021, 9, 253. [Google Scholar] [CrossRef]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Drugs for Prevention or Treatment of Vascular Dementia. JP Patent 5,382,633, 11 October 2013. [Google Scholar]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Drugs for Prevention or Treatment of Vascular Dementia. JP Patent 5,294,194, 21 June 2013. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).