
The Role of Autophagy in Chemical Proteasome Inhibition Model of Retinal Degeneration




Abstract
:1. Introduction
2. Results
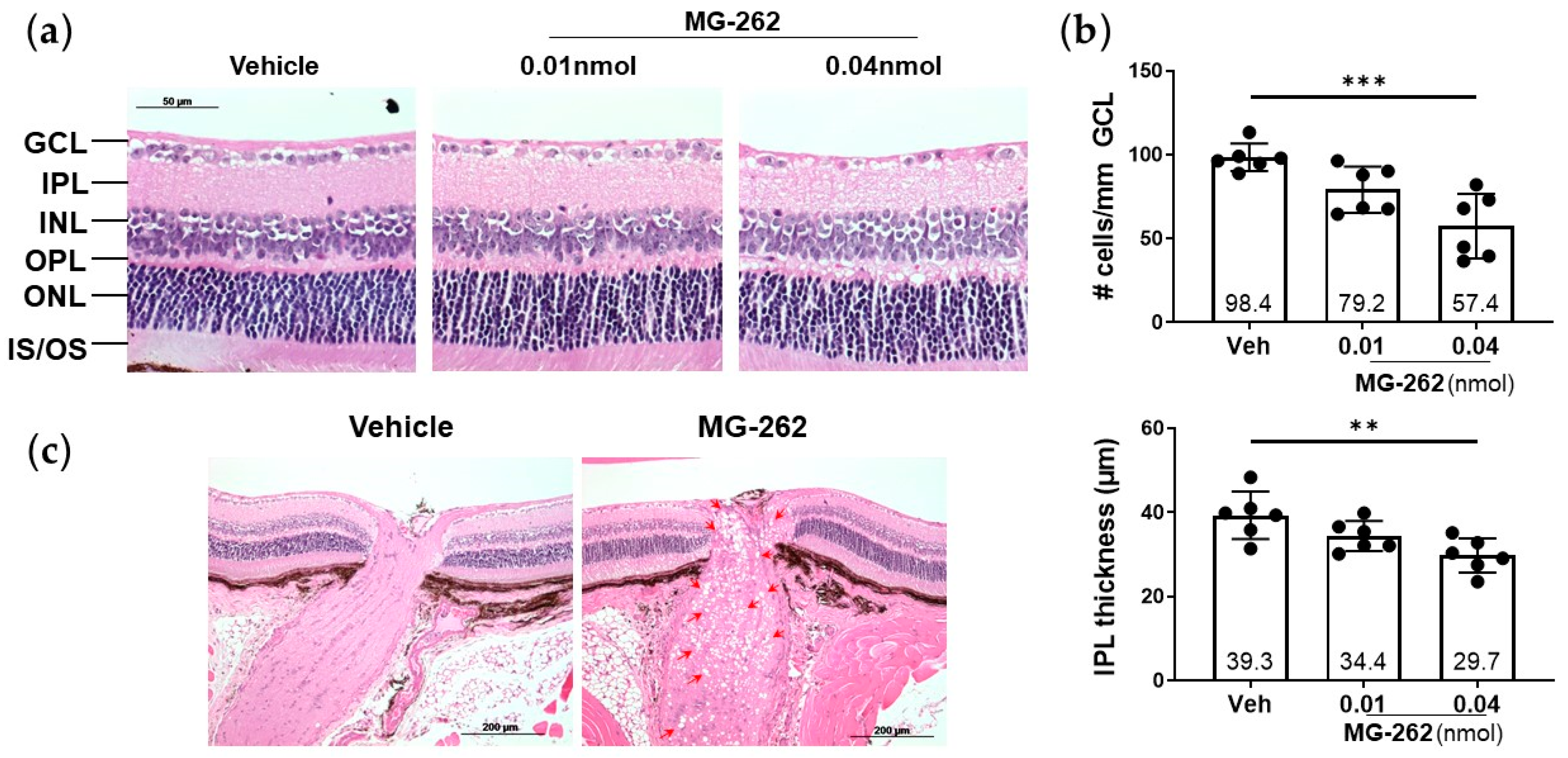
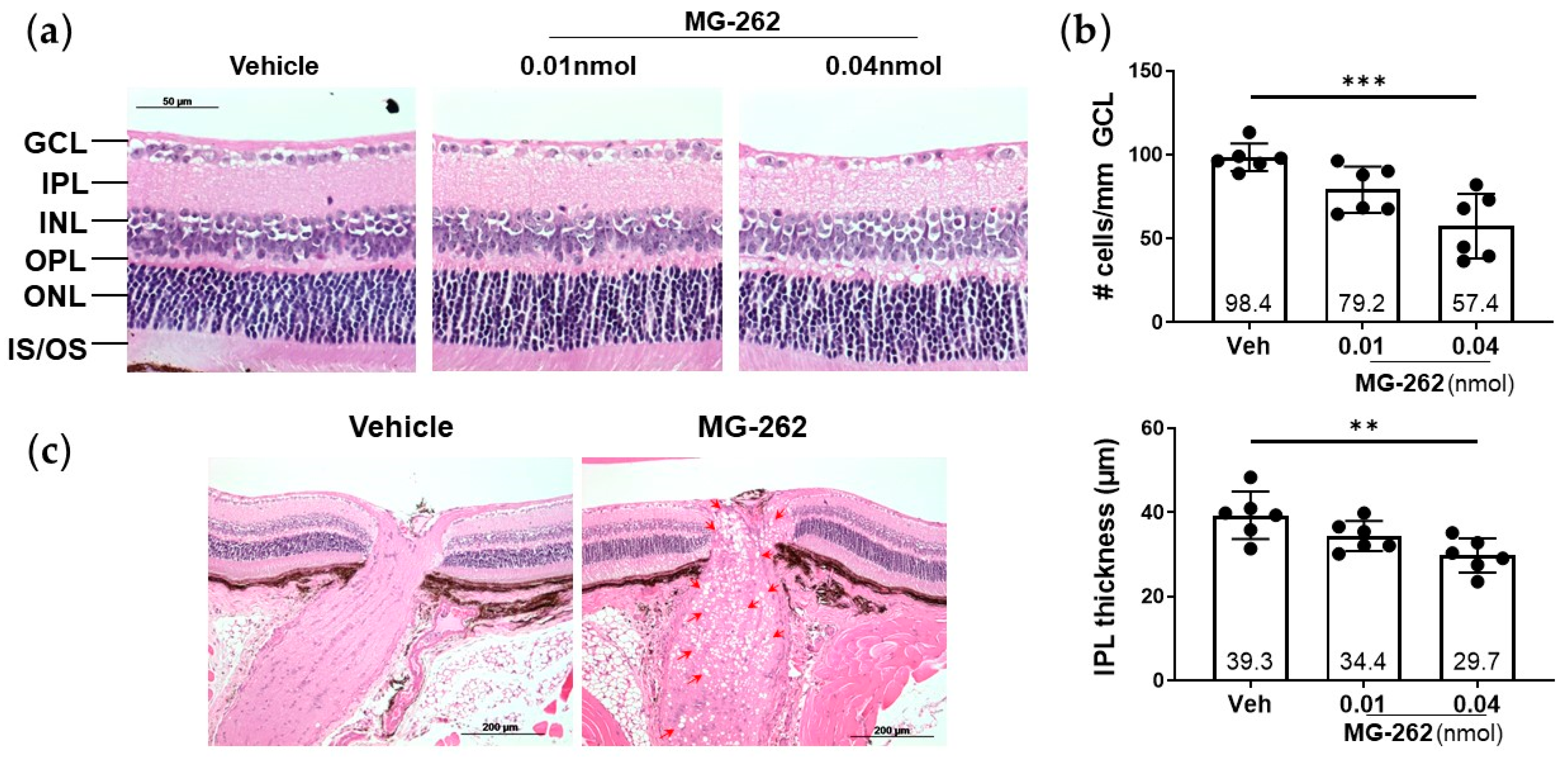
2.1. Intravitreal (IVT) Injection of a Proteasome Inhibitor Induced Inner Retinal Degeneration in Mice
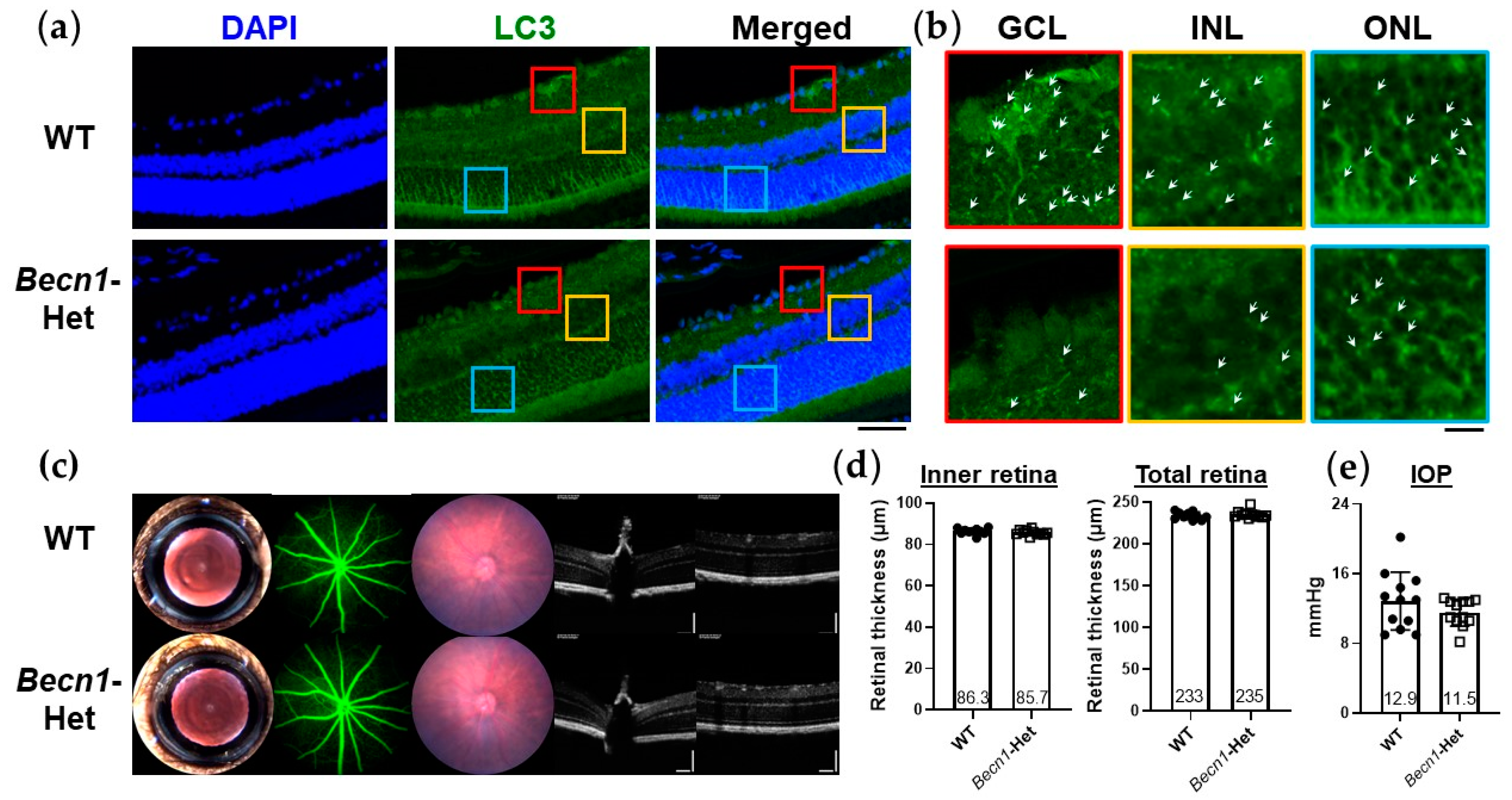
2.2. Characterization of Ocular Phenotypes of Beclin1-Heterozygous (Becn1-Het) Mice with Reduced Level of Retinal Autophagy
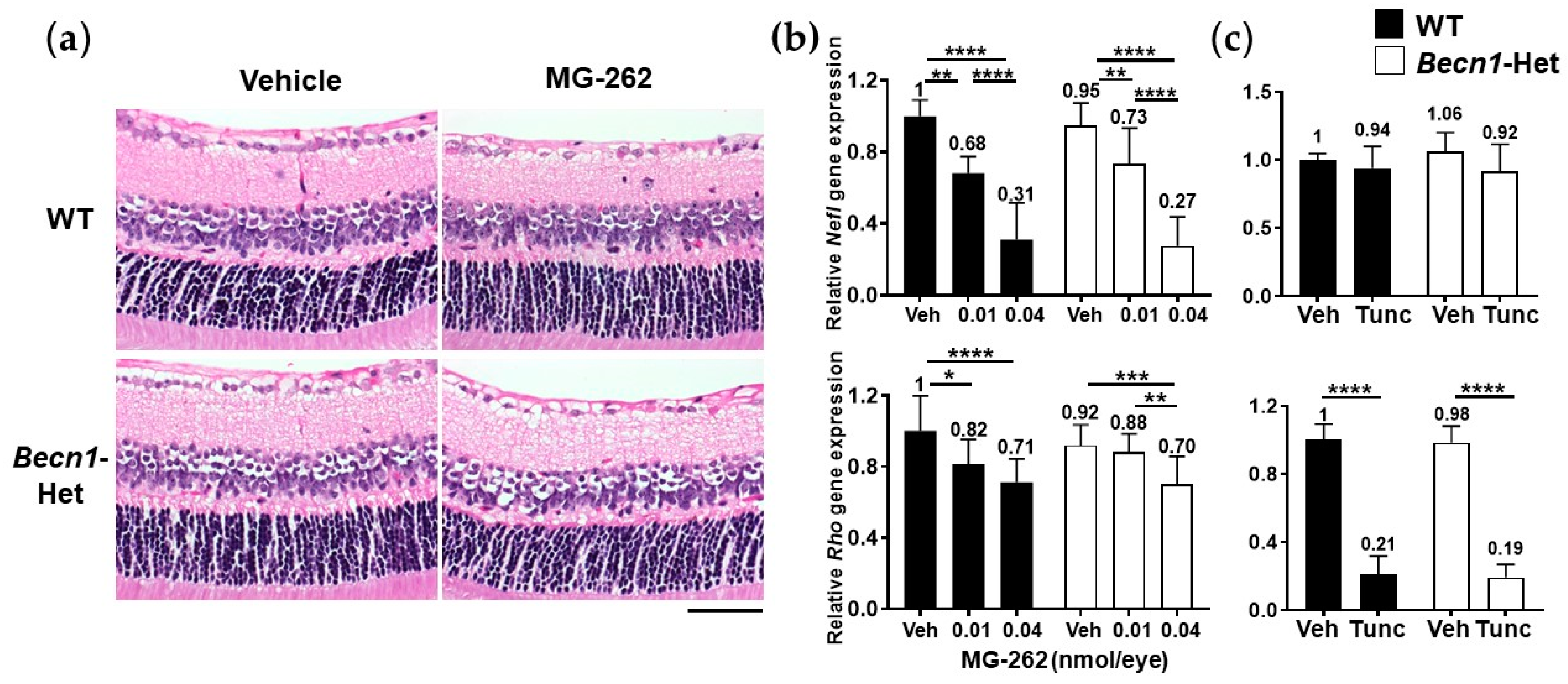
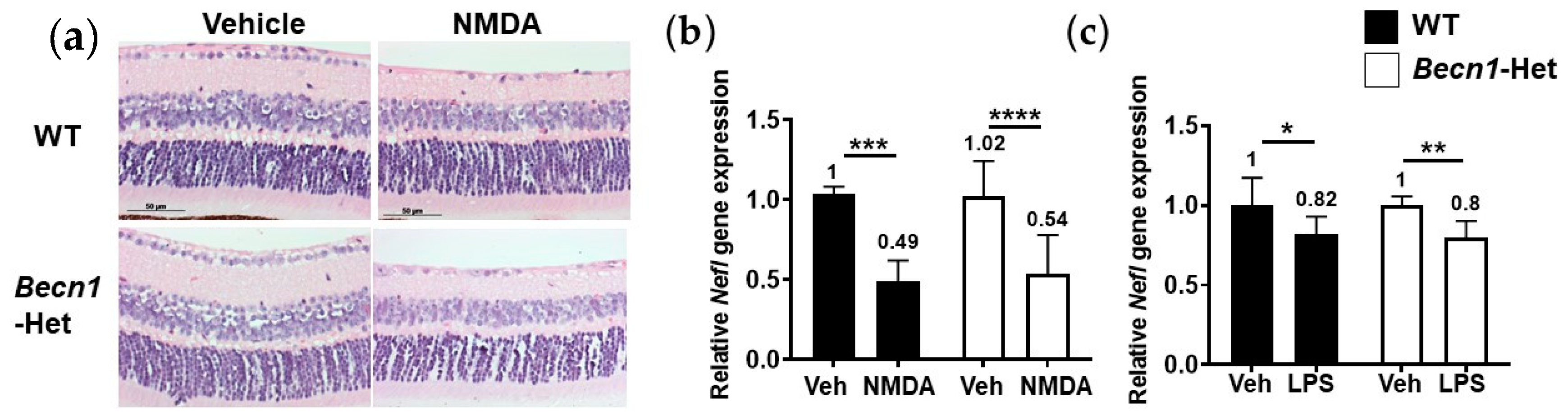
2.3. Becn1-Het and Wild-Type (WT) Mice Possess Equal Vulnerability to Proteasome Inhibition and Endoplasmic Reticulum (ER) Stress
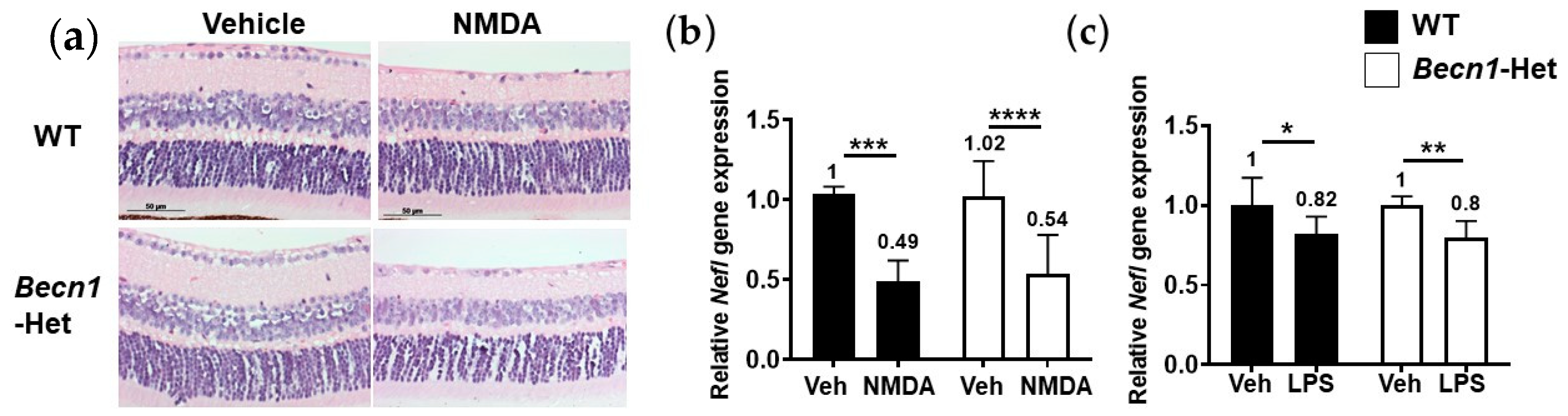
2.4. Retinal Degenerative Effects of Other Pharmacological Insults in Becn1-Het Mice
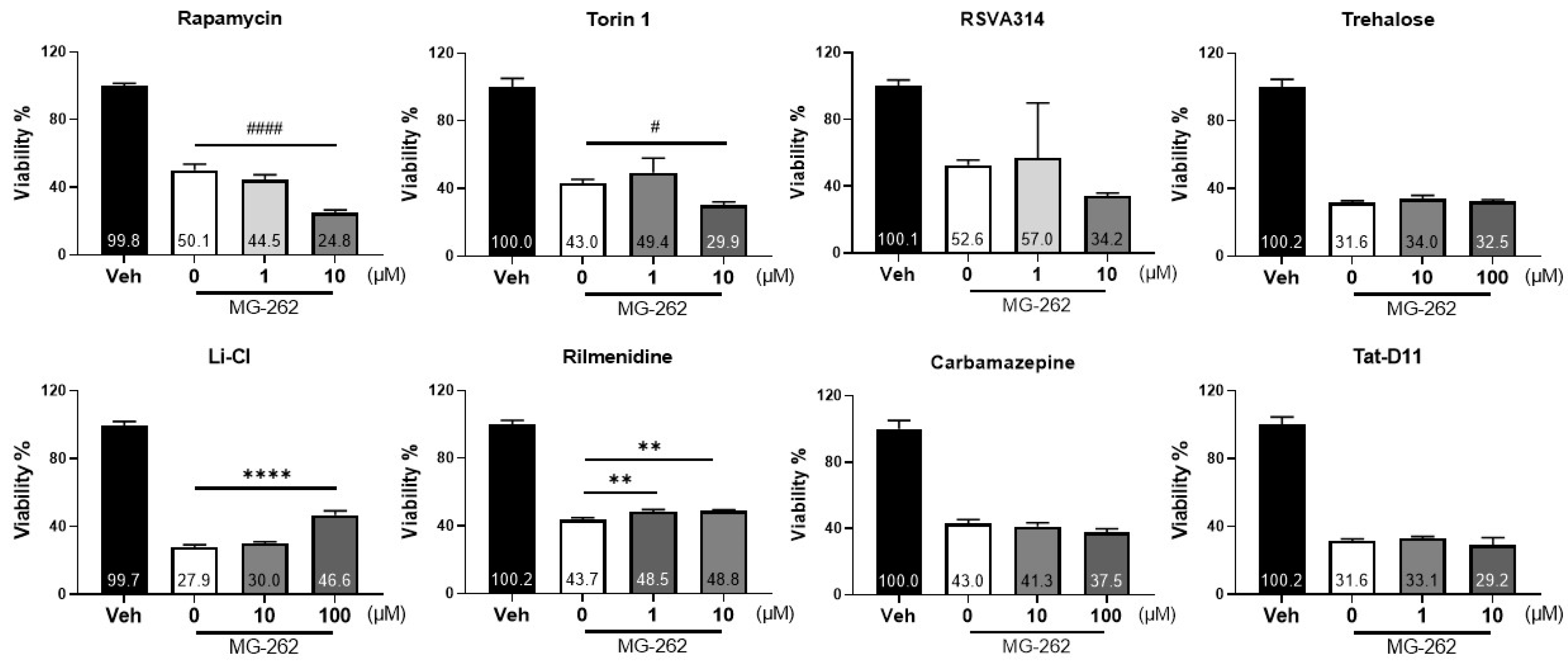
2.5. Specific Autophagy Activators Could Protect against MG-262-Induced Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Mice
4.3. IVT Injection
4.4. Intraocular Pressure (IOP) Measurement and Imaging
4.5. Histological Evaluation and Immunostaining
4.6. Quantitative PCR
4.7. Cell Culture and Viability Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, H.A.; Gonzalez, A.E.; Cerda-Troncoso, C.; Shaugnessy, R.; Otth, C.; Soza, A.; Burgos, P.V. Interplay between the autophagy-lysosomal pathway and the ubiquitin-proteasome system: A target for therapeutic development in Alzheimer’s disease. Front. Cell. Neurosci. 2018, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Raynes, R.; Pomatto, L.C.; Davies, K.J. Degradation of oxidized proteins by the proteasome: Distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol. Asp. Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.P.; Björklund, L.M.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. NeuroReport 2002, 13, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.P.; Belizaire, R.; Jenner, P.; Olanow, C.; Isacson, O. Selective loss of 20S proteasome α-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Xie, W.; Li, X.; Li, C.; Zhu, W.; Jankovic, J.; Le, W. Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. J. Neurochem. 2010, 115, 188–199. [Google Scholar] [CrossRef]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Arnaud, L.; Rockwell, P.; Figueiredo-Pereira, M.E. A single amino acid substitution in a proteasome subunit triggers aggregation of ubiquitinated proteins in stressed neuronal cells. J. Neurochem. 2004, 90, 19–28. [Google Scholar] [CrossRef]
- Lim, J.; Yue, Z. Neuronal Aggregates: Formation, Clearance, and Spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Ueno, T.; Waguri, S.; Uchiyama, Y.; Kominami, E.; Tanaka, K. Constitutive autophagy: Vital role in clearance of unfavorable proteins in neurons. Cell Death Differ. 2007, 14, 887–894. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Leger, F.; Fernagut, P.-O.; Canron, M.-H.; Léoni, S.; Vital, C.; Tison, F.; Bezard, E.; Vital, A. Protein Aggregation in the Aging Retina. J. Neuropathol. Exp. Neurol. 2011, 70, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapphahn, R.J.; Bigelow, E.J.; Ferrington, D.A. Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp. Eye Res. 2007, 84, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, J.L.; Kapphahn, R.J.; Ferrington, D.A. Proteasome function and protein oxidation in the aged retina. Exp. Eye Res. 2002, 75, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; De La Villa, P.; De La Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 2013, 12, 478–4882. [Google Scholar] [CrossRef] [Green Version]
- Lobanova, E.; Finkelstein, S.; Skiba, N.P.; Arshavsky, V.Y. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9986–9991. [Google Scholar] [CrossRef] [Green Version]
- Ando, R.; Noda, K.; Tomaru, U.; Kamoshita, M.; Ozawa, Y.; Notomi, S.; Hisatomi, T.; Noda, M.; Kanda, A.; Ishibashi, T.; et al. Decreased Proteasomal Activity Causes Photoreceptor Degeneration in Mice. Investig. Opthalmol. Vis. Sci. 2014, 55, 4682–4690. [Google Scholar] [CrossRef] [Green Version]
- Lobanova, E.; Finkelstein, S.; Li, J.; Travis, A.M.; Hao, Y.; Klingeborn, M.; Skiba, N.P.; Deshaies, R.J.; Arshavsky, V.Y. Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat. Commun. 2018, 9, 1738. [Google Scholar] [CrossRef]
- Besirli, C.; Chinskey, N.D.; Zheng, Q.-D.; Zacks, D.N. Autophagy Activation in the Injured Photoreceptor Inhibits Fas-Mediated Apoptosis. Investig. Opthalmol. Vis. Sci. 2011, 52, 4193–4199. [Google Scholar] [CrossRef] [Green Version]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and Autophagy in Photoreceptors Exposed to Oxidative Stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Rodríiguez-Muela, N.; Hernández-Pinto, A.M.; Serrano-Puebla, A.; Garcíaledo, L.; Latorre, S.; de la Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015, 22, 476–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A.; Grant, M.B.; Boulton, M.E. Autophagy in the Retina: A Potential Role in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2011, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef]
- Fernández-Albarral, J.A. The Role of Autophagy in Eye Diseases. Life 2021, 11, 189. [Google Scholar] [CrossRef]
- Kageyama, M.; Ota, T.; Sasaoka, M.; Katsuta, O.; Shinomiya, K. Chemical proteasome inhibition as a novel animal model of inner retinal degeneration in rats. PLoS ONE 2019, 14, e0217945. [Google Scholar] [CrossRef]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, Y. Beclin–phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimnah, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, B.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.-Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy Protects the Retina from Light-induced Degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, I.; Oglesby, E.; Buckingham, B.P.; Son, J.L.; Roberson, E.D.O.; Steele, M.R.; Inman, D.M.; Vetter, M.L.; Horner, P.J.; Marsh-Armstrong, N. Retinal Ganglion Cells Downregulate Gene Expression and Lose Their Axons within the Optic Nerve Head in a Mouse Glaucoma Model. J. Neurosci. 2008, 28, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Luna, R.I.M.-D.; Lou, C.-H.; Nekkalapudi, S.; Kelly, L.E.; Sater, A.K.; El-Hodiri, H.M. Regulation of photoreceptor gene expression by the retinal homeobox (Rx) gene product. Dev. Biol. 2010, 339, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, A.; Massieu, L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch. Med Res. 2006, 37, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Manev, H.; Favaron, M.; Guidotti, A.; Costa, E. Delayed increase of Ca2+ influx elicited by glutamate: Role in neuronal death. Mol. Pharmacol. 1989, 36, 106–112. [Google Scholar]
- Jang, S.; Lee, J.-H.; Choi, K.-R.; Kim, N.; Yoo, H.-S.; Oh, S. Cytochemical Alterations in the Rat Retina by LPS Administration. Neurochem. Res. 2006, 32, 1–10. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nat. Cell Biol. 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; d’Abramo, C.; Davies, P.; Marambaud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2011, 25, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, K.; Rosingol, I.; Sierra-Filardi, E.; Schmelter, C.; Grus, F.; Boya, P. Age related RGC vulnerability in an autophagy deficient mouse model. Investig. Ophthalmol. Vis. Sci. 2020, 61, 2360. [Google Scholar]
- Rodríguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2011, 19, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Varano, G.P.; Adornetto, A.; Nazio, F.; Tettamanti, G.; Girardello, R.; Cianfanelli, V.; Cavaliere, F.; Morrone, L.A.; Corasaniti, M.T.; et al. Rapamycin and fasting sustain autophagy response activated by ischemia/reperfusion injury and promote retinal ganglion cell survival. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting Axon Regeneration in the Adult CNS by Modulation of the PTEN/mTOR Pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.A.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Yao, J.; Jia, L.; Thompson, D.A.; Zacks, D.N. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin Is Neuroprotective in a Rat Chronic Hypertensive Glaucoma Model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef]
- Lim, J.-H.A.; Stafford, B.K.; Nguyen, B.K.S.P.L.; Lien, J.-H.A.L.B.V.; Wang, C.; Zukor, K.; He, C.W.Z.; Huberman, A.D. Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nat. Neurosci. 2016, 19, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Tang, L.; Wei, W.; Li, Z.; Li, Y.; Duan, X.; Chen, H. mTOR regulates neuroprotective effect of immunized CD4+Foxp3+ T cells in optic nerve ischemia. Sci. Rep. 2016, 6, 37805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, A.; Gianetto, D.; Conte, D.; Bosone, A.; Vercelli, A. Activation of Autophagy in a Rat Model of Retinal Ischemia following High Intraocular Pressure. PLoS ONE 2011, 6, e22514. [Google Scholar] [CrossRef] [Green Version]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology; Springer Science and Business Media LLC: New York, NY, USA, 2013; Volume 1078, pp. 9–21. [Google Scholar]
- Bell, M.; Zempel, H. SH-SY5Y-derived neurons: A human neuronal model system for investigating TAU sorting and neuronal subtype-specific TAU vulnerability. Rev. Neurosci. 2021. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Kato, Y.; Isobe, K.-i.; Tanaka, M.; Naoi, M.; Osawa, T. An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH-SY5Y cells. J. Neurosci. Res. 2003, 74, 589–597. [Google Scholar] [CrossRef]
- Cheng, B.; Maffi, S.K.; Martinez, A.A.; Acosta, Y.P.V.; Morales, L.D.; Roberts, J.L. Insulin-like growth factor-I mediates neuroprotection in proteasome inhibition-induced cytotoxicity in SH-SY5Y cells. Mol. Cell. Neurosci. 2011, 47, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.; Bruelle, C.; Scifo, E.; Pham, D.D.; Soliymani, R.; Lalowski, M.; Lindholm, D. Dynamic Interaction of USP14 with the Chaperone HSC70 Mediates Crosstalk between the Proteasome, ER Signaling, and Autophagy. iScience 2020, 23, 100790. [Google Scholar] [CrossRef] [Green Version]
- Pantazopoulou, M.; Brembati, V.; Kanellidi, A.; Bousset, L.; Melki, R.; Stefanis, L. Distinct alpha-Synuclein species induced by seeding are selectively cleared by the Lysosome or the Proteasome in neuronally differentiated SH-SY5Y cells. J. Neurochem. 2021, 156, 880–896. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renna, M. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative dis-eases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [Green Version]
- Williams, A. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; A Fiebig, A.; Andrews, D.; Molgó, J.; Díaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and Autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Xia, H.-G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, N.A. iDrugs and iDevices Discovery Research: Preclinical Assays, Techniques, and Animal Model Studies for Ocular Hypotensives and Neuroprotectants. J. Ocul. Pharmacol. Ther. 2018, 34, 7–39. [Google Scholar] [CrossRef]
- Sharif, N.A. Glaucomatous optic neuropathy treatment options: The promise of novel therapeutics, techniques and tools to help preserve vision. Neural Regen. Res. 2018, 13, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, M.; Kageyama, M.; Ohashi, K.; Sasaoka, M.; Sato, R.; Tanaka, M.; Tashiro, K. Nafamostat and sepimostat identified as novel neuroprotective agents via NR2B N-methyl-D-aspartate receptor antagonism using a rat retinal excitotoxicity model. Sci. Rep. 2019, 9, 20409–20412. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, M.; Ota, T.; Kageyama, M. Rotenone-induced inner retinal degeneration via presynaptic activation of voltage-dependent sodium and L-type calcium channels in rats. Sci. Rep. 2020, 10, 1–16. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| neurofilament light chain (Nefl) | CCGTACTTTTCGACCTCCTACA | CTTGTGTGCGGATAGACTTGAG |
| rhodopsin (Rho) | CCCTTCTCCAACGTCACAGG | TGAGGAAGTTGATGGGGAAGC |
| glyceraldehyde-3-hosphate dehydrogenase (Gapdh) 1 | TGGCCTTCCGTGTTCCTAC | GAGTTGCTGTTGAAGTCGCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunawan, M.; Low, C.; Neo, K.; Yeo, S.; Ho, C.; Barathi, V.A.; Chan, A.S.; Sharif, N.A.; Kageyama, M. The Role of Autophagy in Chemical Proteasome Inhibition Model of Retinal Degeneration. Int. J. Mol. Sci. 2021, 22, 7271. https://doi.org/10.3390/ijms22147271
Gunawan M, Low C, Neo K, Yeo S, Ho C, Barathi VA, Chan AS, Sharif NA, Kageyama M. The Role of Autophagy in Chemical Proteasome Inhibition Model of Retinal Degeneration. International Journal of Molecular Sciences. 2021; 22(14):7271. https://doi.org/10.3390/ijms22147271
Chicago/Turabian StyleGunawan, Merry, Choonbing Low, Kurt Neo, Siawey Yeo, Candice Ho, Veluchamy A. Barathi, Anita Sookyee Chan, Najam A. Sharif, and Masaaki Kageyama. 2021. "The Role of Autophagy in Chemical Proteasome Inhibition Model of Retinal Degeneration" International Journal of Molecular Sciences 22, no. 14: 7271. https://doi.org/10.3390/ijms22147271
APA StyleGunawan, M., Low, C., Neo, K., Yeo, S., Ho, C., Barathi, V. A., Chan, A. S., Sharif, N. A., & Kageyama, M. (2021). The Role of Autophagy in Chemical Proteasome Inhibition Model of Retinal Degeneration. International Journal of Molecular Sciences, 22(14), 7271. https://doi.org/10.3390/ijms22147271