
Living Proof of Activity of Extracellular Vesicles in the Central Nervous System

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Endogenous EVs in the CNS and Neurodevelopment
2.1. Neuron–Astrocyte Communication
2.2. Synaptic Development and Signaling
2.3. Oligodendrocyte-Derived EVs in the CNS
2.4. EVs in Neurodevelopment
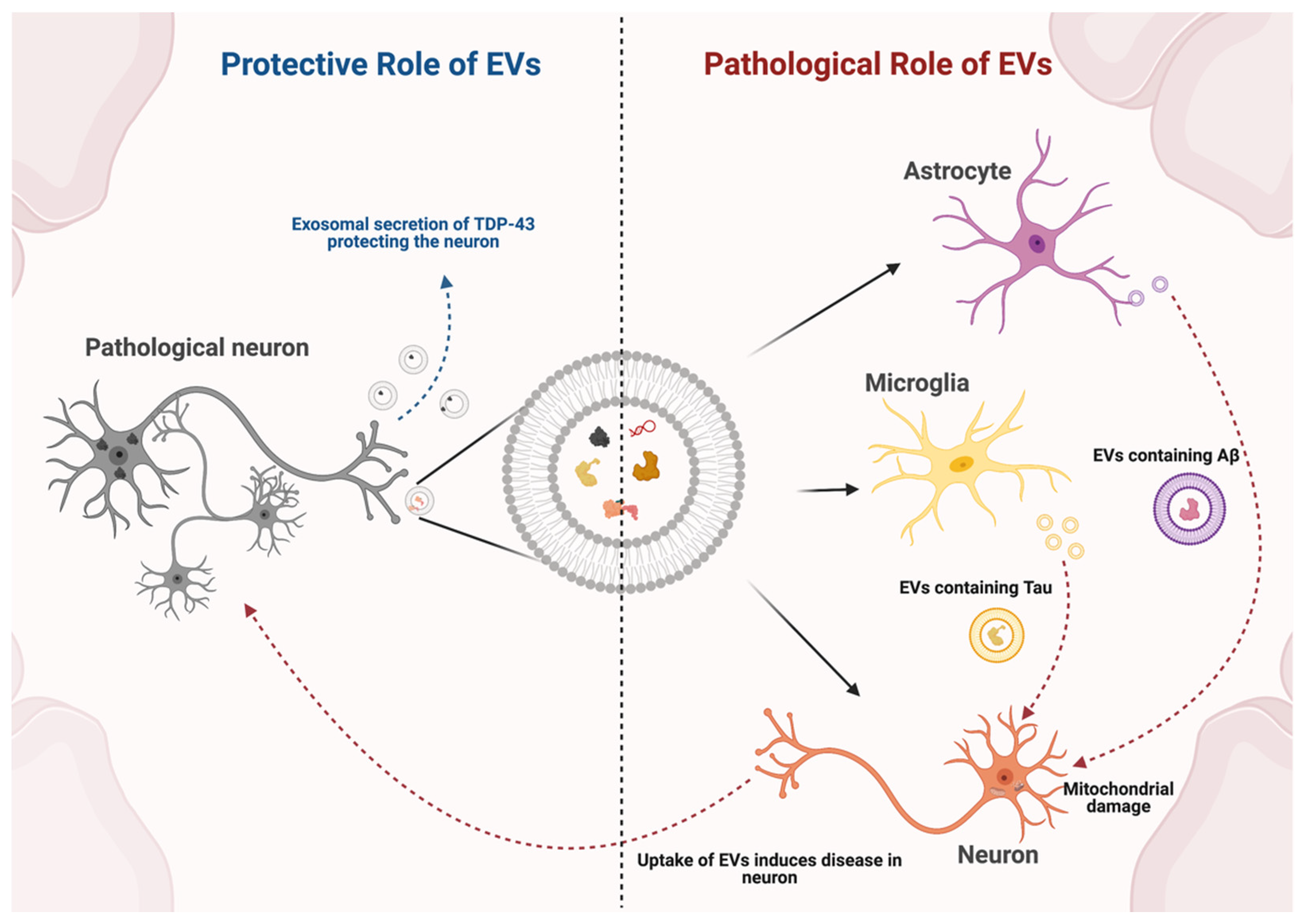
3. Role of Endogenous EVs in Neurodegenerative Disease
3.1. Parkinson’s Disease
3.2. Alzheimer’s Disease
3.3. Amyotrophic Lateral Sclerosis
4. Stroke and Traumatic Brain Injury
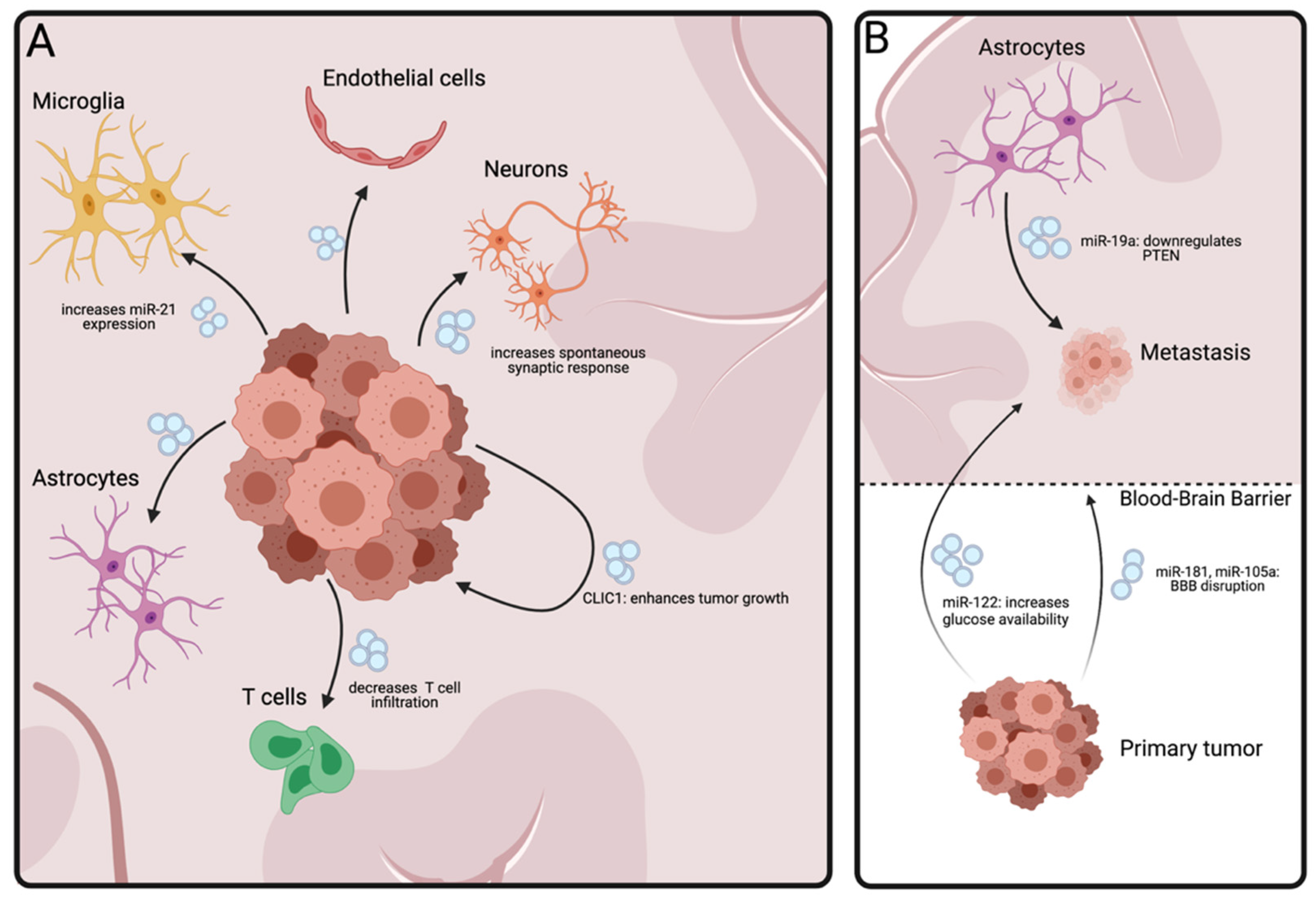
5. Brain Tumors
5.1. Glial Tumors
5.2. Brain Metastasis
6. Conclusions and Future Prospects
7. Material and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garden, G.A.; La Spada, A.R. Intercellular (Mis)communication in Neurodegenerative Disease. Neuron 2012, 73, 886–901. [Google Scholar] [CrossRef] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Gasnier, B. The loading of neurotransmitters into synaptic vesicles. Biochimie 2000, 82, 327–337. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The synaptic vesicle cycle revisited: New insights into the modes and mechanisms. J. Neurosci. Soc. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal Exosomal miRNA-dependent Translational Regulation of Astroglial Glutamate Transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [Green Version]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte-Neuron Communication. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012, 31, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; De Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. Precursor Protein Carboxyl-terminal Fragments from the Cell into the Brain Extracellular Space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, L.J.; Scicluna, B.J.; Cheng, L.; Bawden, E.G.; Masters, C.L.; Ang, C.S.; Willamson, N.; McLean, C.; Barnham, K.J.; Hill, A.F. A rigorous method to enrich for exosomes from brain tissue. J. Extracell. Vesicles 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gallart-Palau, X.; Serra, A.; Sze, S.K. Enrichment of extracellular vesicles from tissues of the central nervous system by PROSPR. Mol. Neurodegener. 2016, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Badhwar, A.P.; Haqqani, A.S. Biomarker potential of brain-secreted extracellular vesicles in blood in Alzheimer’s disease. Alzheimer’s Dement. Diag. Assess. Dis. Monit. 2020, 12. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Andrews, A.M.; Paul, D.; Pachter, J.S. Extracellular vesicles: Mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 2018, 15, 1–21. [Google Scholar] [CrossRef]
- Record, M.; Subra, C.; Silvente-Poirot, S.; Poirot, M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 2011, 81, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalheiser, N.R. Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol. Direct 2007, 2, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bakhti, M.; Winter, C.; Simons, M. Inhibition of myelin membrane sheath formation by oligodendrocyte-derived exosome-like vesicles. J. Biol. Chem. 2011, 286, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Feliciano, D.M.; Zhang, S.; Nasrallah, C.M.; Lisgo, S.N.; Bordey, A. Embryonic cerebrospinal fluid nanovesicles carry evolutionarily conserved molecules and promote neural stem cell amplification. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, J.; Choi, Y.K. The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Rimmele, T.S.; Rosenberg, P.A. GLT-1: The elusive presynaptic glutamate transporter. Neurochem. Int. 2016, 98, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Malmevik, J.; Petri, R.; Knauff, P.; Brattås, P.L.; Åkerblom, M.; Jakobsson, J. Distinct cognitive effects and underlying transcriptome changes upon inhibition of individual miRNAs in hippocampal neurons. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Vidensky, S.; Jin, L.; Jie, C.; Lorenzini, I.; Frankl, M.; Rothstein, J.D. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia 2011, 59, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Koles, K.; Nunnari, J.; Korkut, C.; Barria, R.; Brewer, C.; Li, Y.; Leszyk, J.; Zhang, B.; Budnik, V. Mechanism of evenness interrupted (Evi)-exosome release at synaptic boutons. J. Biol. Chem. 2012, 287, 16820–16834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patapoutian, A.; Reichardt, L.F. Roles of Wnt proteins in neural development and maintenance. Curr. Opin. Neurobiol. 2000, 10, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Frühbeis, C.; Kuo-Elsner, W.P.; Barth, K.; Peris, L.; Tenzer, S.; Möbius, W.; Werner, H.B.; Nave, K.A.; Fröhlich, D.; Krämer-Albers, E.M. Oligodendrocyte-derived exosomes promote axonal transport and axonal long-term maintenance. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Mensch, S.; Baraban, M.; Almeida, R.; Czopka, T.; Ausborn, J.; El Manira, A.; Lyons, D.A. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 2015, 18, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, I.; Klugmann, M.; Anderson, T.; Yool, D.; Thomson, C.; Schwab, M.H.; Schneider, A.; Zimmermann, F.; McCulloch, M.; Nadon, N.; et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mesci, P.; Carromeu, C.; McClatchy, D.R.; Schiapparelli, L.; Yates, J.R.; Muotri, A.R.; Cline, H.T. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 16086–16094. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Elsherbini, A.; Kirov, A.S.; Dinkins, M.B.; Wang, G.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. Association of A with ceramide-enriched astrosomes mediates A neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 60. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef]
- You, Y.; Ikezu, T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol. Dis. 2019, 104512. [Google Scholar] [CrossRef]
- Maniecka, Z.; Polymenidou, M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res. 2015, 207, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, S.; Kim, W.H.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Kim, C.; Gonzalez, T.; Bisquertt, A.; Patrick, C.; Rockenstein, E.; Adame, A.; Lee, S.J.; Desplats, P.; Masliah, E. α-synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum. Mol. Genet. 2015, 25, 1100–1115. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.; Sekiguchi, M.; Akagi, T.; Izumi, S.; Komori, T.; Hui, K.; Sörgjerd, K.; Tanaka, M.; Saito, T.; Iwata, N.; et al. Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the golgi. Am. J. Pathol. 2015, 185, 305–313. [Google Scholar] [CrossRef]
- Han, J.-H.; Ryu, H.-H.; Jun, M.-H.; Jang, D.-J.; Lee, J.-A. The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem. Biophys. Res. Commun. 2012, 421, 544–549. [Google Scholar] [CrossRef]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xu, Y.; Deng, W.; Zhang, L.; Zhu, H.; Yu, P.; Qu, Y.; Zhao, W.; Han, Y.; Qin, C. Brain Derived Exosomes Are a Double-Edged Sword in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse Formation and Function Is Modulated by the Amyloid Precursor Protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef]
- Albayram, O.; Angeli, P.; Bernstein, E.; Baxley, S.; Gao, Z.; Ping Lu, K.; Zhou, X.Z. Targeting Prion-like Cis Phosphorylated Tau Pathology in Neurodegenerative Diseases. J. Alzheimer’s Dis. Park. 2018, 8. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.; Polanco, J.C.; Götz, J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. J. Alzheimer’s Dis. 2016, 54, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Wolozin, B.; Butovsky, O.; Ikezu, T.; Therapeutics, E. Depletion of microglia and inhibition of exosome synthe. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction invivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [Green Version]
- Elsherbini, A.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. In vivo evidence of exosome-mediated Aβ neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Lackland, D.T.; Roccella, E.J.; Deutsch, A.F.; Fornage, M.; George, M.G.; Howard, G.; Kissela, B.M.; Kittner, S.J.; Lichtman, J.H.; Lisabeth, L.D.; et al. Factors influencing the decline in stroke mortality a statement from the american heart association/american stroke association. Stroke 2014, 45, 315–353. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Chopp, M. Exosomes in stroke pathogenesis and therapy. J. Clin. Investig. 2016, 126, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Chiva-Blanch, G.; Suades, R.; Crespo, J.; Peña, E.; Padró, T.; Jiménez-Xarrié, E.; Martí-Fàbregas, J.; Badimon, L. Microparticle shedding from neural progenitor cells and vascular compartment cells is increased in ischemic stroke. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Couch, Y.; Akbar, N.; Davis, S.; Fischer, R.; Dickens, A.M.; Neuhaus, A.A.; Burgess, A.I.; Rothwell, P.M.; Buchan, A.M. Inflammatory Stroke Extracellular Vesicles Induce Macrophage Activation. Stroke 2017, 48, 2292–2296. [Google Scholar] [CrossRef]
- Bang, O.Y.; Kim, E.H. Mesenchymal Stem Cell-Derived Extracellular Vesicle Therapy for Stroke: Challenges and Progress. Front. Neurol. 2019, 10, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenz, K.T.; Just, J.; Blauenfeldt, R.A.; Drasbek, K.R. Extracellular Vesicles in Acute Stroke Diagnostics. Biomedicines 2020, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Beard, K.; Meaney, D.F.; Issadore, D. Clinical Applications of Extracellular Vesicles in the Diagnosis and Treatment of Traumatic Brain Injury. J. Neurotrauma 2020, 37, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.B.; Hochfelder, C.G.; Lamberty, B.G.; Meays, B.M.; Morsey, B.M.; Kelso, M.L.; Fox, H.S.; Yelamanchili, S.V. Traumatic brain injury increases levels of miR-21 in extracellular vesicles: Implications for neuroinflammation. FEBS Open Bio 2016, 6, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Buller, B.; Liu, X.; Wang, X.; Zhang, R.L.; Zhang, L.; Hozeska-Solgot, A.; Chopp, M.; Zhang, Z.G. MicroRNA-21 protects neurons from ischemic death. FEBS J. 2010, 277, 4299–4307. [Google Scholar] [CrossRef] [Green Version]
- Bhalala, O.G.; Pan, L.; Sahni, V.; McGuire, T.L.; Gruner, K.; Tourtellotte, W.G.; Kessler, J.A. microRNA-21 regulates astrocytic response following spinal cord injury. J. Neurosci. 2012, 32, 17935–17947. [Google Scholar] [CrossRef]
- Schulz, M.; Salamero-Boix, A.; Niesel, K.; Alekseeva, T.; Sevenich, L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019, 10, 1713. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro. Oncol. 2019, 21, V1–V100. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Zeng, A.; Wei, Z.; Rabinovsky, R.; Jun, H.J.; El Fatimy, R.; Deforzh, E.; Arora, R.; Yao, Y.; Yao, S.; Yan, W.; et al. Glioblastoma-Derived Extracellular Vesicles Facilitate Transformation of Astrocytes via Reprogramming Oncogenic Metabolism. iScience 2020, 23. [Google Scholar] [CrossRef] [PubMed]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [Green Version]
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Brescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.I.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Acharya, S.; Gravanis, I.; Mehmood, S.; Seidman, R.J.; Shroyer, K.R.; Hajjar, K.A.; Tsirka, S.E. Annexin A2 promotes glioma cell invasion and tumor progression. J. Neurosci. 2011, 31, 14346–14360. [Google Scholar] [CrossRef] [Green Version]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre Repooting and characterization. Nat Neurosci 2010, 13, 133–140. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Z.; Mashimo, T.; Shen, B.; Nyagilo, J.; Wang, H.; Wang, Y.; Liu, Z.; Mulgaonkar, A.; Hu, X.L.; et al. Gliomas Interact with Non-glioma Brain Cells via Extracellular Vesicles. Cell Rep. 2020, 30, 2489–2500.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Myskiw, C.; Rommel, A.; Verma, I.M. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J. Mol. Med. 2013, 91, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, H.L.; Hu, G.W.; Zhang, B.; Kuang, W.; Chen, Y.; Wu, L.; Xu, G.H. Glioma cells enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep. 2017, 38, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef] [Green Version]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-derived miRNA-containing extracellular vesicles induce angiogenesis by reprogramming brain endothelial cells. Cell Rep. 2020, 30, 2065–2074.e4. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Lan, F.; Xia, T. Hypoxic Glioma Cell-Secreted Exosomal miR-301a Activates Wnt/β-catenin Signaling and Promotes Radiation Resistance by Targeting TCEAL7. Mol. Ther. 2019, 27, 1939–1949. [Google Scholar] [CrossRef]
- Abels, E.R.; Maas, S.L.N.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119.e7. [Google Scholar] [CrossRef] [Green Version]
- Van Der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.W.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro. Oncol. 2016, 18, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of Fractalkine Receptor CX 3 CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.M.; Wang, Y.B.; Yuan, X.H. Exosomes from murine-derived Gl26 cells promote glioblastoma tumor growth by reducing number and function of CD8+T cells. Asian Pac. J. Cancer Prev. 2013, 14, 309–314. [Google Scholar] [CrossRef] [Green Version]
- van Solinge, T.S.; Abels, E.R.; van de Haar, L.L.; Hanlon, K.S.; Maas, S.L.N.; Schnoor, R.; de Vrij, J.; Breakefield, X.O.; Broekman, M.L.D. Versatile Role of Rab27a in Glioma: Effects on Release of Extracellular Vesicles, Cell Viability, and Tumor Progression. Front. Mol. Biosci. 2020, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Lee, E.Q.; Wen, P.Y. Epidemiology of brain metastases. Curr. Oncol. Rep. 2012, 14, 48–54. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, L.; Li, L.; Cao, Y. Exosomes Derived from Brain Metastatic Breast Cancer Cells Destroy the Blood-Brain Barrier by Carrying lncRNA GS1-600G8.5. Biomed Res. Int. 2020, 2020. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted Deletion Reveals Essential and Overlapping Functions of the miR-17∼92 Family of miRNA Clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahjoum, S.; Rufino-Ramos, D.; Pereira de Almeida, L.; Broekman, M.L.D.; Breakefield, X.O.; van Solinge, T.S. Living Proof of Activity of Extracellular Vesicles in the Central Nervous System. Int. J. Mol. Sci. 2021, 22, 7294. https://doi.org/10.3390/ijms22147294
Mahjoum S, Rufino-Ramos D, Pereira de Almeida L, Broekman MLD, Breakefield XO, van Solinge TS. Living Proof of Activity of Extracellular Vesicles in the Central Nervous System. International Journal of Molecular Sciences. 2021; 22(14):7294. https://doi.org/10.3390/ijms22147294
Chicago/Turabian StyleMahjoum, Shadi, David Rufino-Ramos, Luís Pereira de Almeida, Marike L. D. Broekman, Xandra O. Breakefield, and Thomas S. van Solinge. 2021. "Living Proof of Activity of Extracellular Vesicles in the Central Nervous System" International Journal of Molecular Sciences 22, no. 14: 7294. https://doi.org/10.3390/ijms22147294
APA StyleMahjoum, S., Rufino-Ramos, D., Pereira de Almeida, L., Broekman, M. L. D., Breakefield, X. O., & van Solinge, T. S. (2021). Living Proof of Activity of Extracellular Vesicles in the Central Nervous System. International Journal of Molecular Sciences, 22(14), 7294. https://doi.org/10.3390/ijms22147294