
Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression

 , and
, and
Abstract
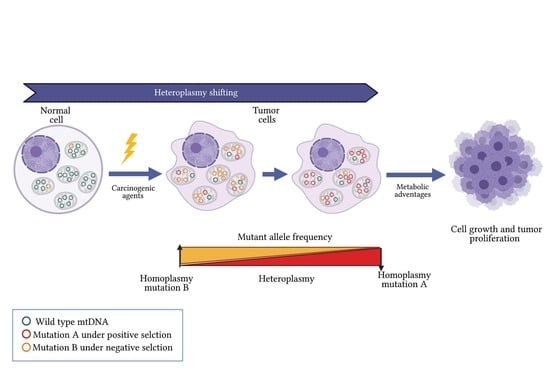
1. Introduction
2. Mitochondrial Genome
3. Origins of Heteroplasmy and Its Impact on Disease
4. Mitochondrial Alterations in Cancer
5. Heteroplasmy Shifting in Cancer: Potentially Unique Profiles
6. Tumor Microenvironment and Heteroplasmy
7. Heteroplasmy as a Potential Biomarker of Development and Progression in Cancer
8. Heteroplasmy in Epigenetic Modulation
9. Heteroplasmy Shifting as Therapeutic Strategy
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, L.; Li, Y.; Wu, Y.; Li, X.; Wu, X. Four long noncoding RNAs act as biomarkers in lung adenocarcinoma. Open Med. 2021, 16, 660–671. [Google Scholar] [CrossRef]
- Ghafarpour, V.; Khansari, M.; Banaei-Moghaddam, A.M.; Najafi, A.; Masoudi-Nejad, A. DNA methylation association with stage progression of head and neck squamous cell carcinoma. Comput. Biol. Med. 2021, 134, 104473. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Hägg, S.; Jylhävä, J.; Wang, Y.; Czene, K.; Grassmann, F. Correction to: Deciphering the genetic and epidemiological landscape of mitochondrial DNA abundance. Qual. Life Res. 2021, 140, 863. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Basu, S.; Xie, X.; Uhler, J.P.; Hedberg-Oldfors, C.; Milenkovic, D.; Baris, O.R.; Kimoloi, S.; Matic, S.; Stewart, J.B.; Larsson, N.-G.; et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLoS Genet. 2020, 16, e1009242. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Rao, S.G. Mitochondrial Changes in Cancer. Organotypic Models Drug Dev. 2016, 240, 211–227. [Google Scholar] [CrossRef]
- Bussard, K.M.; Siracusa, L.D. Understanding Mitochondrial Polymorphisms in Cancer. Cancer Res. 2017, 77, 6051–6059. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martin, S.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Li, J.; Zhang, W.; Liao, Y.; Chen, J.; Sun, Z. Correlational study on mitochondrial DNA mutations as potential risk factors in breast cancer. Oncotarget 2016, 7, 31270–31283. [Google Scholar] [CrossRef]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 2017, 26, 2912–2922. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240. [Google Scholar] [CrossRef]
- Tikochinski, Y.; Carreras, C.; Tikochinski, G.; Vilaca, S.T. Population-specific signatures of intra-individual mitochondrial DNA heteroplasmy and their potential evolutionary advantages. Sci. Rep. 2020, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wei, Y.; Wang, Q.; Xu, H.; Wang, Y.; Yao, A.; Yang, H.; Gao, Y.; Zhou, F. Heteroplasmy of mutant mitochondrial DNA A10398G and analysis of its prognostic value in non-small cell lung cancer. Oncol. Lett. 2016, 12, 3081–3088. [Google Scholar] [CrossRef]
- Fendt, L.; Fazzini, F.; Weissensteiner, H.; Bruckmoser, E.; Schönherr, S.; Schäfer, G.; Losso, J.L.; Streiter, G.A.; Lamina, C.; Rasse, M.; et al. Profiling of Mitochondrial DNA Heteroplasmy in a Prospective Oral Squamous Cell Carcinoma Study. Cancers 2020, 12, 1933. [Google Scholar] [CrossRef]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic Mutations throughout the Entire Mitochondrial Genome Are Associated with Elevated PSA Levels in Prostate Cancer Patients. Am. J. Hum. Genet. 2010, 87, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol. 1975, 29, 21–38. [Google Scholar]
- Yamauchi, A. Rate of Gene Transfer From Mitochondria to Nucleus: Effects of Cytoplasmic Inheritance System and Intensity of Intracellular Competition. Genetics 2005, 171, 1387–1396. [Google Scholar] [CrossRef][Green Version]
- Anderson, S.E.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef]
- Hou, X.-S.; Wang, H.-S.; Mugaka, B.P.; Yang, G.-J.; Ding, Y. Mitochondria: Promising organelle targets for cancer diagnosis and treatment. Biomater. Sci. 2018, 6, 2786–2797. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, R.; Tedone, E.; Ludlow, A.T.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Dong, J.; Wong, L.-J.; Mims, M.P. Mitochondrial inheritance and cancer. Transl. Res. 2018, 202, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Lajbner, Z.; Pnini, R.; Camus, M.F.; Miller, J.; Dowling, D. Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci. Rep. 2018, 8, 9500. [Google Scholar] [CrossRef] [PubMed]
- Motoi, M.; Nishimura, T.; Egashira, Y.; Kishida, F.; Watanuki, S. Relationship between mitochondrial haplogroup and physiological responses to hypobaric hypoxia. J. Physiol. Anthr. 2016, 35, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gong, L.; Liu, X.; Chen, X.; Yang, S.; Luo, Y. Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Toncheva, D.; Serbezov, D.; Karachanak-Yankova, S.; Nesheva, D. Ancient mitochondrial DNA pathogenic variants putatively associated with mitochondrial disease. PLoS ONE 2020, 15, e0233666. [Google Scholar] [CrossRef]
- Xiao, F.; Li, M.; Wang, J.; Liu, J.; Li, J.; Fang, H.; Lyu, J.; Shen, L. Association between mitochondrial DNA haplogroup variation and coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 960–966. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van De Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int. J. Cancer 2018, 142, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. The second genome: Effects of the mitochondrial genome on cancer progression. Adv. Cancer Res. 2019, 142, 63–105. [Google Scholar] [CrossRef]
- Wu, C.-W.; Yin, P.-H.; Hung, W.-Y.; Li, A.F.-Y.; Li, S.-H.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosom. Cancer 2005, 44, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1836, 336–344. [Google Scholar] [CrossRef]
- Wallace, D.C. Bioenergetic Origins of Complexity and Disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2021, 22, 106–118. [Google Scholar] [CrossRef]
- Zheng, W.; Khrapko, K.; Coller, H.A.; Thilly, W.G.; Copeland, W.C. Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat. Res. 2006, 599, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wisnovsky, S.; Sack, T.; Pagliarini, D.J.; Laposa, R.R.; Kelley, S.O. DNA Polymerase theta Increases Mutational Rates in Mitochondrial DNA. ACS Chem. Biol. 2018, 13, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916. [Google Scholar] [CrossRef] [PubMed]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Morales, S.; Perez-Amado, C.J.; Langley, E.; Hidalgo-Miranda, A. Overview of mitochondrial germline variants and mutations in human disease: Focus on breast cancer (Review). Int. J. Oncol. 2018, 53, 923–936. [Google Scholar]
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Adv. Exp. Med. Biol. 2017, 982, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Hirohata, K.; Kohrn, B.F.; Fox, E.J.; Chang, C.-C.; Loeb, L.A. Detection of Ultra-Rare Mitochondrial Mutations in Breast Stem Cells by Duplex Sequencing. PLoS ONE 2015, 10, e0136216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burr, S.; Chinnery, P.F. The mitochondrial DNA genetic bottleneck: Inheritance and beyond. Essays Biochem. 2018, 62, 225–234. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef] [PubMed]
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.W.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chinnery, P.F. Inheritance of mitochondrial DNA in humans: Implications for rare and common diseases. J. Intern. Med. 2020, 287, 634–644. [Google Scholar] [CrossRef]
- Johnston, I.; Burgstaller, J.P.; Havlicek, V.; Kolbe, T.; Rülicke, T.; Brem, G.; Poulton, J.; Jones, N.S. Stochastic modelling, Bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism. eLife 2015, 4, e07464. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.M.; Sondheimer, N. Heteroplasmy Shifting as Therapy for Mitochondrial Disorders. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 257–267. [Google Scholar] [CrossRef]
- Jackson, M.V.; Krasnodembskaya, A.D. Analysis of Mitochondrial Transfer in Direct Co-cultures of Human Monocyte-derived Macrophages (MDM) and Mesenchymal Stem Cells (MSC). Bio-Protocol 2017, 7, e2255. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab. 2021, 33, 270–282.e8. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zheng, X.; Wang, X.; Jin, T.; Cui, L.; Chen, Z. Mesenchymal Stem/Stromal Cell-Mediated Mitochondrial Transfer and the Therapeutic Potential in Treatment of Neurological Diseases. Stem Cells Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- She, Z.; Xie, M.; Hun, M.; Abdirahman, A.S.; Li, C.; Wu, F.; Luo, S.; Wan, W.; Wen, C.; Tian, J. Immunoregulatory Effects of Mitochondria Transferred by Extracellular Vesicles. Front. Immunol. 2021, 11, 628576. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, J.P.; Kolbe, T.; Havlicek, V.; Hembach, S.; Poulton, J.; Piálek, J.; Steinborn, R.; Rülicke, T.; Brem, G.; Jones, N.S.; et al. Large-scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations. Nat. Commun. 2018, 9, 2488. [Google Scholar] [CrossRef] [PubMed]
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Johnston, I.G.; Prota, G.; Gileadi, U.; Justo-Méndez, R.; Acín-Pérez, R.; Martínez-De-Mena, R.; Fernández-Toro, J.M.; Jimenez-Blasco, D.; et al. Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci. Adv. 2020, 6, eaba5345. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.A.; Wilton, P.R.; Su, M.S.-W.; Paul, I.M.; Arbeithuber, B.; Anthony, K.; Nekrutenko, A.; Nielsen, R.; Makova, K.D. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc. Natl. Acad. Sci. USA 2019, 116, 25172–25178. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuziodonahue, C.A.; Markowitz, S.D.; Velculescu, V.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nat. Cell Biol. 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [PubMed]
- Zekonyte, U.; Bacman, S.R.; Moraes, C.T. DNA-editing enzymes as potential treatments for heteroplasmic mtDNA diseases. J. Intern. Med. 2020, 287, 685–697. [Google Scholar] [CrossRef]
- Sacconi, S.; Salviati, L.; Nishigaki, Y.; Walker, W.F.; Hernandez-Rosa, E.; Trevisson, E.; Delplace, S.; Desnuelle, C.; Shanske, S.; Hirano, M.; et al. A functionally dominant mitochondrial DNA mutation. Hum. Mol. Genet. 2008, 17, 1814–1820. [Google Scholar] [CrossRef]
- Yapa, N.M.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Ameele, J.V.D.; Li, A.Y.; Ma, H.; Chinnery, P.F. Mitochondrial heteroplasmy beyond the oocyte bottleneck. Semin. Cell Dev. Biol. 2020, 97, 156–166. [Google Scholar] [CrossRef]
- Stefano, G.B.; Kream, R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016, 4, 259–262. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Janssen, K.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.; Campbell, S.L.; et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.P.; Stewart, S.; Budnick, J.A.; Caswell, C.C.; Hulver, M.W.; Mukherjee, K.; Srivastava, S. Quantitative Variation in m.3243A > G Mutation Produce Discrete Changes in Energy Metabolism. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, W.; Thompson, P.; Hannun, Y.A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9, 3490. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef]
- Wang, B.; Qiao, L.; Wang, Y.; Zeng, J.; Chen, D.; Guo, H.; Zhang, Y. Mitochondrial DNA D-loop lesions with the enhancement of DNA repair contribute to gastrointestinal cancer progression. Oncol. Rep. 2018, 40, 3694–3704. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Chang, H.-S.; Wu, Y.-C.; Cheng, W.-L.; Lin, T.-T.; Chang, H.-J.; Kuo, S.-J.; Chen, S.-T.; Liu, C.-S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Amado, C.J.; Tovar, H.; Gómez-Romero, L.; Beltrán-Anaya, F.O.; Bautista-Piña, V.; Dominguez-Reyes, C.; Villegas-Carlos, F.; Tenorio-Torres, A.; Alfaro-Ruíz, L.A.; Hidalgo-Miranda, A.; et al. Mitochondrial DNA Mutation Analysis in Breast Cancer: Shifting From Germline Heteroplasmy Toward Homoplasmy in Tumors. Front. Oncol. 2020, 10, 572954. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Chu, L.-H.; John, E.M.; Ding, Y.C.; Ingles, S.A.; Bernstein, L.; Press, M.F.; Ursin, G.; Haiman, C.A.; Neuhausen, S.L. Mitochondrial DNA G10398A variant is not associated with breast cancer in African-American women. Cancer Genet. Cytogenet. 2008, 181, 16–19. [Google Scholar] [CrossRef][Green Version]
- Canter, J.A.; Kallianpur, A.R.; Parl, F.F.; Millikan, R.C. Mitochondrial DNA G10398A Polymorphism and Invasive Breast Cancer in African-American Women. Cancer Res. 2005, 65, 8028–8033. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Krawczyk, T.; Zdrożny, M.; Lubiński, J.; Arnold, R.S.; Kukwa, W.; Ścińska, A.; Golik, P.; Bartnik, E.; Petros, J.A. Mitochondrial NADH-dehydrogenase subunit 3 (ND3) polymorphism (A10398G) and sporadic breast cancer in Poland. Breast Cancer Res. Treat. 2009, 121, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, W.; Jiang, H.-G.; Zhao, H.; Peng, X.-H.; Wei, Y.-H.; Wei, J.-N.; Xie, C.-H.; Liang, C.; Zhong, Y.-H.; et al. Prognostic value of mitochondrial DNA content and G10398A polymorphism in non-small cell lung cancer. Oncol. Rep. 2013, 30, 3006–3012. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, K.; Malyarchuk, B.; Jawień, A.; Marszałek, A.; Banaszkiewicz, Z.; Jarmocik, P.; Grzybowski, T. Mitogenomic differences between the normal and tumor cells of colorectal cancer patients. Hum. Mutat. 2018, 39, 691–701. [Google Scholar] [CrossRef]
- Palodhi, A.; Ghosh, S.; Biswas, N.K.; Basu, A.; Majumder, P.P.; Maitra, A. Profiling of genomic alterations of mitochondrial DNA in gingivobuccal oral squamous cell carcinoma: Implications for disease progress. Mitochondrion 2019, 46, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Chang, S.-C.; Ou, L.-H.; Chen, C.-M.; Hsieh, S.S.-W.; Chung, Y.-P.; King, K.-L.; Lin, S.-L.; Wei, Y.-H. Mitochondrial DNA alterations correlate with the pathological status and the immunological ER, PR, HER-2/neu, p53 and Ki-67 expression in breast invasive ductal carcinoma. Oncol. Rep. 2015, 33, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Scholle, L.M.; Zierz, S.; Mawrin, C.; Wickenhauser, C.; Urban, D.L. Heteroplasmy and Copy Number in the Common m.3243A>G Mutation—A Post-Mortem Genotype–Phenotype Analysis. Genes 2020, 11, 212. [Google Scholar] [CrossRef]
- Gentiluomo, M.; Katzke, V.A.; Kaaks, R.; Tjønneland, A.; Severi, G.; Perduca, V.; Boutron-Ruault, M.-C.; Weiderpass, E.; Ferrari, P.; Johnson, T.; et al. Mitochondrial DNA Copy-Number Variation and Pancreatic Cancer Risk in the Prospective EPIC Cohort. Cancer Epidemiol. Biomark. Prev. 2020, 29, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Cui, N.-H.; Zhang, S.; Wang, X.-B.; Ming, L. Leukocyte Mitochondrial DNA Copy Number and Risk of Thyroid Cancer: A Two-Stage Case-Control Study. Front. Endocrinol. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lv, H.; Ji, P.; Zhu, X.; Yuan, H.; Jin, G.; Dai, J.; Hu, Z.; Su, Y.; Ma, H. Mitochondrial DNA copy number is associated with risk of head and neck squamous cell carcinoma in Chinese population. Cancer Med. 2018, 7, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Forman, M.R.; Graham, B.H.; Monahan, P.O.; Giovannucci, E.L.; De Vivo, I.; Chan, A.T.; Nan, H. Association between pre-diagnostic leukocyte mitochondrial DNA copy number and survival among colorectal cancer patients. Cancer Epidemiol. 2020, 68, 101778. [Google Scholar] [CrossRef]
- Nie, H.; Chen, G.; He, J.; Zhang, F.; Li, M.; Wang, Q.; Zhou, H.; Lyu, J.; Bai, Y. Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion 2016, 26, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; He, J.; Shen, L.; Fang, H.; Nie, H.; Jin, T.; Wei, X.; Xin, Y.; Jiang, Y.; Li, H.; et al. The mitochondrial DNA 4,977-bp deletion and its implication in copy number alteration in colorectal cancer. BMC Med. Genet. 2011, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Yusoff, A.A.M.; Abdullah, W.S.W.; Khair, S.Z.N.M.; Radzak, S.M.A. A comprehensive overview of mitochondrial DNA 4977-bp deletion in cancer studies. Oncol. Rev. 2019, 13, 409. [Google Scholar] [CrossRef]
- Beadnell, T.; Fain, C.; Vivian, C.; King, J.; Hastings, R.; Markiewicz, M.; Welch, D. Mitochondrial genetics cooperate with nuclear genetics to selectively alter immune cell development/trafficking. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165648. [Google Scholar] [CrossRef]
- Mambo, E.; Chatterjee, A.; Xing, M.; Tallini, G.; Haugen, B.R.; Yeung, S.-C.J.; Sukumar, S.; Sidransky, D. Tumor-specific changes in mtDNA content in human cancer. Int. J. Cancer 2005, 116, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.D.; Broner, E.C.; Agrawal, N.; London, N.; Pearson, A.; Gupta, A.; Wali, N.; Seiwert, T.Y.; Wheelan, S.; Lingen, M.; et al. Somatic mitochondrial mutation discovery using ultra-deep sequencing of the mitochondrial genome reveals spatial tumor heterogeneity in head and neck squamous cell carcinoma. Cancer Lett. 2020, 471, 49–60. [Google Scholar] [CrossRef]
- Crooks, D.R.; Maio, N.; Lang, M.; Ricketts, C.J.; Vocke, C.D.; Gurram, S.; Turan, S.; Kim, Y.-Y.; Cawthon, G.M.; Sohelian, F.; et al. Mitochondrial DNA alterations underlie an irreversible shift to aerobic glycolysis in fumarate hydratase–deficient renal cancer. Sci. Signal. 2021, 14, eabc4436. [Google Scholar] [CrossRef]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-Yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, J.-H.; Kim, D.-K.; Keum, D.Y. Nuclear and mitochondrial DNAs microsatellite instability and mitochondrial DNA copy number in adenocarcinoma and squamous cell carcinoma of lung: A pilot study. APMIS 2015, 123, 1048–1054. [Google Scholar] [CrossRef]
- Shen, J.; Wan, J.; Song, R.; Zhao, H. Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: A replication study. Carcinogenesis 2015, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alvarez, M.I.; Zorzano, A. Mitochondrial Dynamics and Liver Cancer. Cancers 2021, 13, 2571. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumor progression—A New paradigm with Emerging Importance. Anticancer Agents Med. Chem. 2021. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Zampieri, L.; Silva-Almeida, C.; Rondeau, J.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Gaggar, S.; Gögenur, I. Cancer-Associated Fibroblasts and Tumor-Associated Macrophages in Cancer and Cancer Immunotherapy. Front. Oncol. 2021, 11, 668731. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Huang, D.Q.; Low, S.H.H.; Tan, G.H.; Han, M.J.; Singh, S.; Tang, B.; Chang, S.C.; Lim, J.S.Y.; Omar, M.F.M.; et al. Effect of cell microenvironment on the drug sensitivity of hepatocellular cancer cells. Oncotarget 2021, 12, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ding, Z.; Hu, D.; Sun, F.; Dai, C.; Xie, J.; Hu, X. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J. Pathol. 2012, 227, 189–199. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Targeting extracellular ROS signaling of tumor cells. Anticancer. Res. 2014, 34, 1467–1482. [Google Scholar] [PubMed]
- Bobrowska, J.A.; Awsiuk, K.; Pabijan, J.; Bobrowski, P.; Lekki, J.; Sowa, K.; Rysz, J.; Budkowski, A.; Lekka, M. Biophysical and Biochemical Characteristics as Complementary Indicators of Melanoma Progression. Anal. Chem. 2019, 91, 9885–9892. [Google Scholar] [CrossRef]
- Spinelli, F.M.; Vitale, D.L.; Sevic, I.; Alaniz, L. Hyaluronan in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 67–83. [Google Scholar] [CrossRef]
- Robertson, D. The Development of Tumor Cell Characteristics. J. Cell. Physiol. 2014, 229, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Pezet, M.G.; Gomez-Duran, A.; Klimm, F.; Aryaman, J.; Burr, S.; Wei, W.; Saitou, M.; Prudent, J.; Chinnery, P.F. Oxygen tension modulates the mitochondrial genetic bottleneck and influences the segregation of a heteroplasmic mtDNA variant in vitro. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef]
- Krjutškov, K.; Koltsina, M.; Grand, K.; Võsa, U.; Sauk, M.; Tõnisson, N.; Salumets, A. Tissue-specific mitochondrial heteroplasmy at position 16,093 within the same individual. Curr. Genet. 2013, 60, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiang, X.; Yang, Q.; Zhao, J.; Zhou, Q.; Zhou, Y. The Functions, Methods, and Mobility of Mitochondrial Transfer between Cells. Front. Oncol. 2021, 11, 672781. [Google Scholar] [CrossRef] [PubMed]
- Hekmatshoar, Y.; Nakhle, J.; Galloni, M.; Vignais, M.-L. The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem. J. 2018, 475, 2305–2328. [Google Scholar] [CrossRef]
- Abnet, C.C.; Huppi, K.; Carrera, A.; Armistead, D.; McKenney, K.; Hu, N.; Tang, Z.-Z.; Taylor, P.R.; Dawsey, S.M. Control region mutations and the ’common deletion’ are frequent in the mitochondrial DNA of patients with esophageal squamous cell carcinoma. BMC Cancer 2004, 4, 30. [Google Scholar] [CrossRef]
- Liu, Q.; Lin, D.; Li, M.; Gu, Z.; Zhao, Y. Evidence of Neutral Evolution of Mitochondrial DNA in Human Hepatocellular Carcinoma. Genome Biol. Evol. 2019, 11, 2909–2916. [Google Scholar] [CrossRef]
- Ling, S.; Hu, Z.; Yang, Z.; Yang, F.; Li, Y.; Lin, P.; Chen, K.; Dong, L.; Cao, L.; Tao, Y.; et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef]
- Beadnell, T.C.; Scheid, A.D.; Vivian, C.J.; Welch, D.R. Roles of the mitochondrial genetics in cancer metastasis: Not to be ignored any longer. Cancer Metastasis Rev. 2018, 37, 615–632. [Google Scholar] [CrossRef]
- McCrow, J.; Petersen, D.C.; Louw, M.; Chan, E.K.F.; Harmeyer, K.; Vecchiarelli, S.; Lyons, R.J.; Bornman, M.S.R.; Hayes, V.M. Spectrum of mitochondrial genomic variation and associated clinical presentation of prostate cancer in South African men. Prostate 2015, 76, 349–358. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Denroche, R.E.; Aguiar, J.; Notta, F.; Connor, A.A.; Wilson, J.M.; Stein, L.D.; Gallinger, S.; Boutros, P.C. Mutations in Mitochondrial DNA From Pancreatic Ductal Adenocarcinomas Associate With Survival Times of Patients and Accumulate as Tumors Progress. Gastroenterology 2018, 154, 1620–1624.e5. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Hayashi, J.-I. A novel function of mtDNA: Its involvement in metastasis. Ann. N. Y. Acad. Sci. 2010, 1201, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Weerts, M.; Timmermans, E.; van de Stolpe, A.; Vossen, R.; Anvar, S.; Foekens, J.; Sleijfer, S.; Martens, J. Tumor-Specific Mitochondrial DNA Variants Are Rarely Detected in Cell-Free DNA. Neoplasia 2018, 20, 687–696. [Google Scholar] [CrossRef]
- Guerra, F.; Arbini, A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta BBA Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenet. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Kowal, K.; Tkaczyk, A.; Ząbek, T.; Pierzchała, M.; Ślaska, B. Comparative Analysis of CpG Sites and Islands Distributed in Mitochondrial DNA of Model Organisms. Animals 2020, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Cuenin, C.; Chung, F.; Aguilera, J.R.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, S.Y.; Kim, S.A. Mitochondrial DNA Methylation Is Higher in Acute Coronary Syndrome Than in Stable Coronary Artery Disease. Vivo 2021, 35, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Z.; Cong, X.; Lv, Y.; Li, Z.; Rong, L.; Yang, T.; Yu, D. Mitochondrial gene COX2 methylation and downregulation is a biomarker of aging in heart mesenchymal stem cells. Int. J. Mol. Med. 2020, 47, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Feng, S.; Xiong, L.; Ji, Z.; Cheng, W. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol. Med. Rep. 2012, 6, 125–130. [Google Scholar] [CrossRef]
- Santos, J.H. Mitochondria signaling to the epigenome: A novel role for an old organelle. Free. Radic. Biol. Med. 2021, 170, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Vivian, C.J.; Brinker, A.E.; Graw, S.; Koestler, D.C.; Legendre, C.; Gooden, G.C.; Salhia, B.; Welch, D.R. Mitochondrial Genomic Backgrounds Affect Nuclear DNA Methylation and Gene Expression. Cancer Res. 2017, 77, 6202–6214. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef]
- Hirose, M.; Schilf, P.; Gupta, Y.; Zarse, K.; Künstner, A.; Fähnrich, A.; Busch, H.; Yin, J.; Wright, M.; Ziegler, A.; et al. Low-level mitochondrial heteroplasmy modulates DNA replication, glucose metabolism and lifespan in mice. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Tranah, G.J.; Katzman, S.M.; Lauterjung, K.; Yaffe, K.; Manini, T.M.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Cummings, S.R. Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 2018, 8, 11887. [Google Scholar] [CrossRef]
- Jackson, C.B.; Turnbull, D.M.; Minczuk, M.; Gammage, P.A. Therapeutic Manipulation of mtDNA Heteroplasmy: A Shifting Perspective. Trends Mol. Med. 2020, 26, 698–709. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Mancuso, M. Mitochondrial Diseases: Therapeutic Approaches. Biosci. Rep. 2007, 27, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Hasan-Olive, M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2018, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Tonin, Y.; Heckel, A.-M.; Vyssokikh, M.; Dovydenko, I.; Meschaninova, M.; Rötig, A.; Munnich, A.; Venyaminova, A.; Tarassov, I.; Entelis, N. Modeling of Antigenomic Therapy of Mitochondrial Diseases by Mitochondrially Addressed RNA Targeting a Pathogenic Point Mutation in Mitochondrial DNA. J. Biol. Chem. 2014, 289, 13323–13334. [Google Scholar] [CrossRef]
- Taivassalo, T.; Fu, K.; Johns, T.; Arnold, D.; Karpati, G.; Shoubridge, E.A. Gene shifting: A novel therapy for mitochondrial myopathy. Hum. Mol. Genet. 1999, 8, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.M.; Maheshan, R.; Costford, S.R.; Wahedi, A.; Trajkovski, M.; Plavec, J.; Yatsunyk, L.A.; Ciesielski, G.L.; Kaufman, B.A.; Sondheimer, N. G-quadruplex-mediated reduction of a pathogenic mitochondrial heteroplasmy. Hum. Mol. Genet. 2019, 28, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Duan, N.; Moraes, C.T. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2011, 19, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef]
- Minczuk, M.; Papworth, M.A.; Kolasinska, P.; Murphy, M.P.; Klug, A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. USA 2006, 103, 19689–19694. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.-P.; et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective Elimination of Mitochondrial Mutations in the Germline by Genome Editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2012, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Lightowlers, R.N. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.-I.; Kim, S.; Shin, J.-H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; De Moraes, M.H.; Zeng, J.; Yeh, M.M.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Loutre, R.; Heckel, A.M.; Smirnova, A.; Entelis, N.; Tarassov, I. Can Mitochondrial DNA be CRISPRized: Pro and Contra. IUBMB Life 2018, 70, 1233–1239. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Dickerson, T.; Jauregui, C.E.; Teng, Y. Friend or foe? Mitochondria as a pharmacological target in cancer treatment. Futur. Med. Chem. 2017, 9, 2197–2210. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Name | Function | OXPHOS Complex | Nucleotide Start Position | Nucleotide End Position |
|---|---|---|---|---|---|
| MT-ATP6 | ATP synthase membrane subunit 6 | Proton transmembrane transporter activity | V | 8527 | 9207 |
| MT-ATP8 | ATP synthase membrane subunit 8 | Proton transmembrane transporter activity | 8366 | 8572 | |
| MT-CO1 | Cytochrome c oxidase I | Electron transport Heme binding | 5904 | 7445 | |
| MT-CO2 | Cytochrome c oxidase II | Electron transport | IV | 7586 | 8269 |
| MT-CO3 | Cytochrome c oxidase III | Electron transport | 9207 | 9990 | |
| MT-CYB | Cytochrome b | Electron transport Heme binding | III | 14747 | 15887 |
| MT-ND1 | NADH: ubiquinone oxidoreductase core subunit 1 | Proton pumping Reduction site for ubiquinone | 3307 | 4262 | |
| MT-ND2 | NADH: ubiquinone oxidoreductase core subunit 2 | Proton pumping | 4470 | 5511 | |
| MT-ND3 | NADH: ubiquinone oxidoreductase core subunit 3 | Proton pumping | I | 10059 | 10404 |
| MT-ND4 | NADH: ubiquinone oxidoreductase core subunit 4 | Proton pumping | 10760 | 12137 | |
| MT-ND4L | NADH: ubiquinone oxidoreductase core subunit 4L | Proton pumping | 10470 | 10766 | |
| MT-ND5 | NADH:ubiquinone oxidoreductase core subunit 5 | Proton pumping | 12337 | 14148 | |
| MT-ND6 | NADH:ubiquinone oxidoreductase core subunit 6 | Proton pumping | 14149 | 14673 | |
| MT-RNR1 | 12S rRNA | 12S ribosomal RNA Small subunit | - | 648 | 1601 |
| MT-RNR2 | 16S rRNA | 16S ribosomal RNA Large subunit | - | 1671 | 3229 |
| MT-TA | tRNA-Ala | tRNA for alanine | - | 5587 | 5655 |
| MT-TC | tRNA-Cys | tRNA for cysteine | - | 5761 | 5826 |
| MT-TD | tRNA-Asp | tRNA for aspartic acid | - | 7518 | 7585 |
| MT-TE | tRNA-Glu | tRNA for glutamic acid | - | 14674 | 14742 |
| MT-TF | tRNA-Phe | tRNA for phenylalanine | - | 577 | 647 |
| MT-TG | tRNA-Gly | tRNA for glycine | - | 9991 | 10058 |
| MT-TH | tRNA-His | tRNA for histidine | - | 12138 | 12206 |
| MT-TI | tRNA-Ile | tRNA for isoleucine | - | 4263 | 4331 |
| MT-TK | tRNA-Lys | tRNA for lysine | - | 8295 | 8364 |
| MT-TL1 | tRNA-Leu (UUA/G) 1 | tRNA for leucine 1 | - | 3230 | 3304 |
| MT-TL2 | tRNA-Leu (CUN) 2 | tRNA for leucine 2 | - | 12266 | 12336 |
| MT-TM | tRNA-Met | tRNA for methionine | - | 4402 | 4469 |
| MT-TN | tRNA-Asn | tRNA for asparagine | - | 5657 | 5729 |
| MT-TP | tRNA-Pro | tRNA for proline | - | 15956 | 16023 |
| MT-TQ | tRNA-Gln | tRNA for glutamine | - | 4329 | 4400 |
| MT-TR | tRNA-Arg | tRNA for arginine | - | 10405 | 10469 |
| MT-TS1 | tRNA-Ser (UCN) 1 | tRNA for serine 1 | - | 7446 | 7514 |
| MT-TS2 | tRNA-Ser (AGU/C) 2 | tRNA for serine 2 | - | 12207 | 12265 |
| MT-TT | tRNA-Thr | tRNA for threonine | - | 15888 | 15953 |
| MT-TV | tRNA-Val | tRNA for valine | - | 1602 | 1670 |
| MT-TW | tRNA-Trp | tRNA for tryptophan | - | 5512 | 5579 |
| MT-TY | tRNA-Tyr | tRNA for tyrosine | - | 5826 | 5891 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A.; Jiménez-Morales, S. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. Int. J. Mol. Sci. 2021, 22, 7369. https://doi.org/10.3390/ijms22147369
Pérez-Amado CJ, Bazan-Cordoba A, Hidalgo-Miranda A, Jiménez-Morales S. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. International Journal of Molecular Sciences. 2021; 22(14):7369. https://doi.org/10.3390/ijms22147369
Chicago/Turabian StylePérez-Amado, Carlos Jhovani, Amellalli Bazan-Cordoba, Alfredo Hidalgo-Miranda, and Silvia Jiménez-Morales. 2021. "Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression" International Journal of Molecular Sciences 22, no. 14: 7369. https://doi.org/10.3390/ijms22147369
APA StylePérez-Amado, C. J., Bazan-Cordoba, A., Hidalgo-Miranda, A., & Jiménez-Morales, S. (2021). Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. International Journal of Molecular Sciences, 22(14), 7369. https://doi.org/10.3390/ijms22147369