
Increased Beta-Hydroxybutyrate Level Is Not Sufficient for the Neuroprotective Effect of Long-Term Ketogenic Diet in an Animal Model of Early Parkinson’s Disease. Exploration of Brain and Liver Energy Metabolism Markers


Abstract
:1. Introduction
2. Results
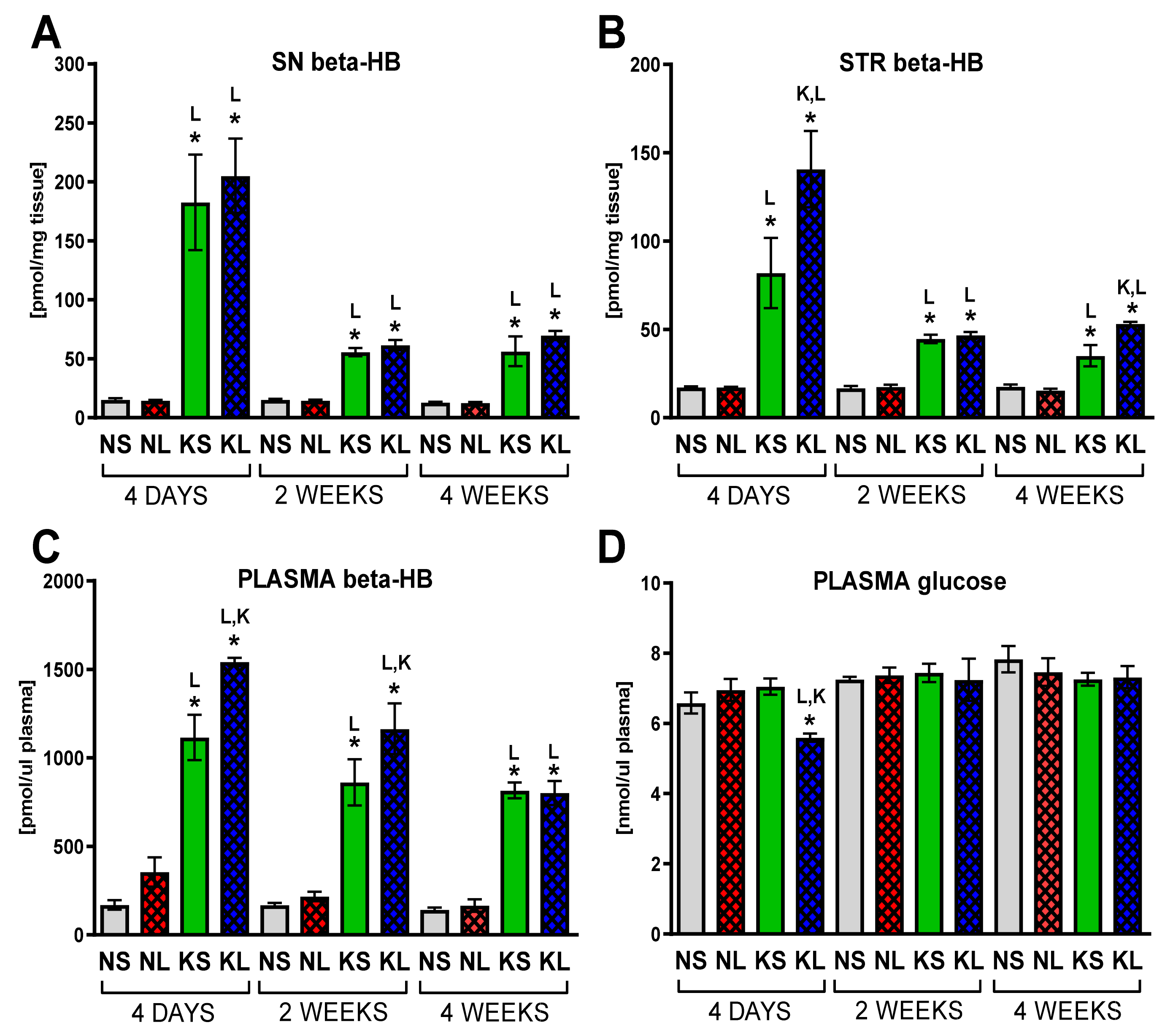
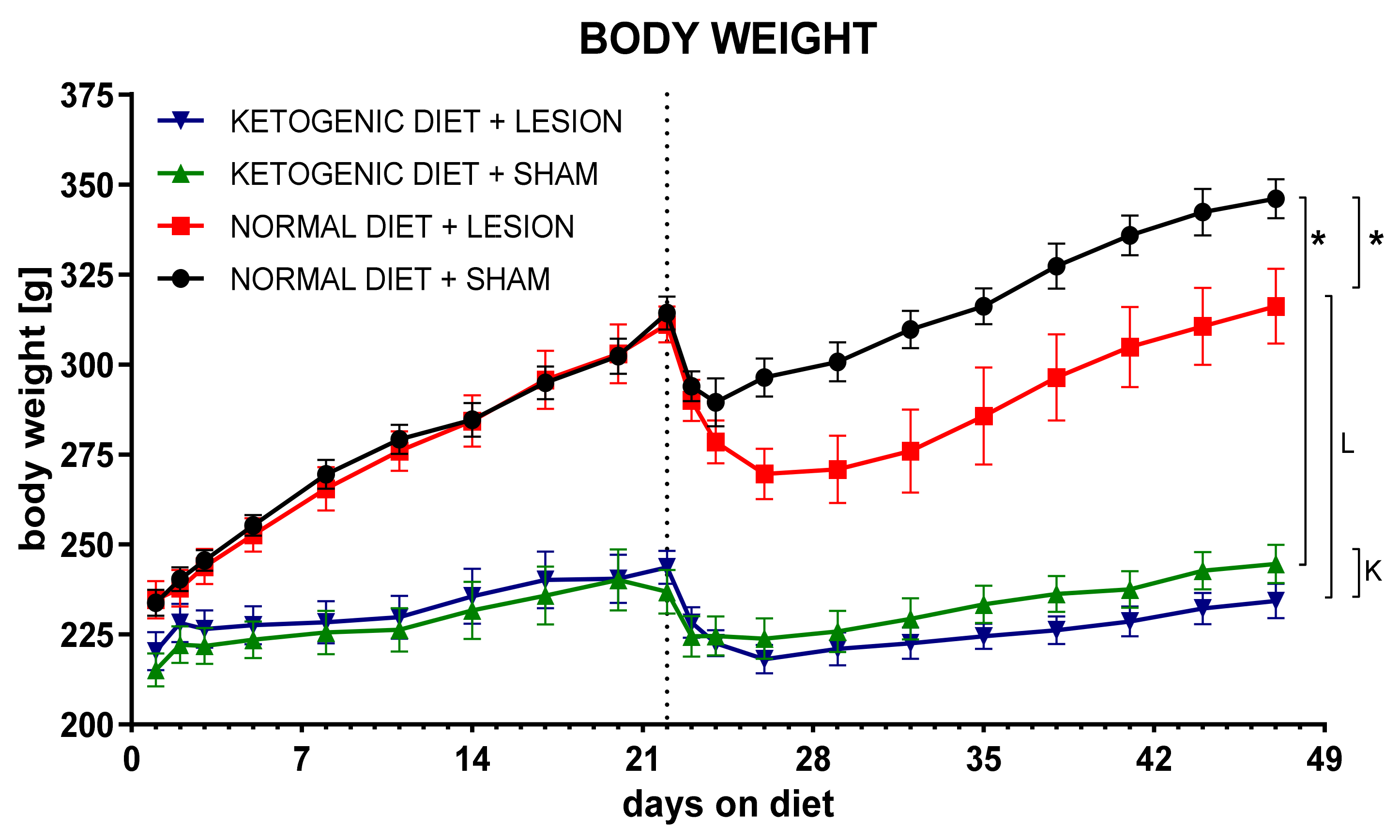
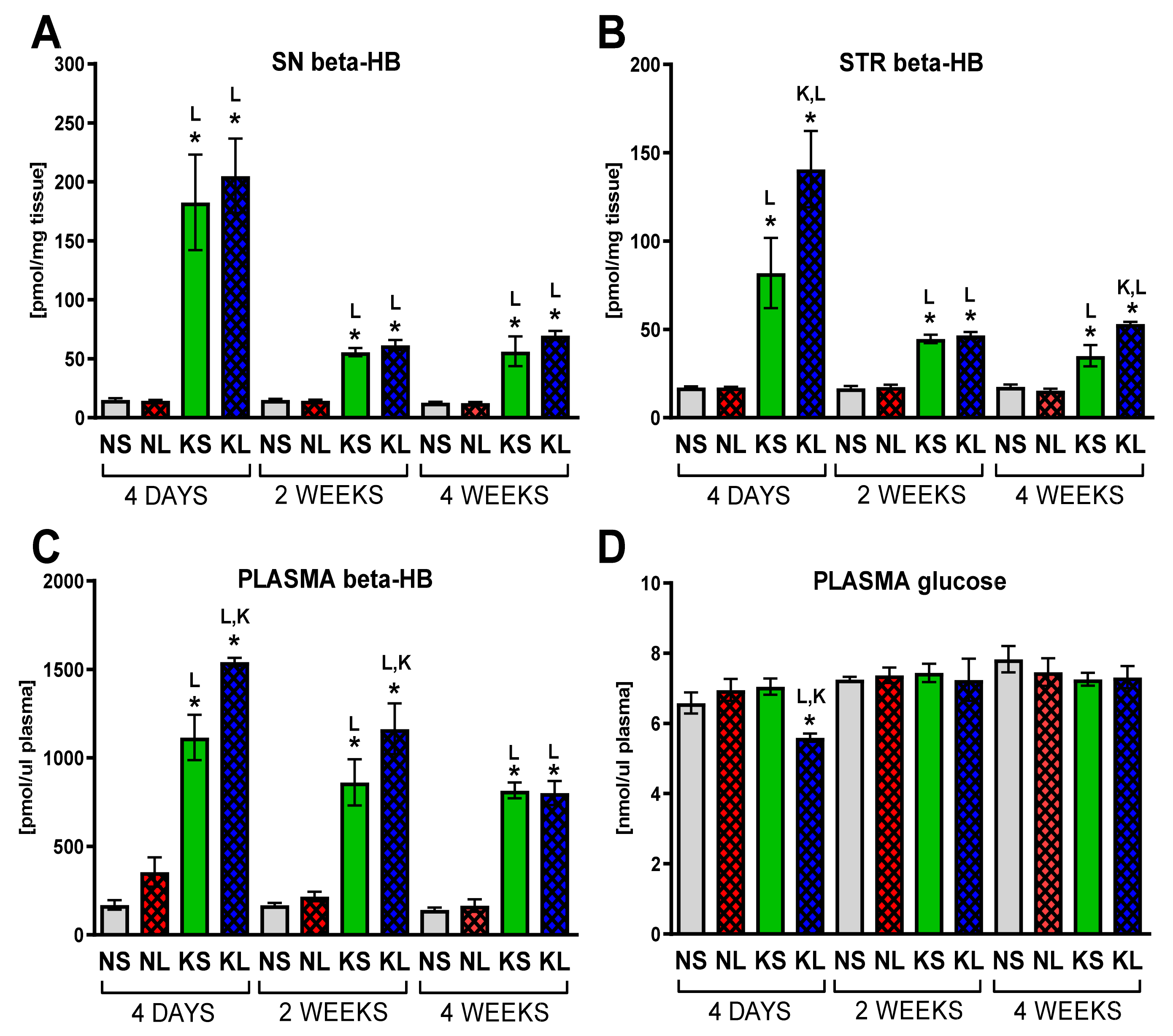
2.1. Chronic Ketogenic Diet Strongly Increases b-HB Both in Plasma and Brain but Does Not Decrease Glucose Levels
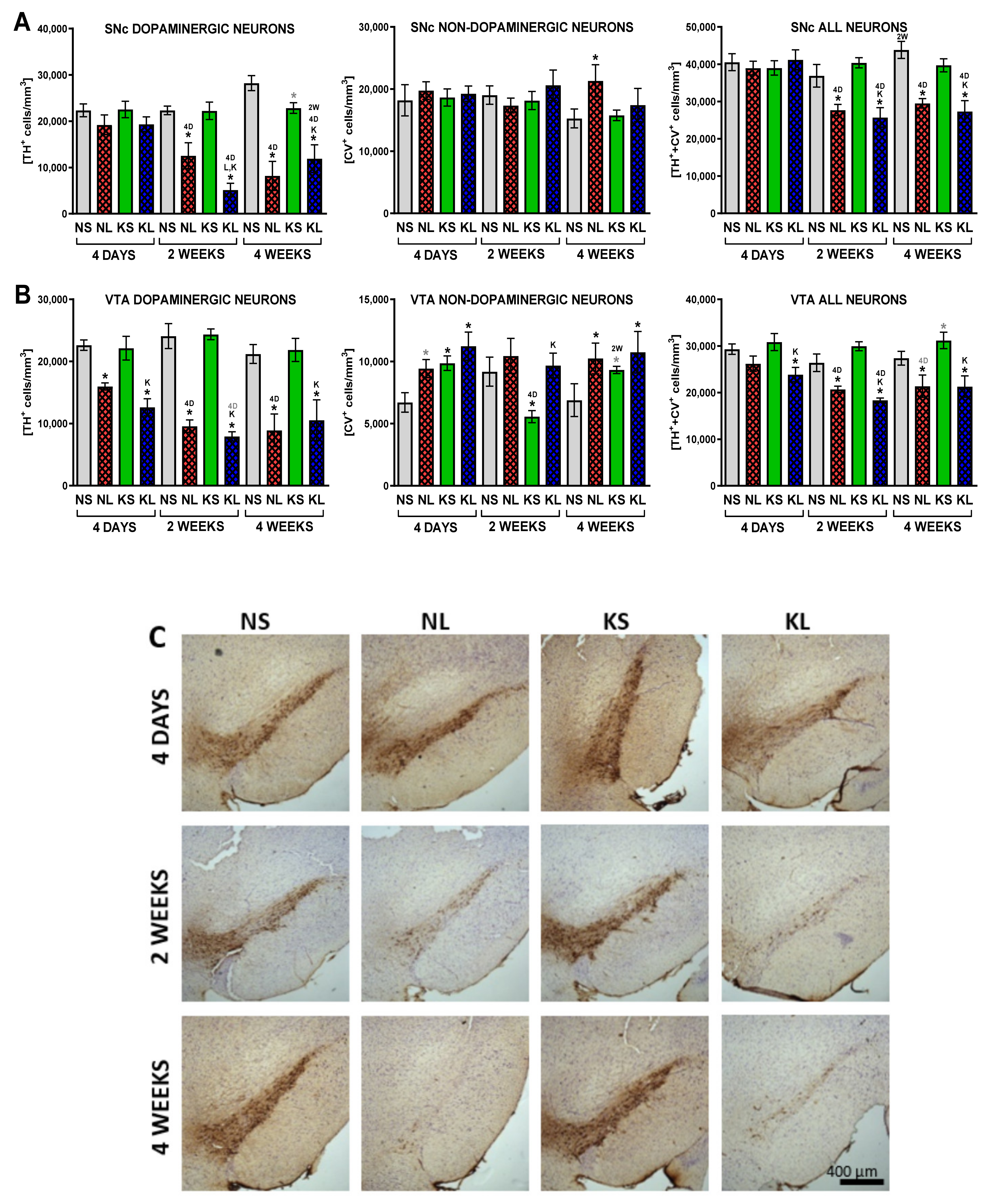
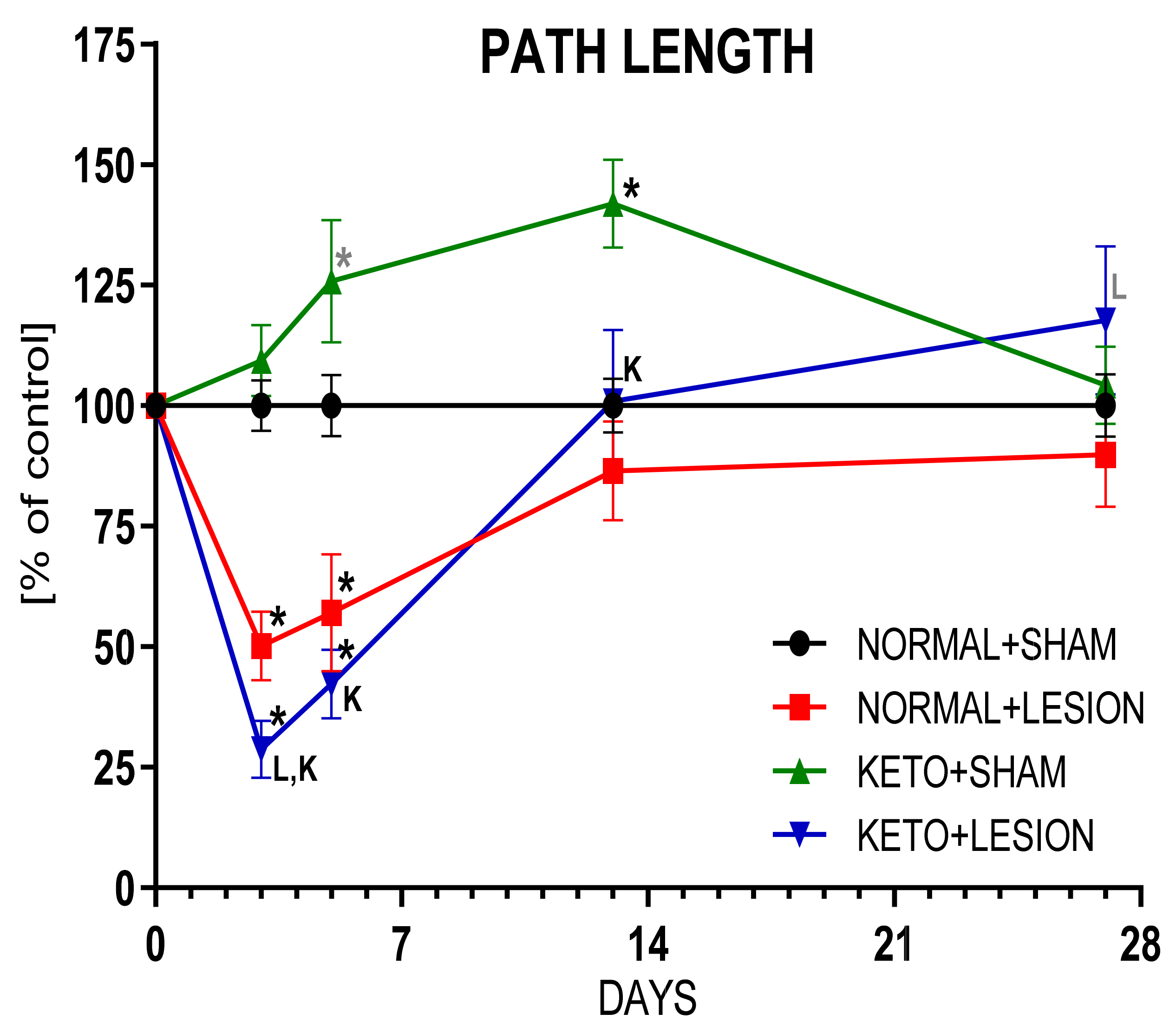
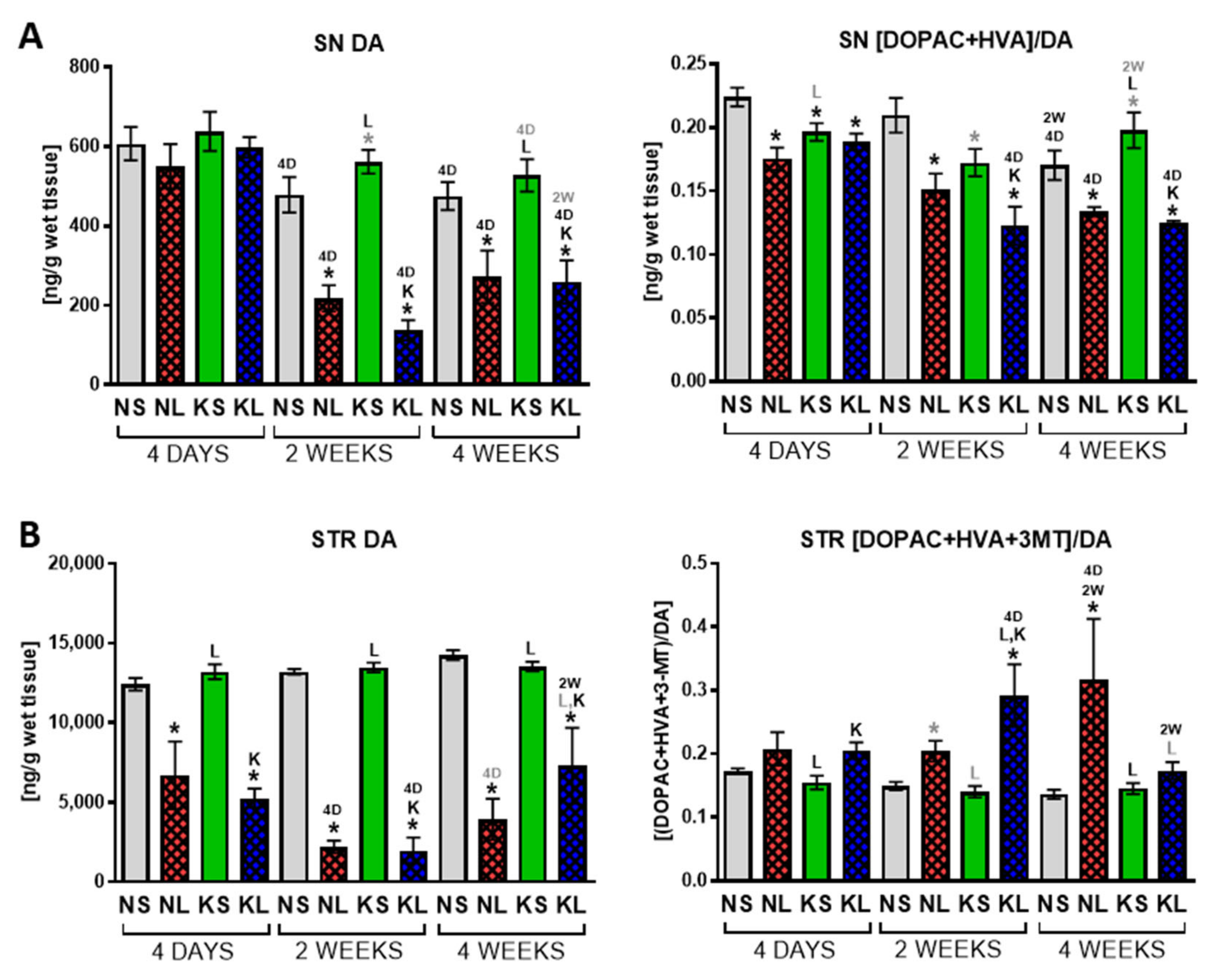
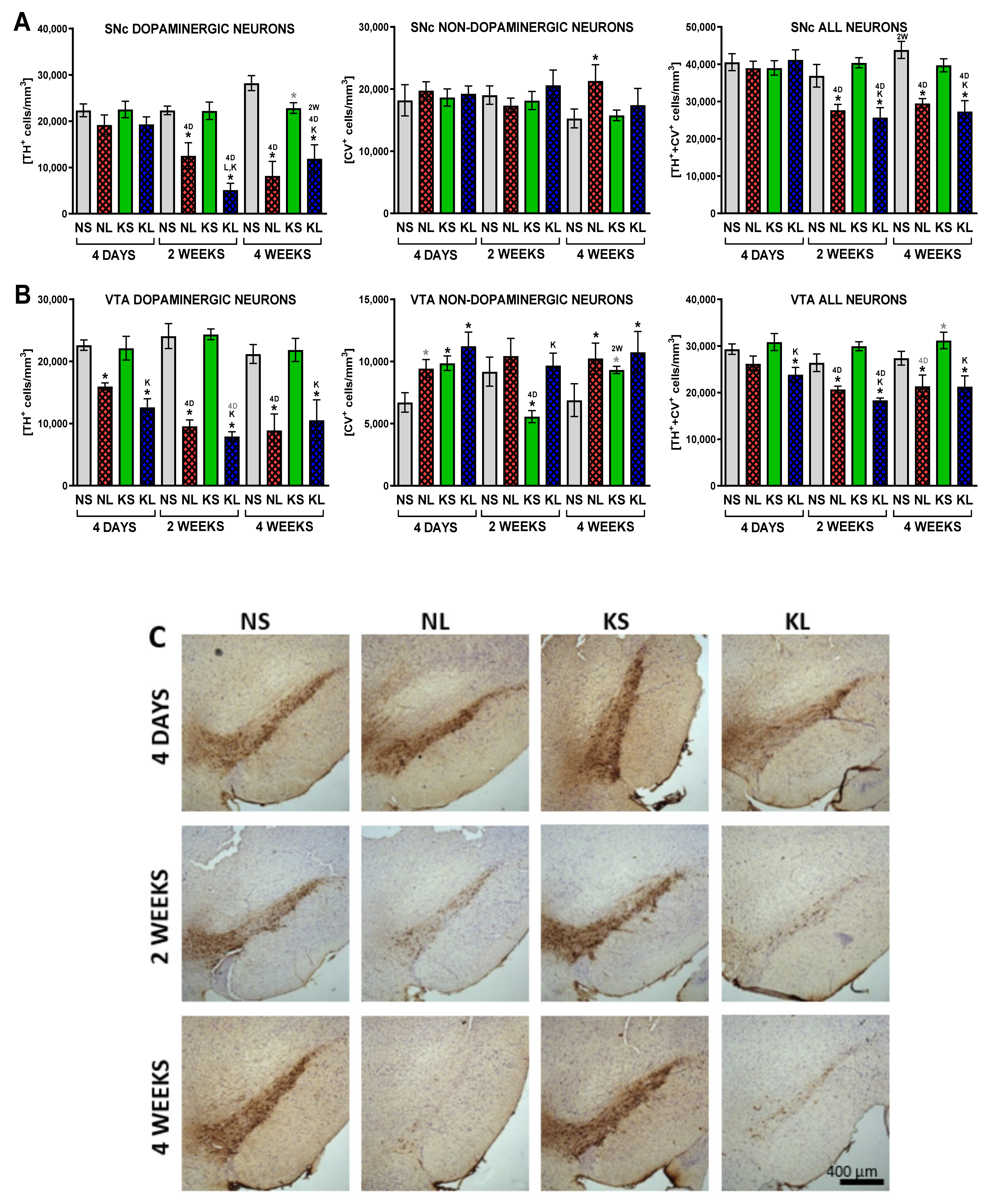
2.2. Ketogenic Diet Does Not Protect DA Neurons from 6-OHDA Induced Degeneration
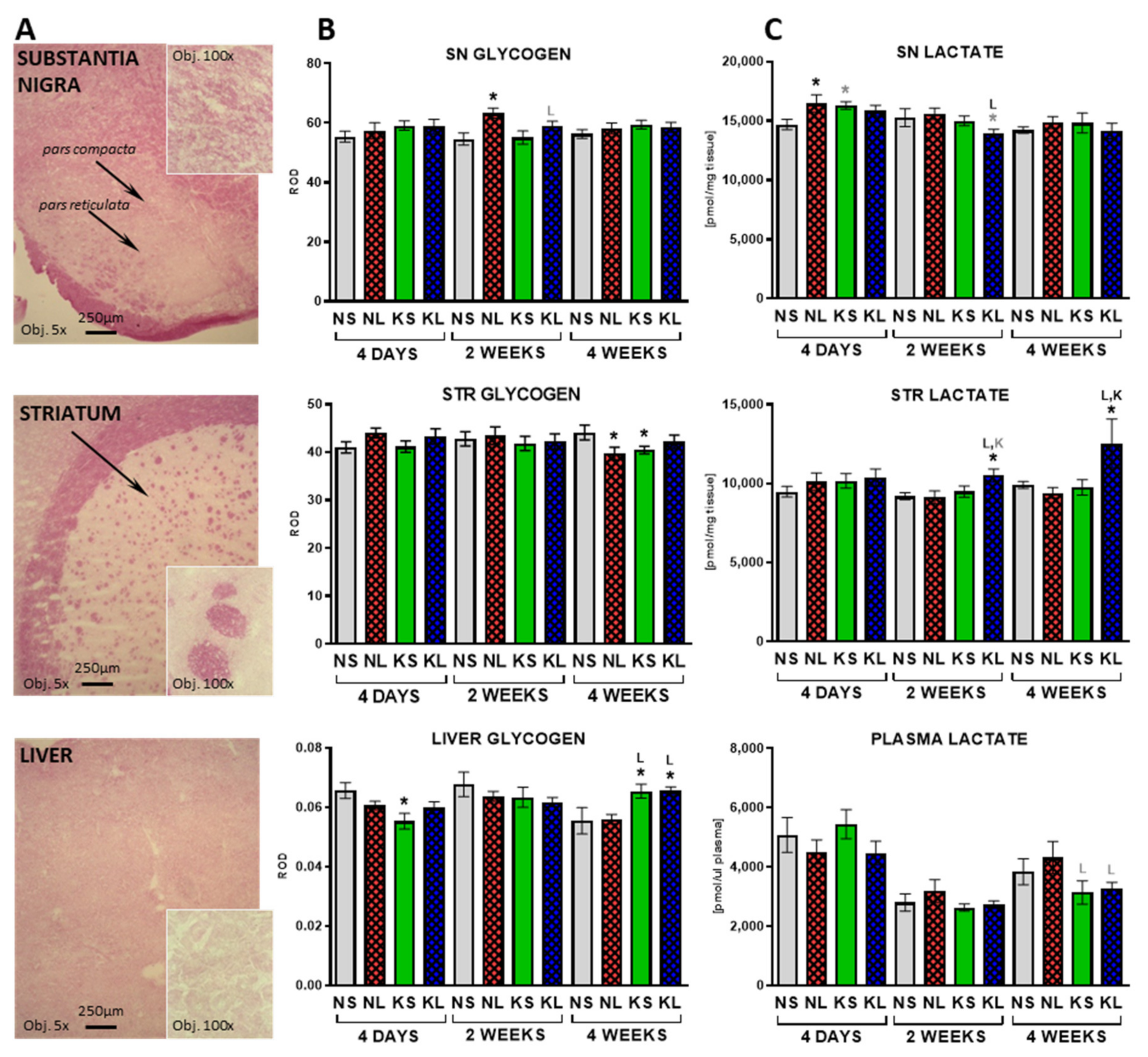
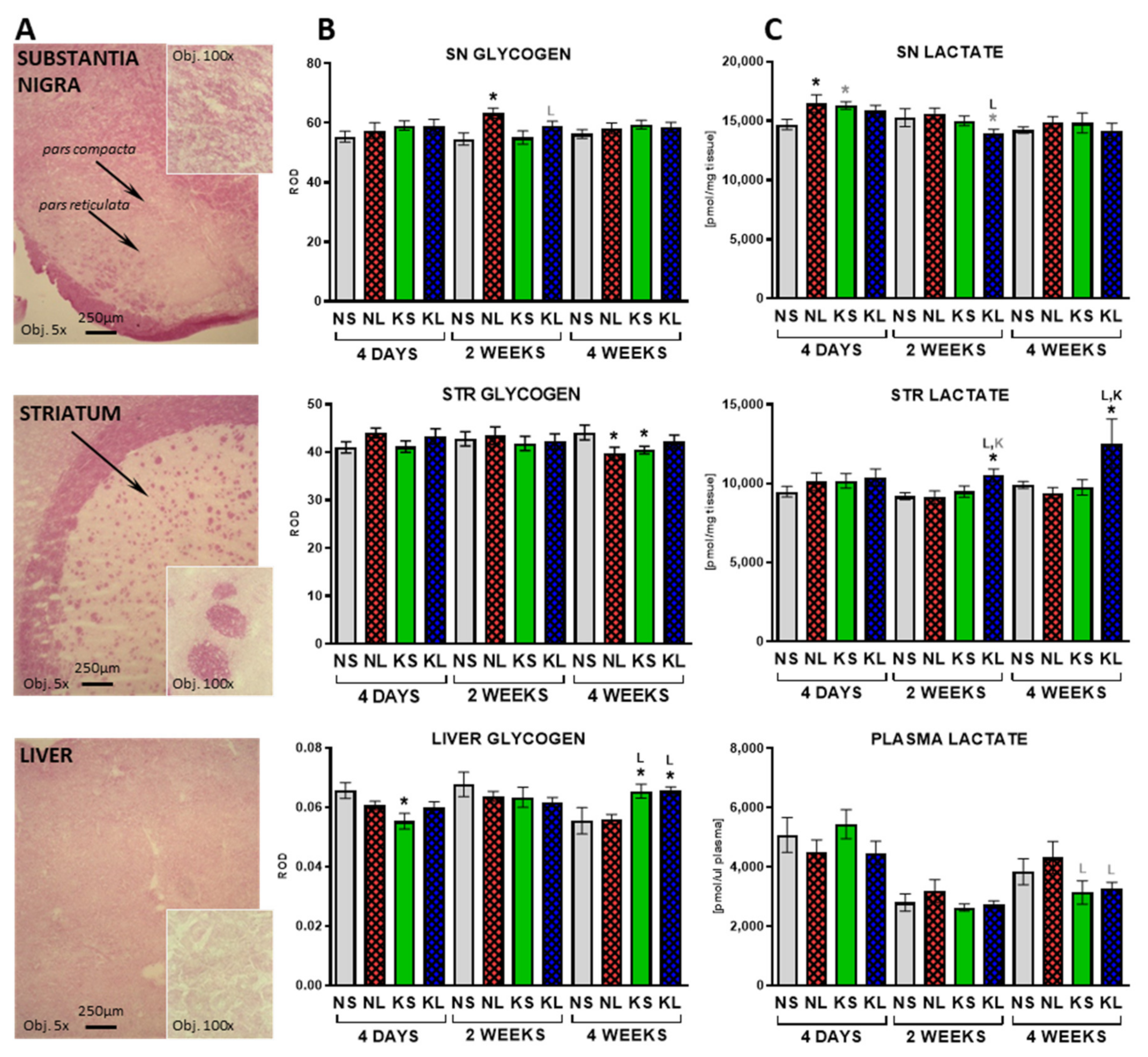
2.3. In the Brain, Glycogen Energy Reserve Does Not Change, but 7 Weeks of Ketogenic Diet Increases It in the Liver
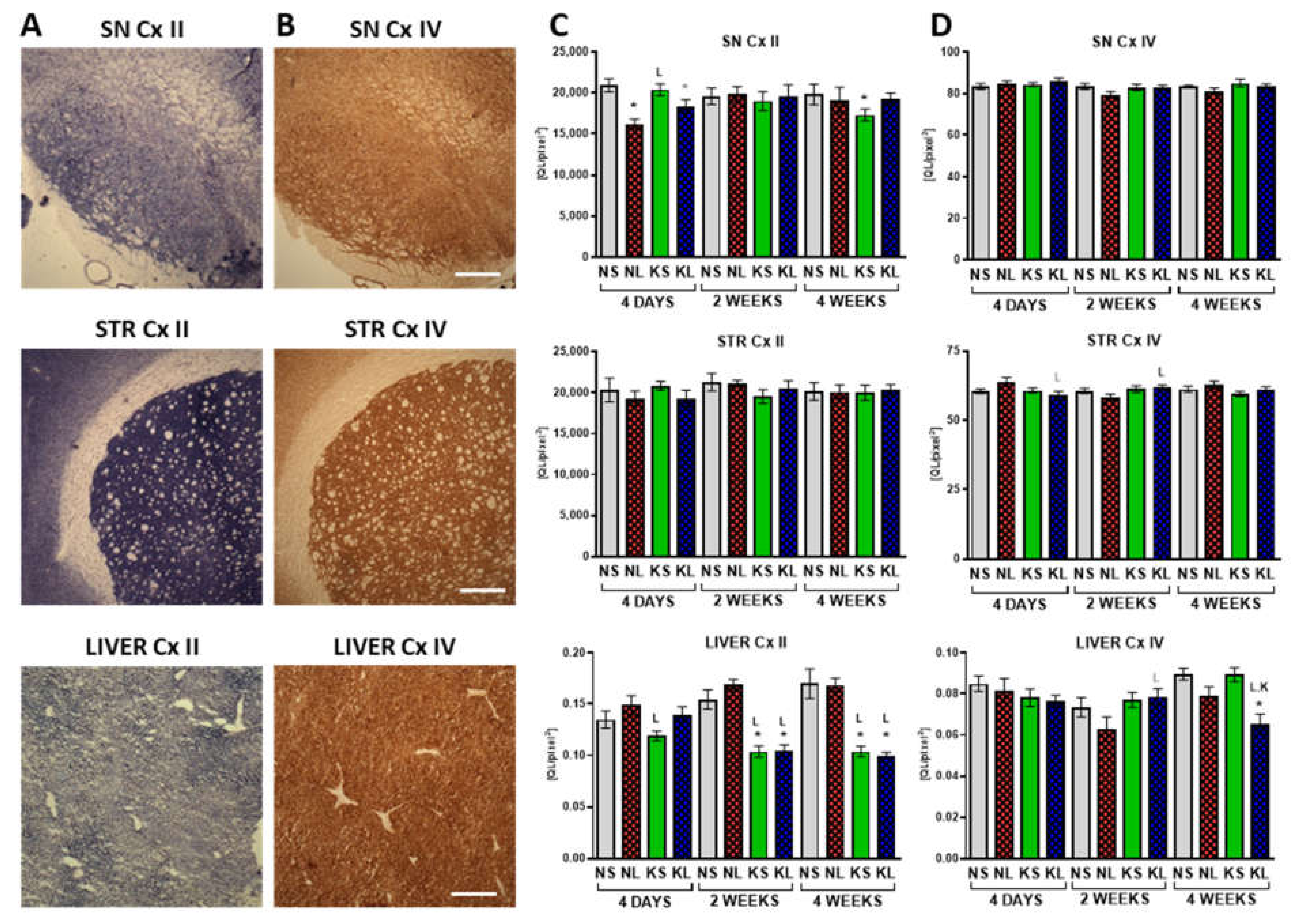
2.4. Mitochondrial Cx II Activity Significantly Decreases in the Liver after Prolonged Ketogenic Diet
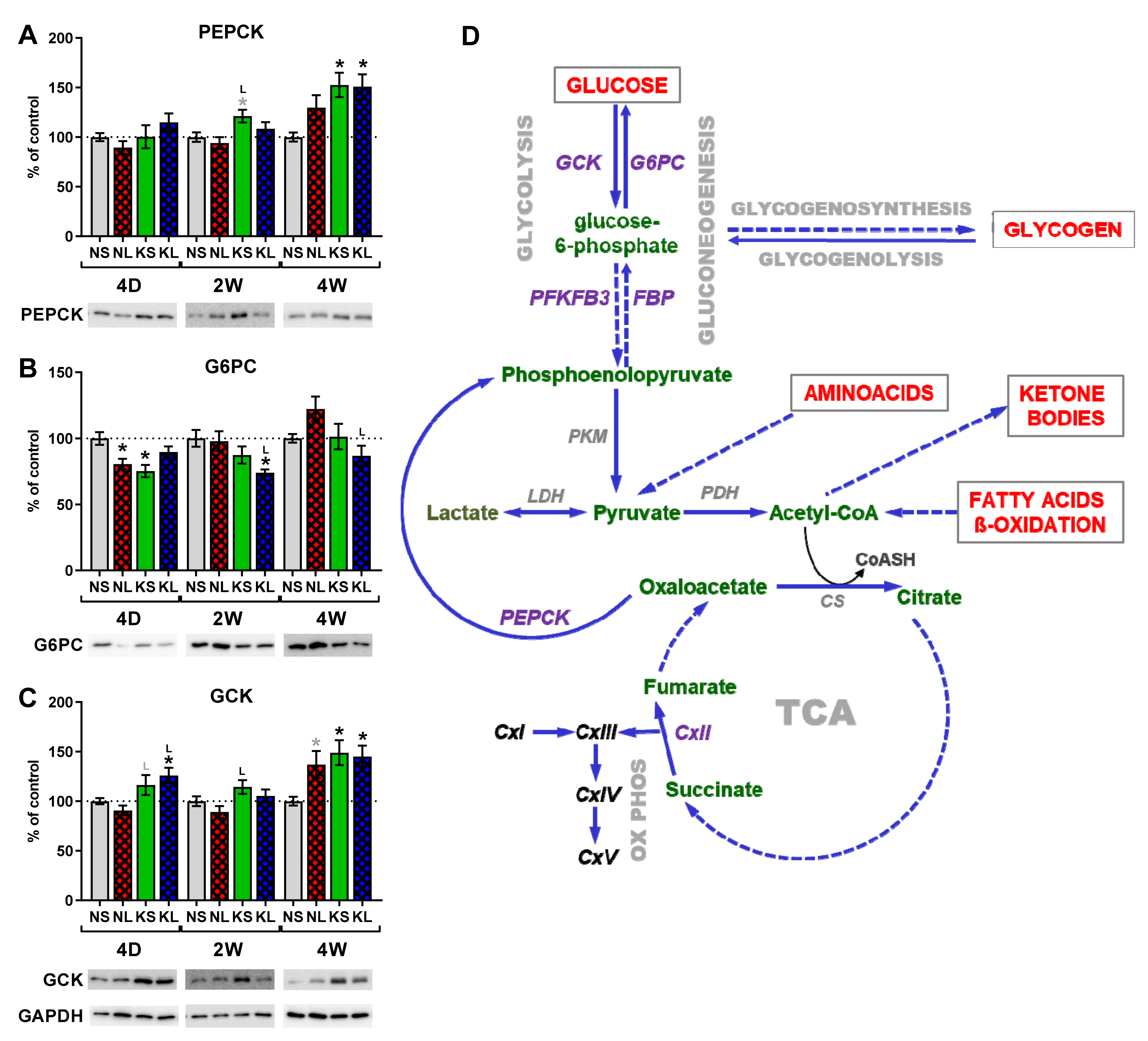
2.5. Gluconeogenesis in the Liver during Ketogenic Diet
3. Discussion
3.1. Proof of Induction of Hyperketonaemia
3.2. Lack of Neuroprotection
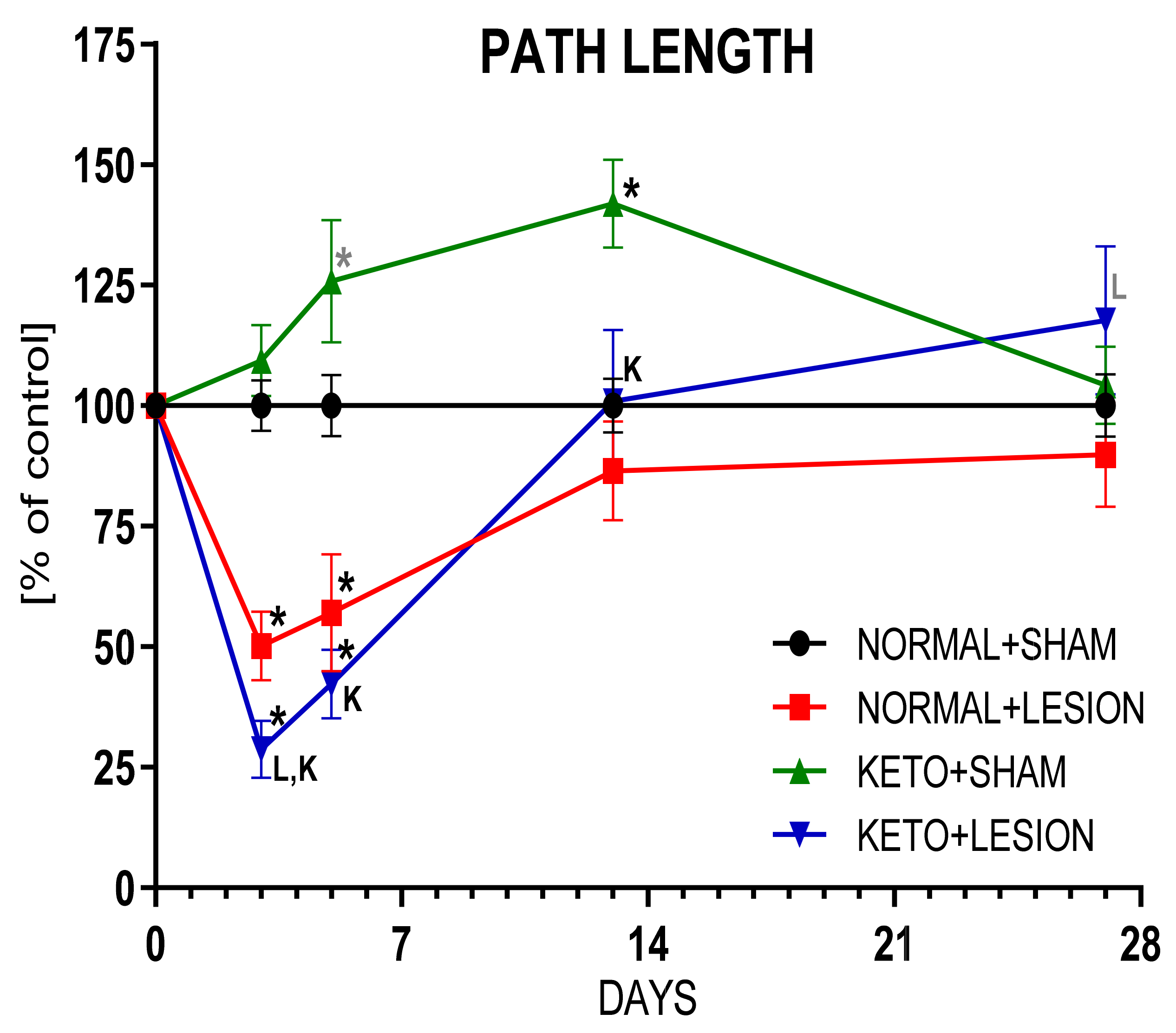
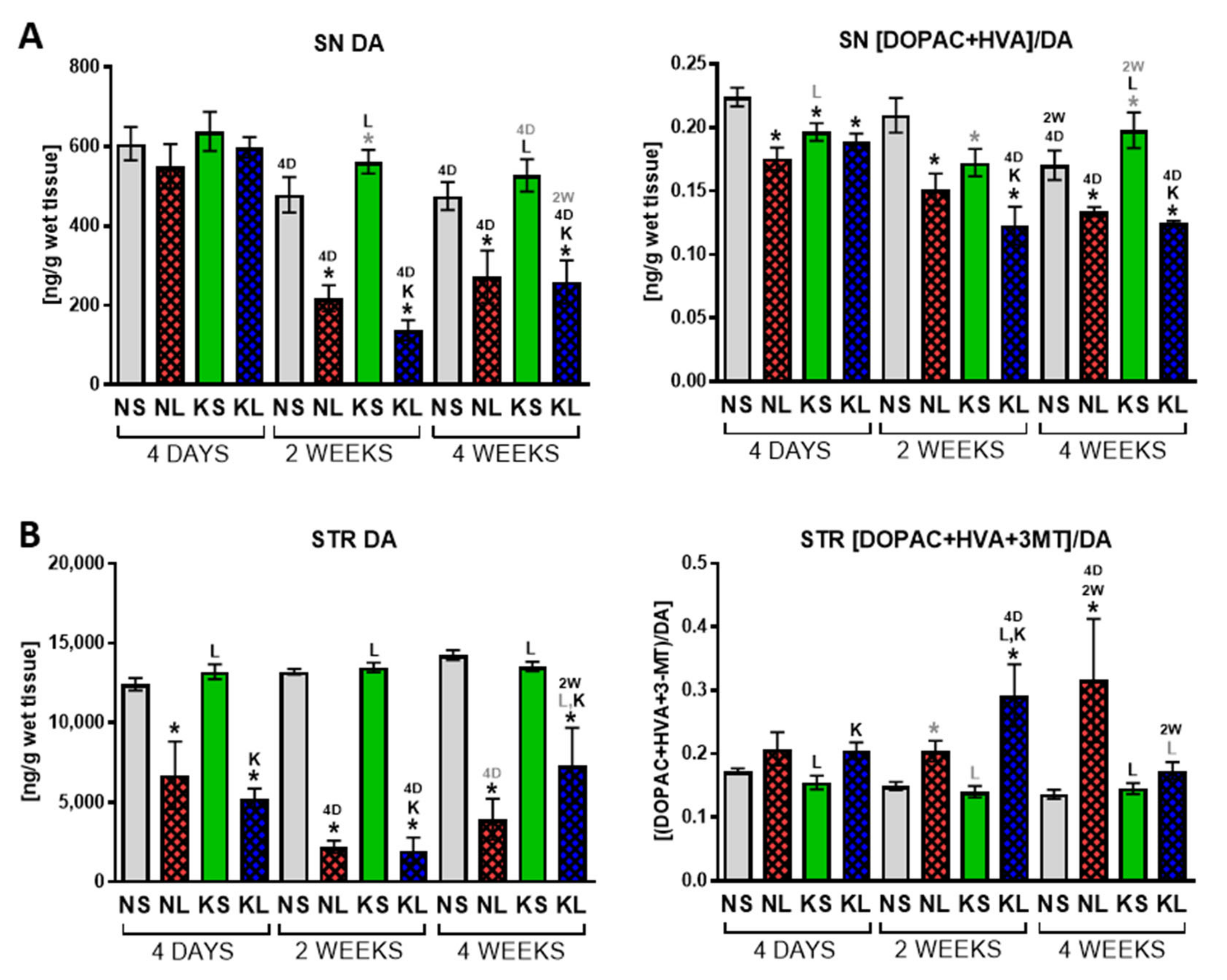
3.3. The Effect on Behaviour and DA Metabolism in the STR
3.4. The Effect on Glucose
3.5. Length of the Diet
3.6. The Effect of Age
3.7. Liver—Glycogenolysis and Gluconeogenesis as Possible Sources of Glucose
3.8. Mitochondria and OxPhos Cxs
3.9. Protein Amount in the Diet
4. Materials and Methods
4.1. Animals and Brain Surgery
4.2. Behavioural Analysis Using Automated Actometers
4.3. HPLC-EC Analysis of DA, Its Metabolites, and Turnover Rates
4.4. Immunohistochemistry and Stereological Counting Neurons
4.5. Quantitative Assays for Energy Metabolism Substrates
4.6. Mitochondrial Cx II and Cx IV Staining in the Tissue Sections
4.7. Glycogen Detection in the Tissue Sections
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hornykiewicz, O. Biochemical aspects of Parkinson’s disease. Neurology 1998, 51, 2–9. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernández-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Iborra, S.F.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Iborra, S.F.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- VanItallie, T.B.; Nonas, C.; DiRocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- McDonald, T.J.W.; Cervenka, M.C. Lessons learned from recent clinical trials of ketogenic diet therapies in adults. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 418–424. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Reynier, P.; Procaccio, V.; Hergesheimer, R.; Corcia, P.; Andres, C.R.; Blasco, H. How Can a Ketogenic Diet Improve Motor Function? Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2016, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rowlands, B.D.; Thomas, D.S.; Housley, G.D.; Rae, C.D. β-Hydroxybutyrate Boosts Mitochondrial and Neuronal Metabolism but is not Preferred over Glucose under Activated Conditions. Neurochem. Res. 2017, 42, 1710–1723. [Google Scholar] [CrossRef]
- Prins, M.L. Cerebral Metabolic Adaptation and Ketone Metabolism after Brain Injury. J. Cereb. Blood Flow Matab. 2008, 28. [Google Scholar] [CrossRef] [Green Version]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-β-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s Disease. Front. Nutr. 2019, 6. [Google Scholar] [CrossRef]
- Christensen, M.G.; Damsgaard, J.; Fink-Jensen, A. Use of ketogenic diets in the treatment of central nervous system diseases: A systematic review. Nord. J. Psychiatry 2020, 75. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic diet in neuromuscular and neurodegenerative diseases. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Rawat, K.; Singh, N.; Kumari, P.; Saha, L. A review on preventive role of ketogenic diet (KD) in CNS disorders from the gut microbiota perspective. Rev. Neurosci. 2020, 32, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczyk, T.; Wick, J.Y. The Ketogenic Diet: Making a Comeback. Consult. Pharm. 2017, 32, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.-C.; Yan, S.-D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X.; Liu, S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 2009, 1286, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Shaafi, S.; Najmi, S.; Aliasgharpour, H.; Mahmoudi, J.; Sadigh-Etemad, S.; Farhoudi, M.; Baniasadi, N. The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: A rat model. Iran. J. Neurol. 2016, 15, 63–69. [Google Scholar] [PubMed]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Elbarbry, F.; Nguyen, V.; Mirka, A.; Zwickey, H.; Rosenbaum, R. A new validated HPLC method for the determination of levodopa: Application to study the impact of ketogenic diet on the pharmacokinetics of levodopa in Parkinson’s participants. Biomed. Chromatogr. 2019, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.A.; Williamson, D.H.; Krebs, H.A. Ketone-body utilization by adult and suckling rat brain in vivo. Biochem. J. 1971, 122, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.A. Uptake of ketone bodies by rat brain in vivo. Biochem. J. 1971, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, M.L.; Matsumoto, J.H. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J. Lipid Res. 2014, 55, 2450–2457. [Google Scholar] [CrossRef] [Green Version]
- Veech, B.C.R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar] [CrossRef]
- LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. Ketones suppress brain glucose consumption. Adv. Exp. Med. Biol. 2009, 645, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steriade, C.; Andrade, D.M.; Faghfoury, H.; Tarnopolsky, M.A.; Tai, P. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr. Neurol. 2014, 50, 498–502. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Kephart, W.C.; Holland, A.M.; Mumford, P.; Mobley, C.B.; Lowery, R.P.; Roberts, M.D.; Wilson, J.M.; Kavazis, A.N. A Ketogenic Diet in Rodents Elicits Improved Mitochondrial Adaptations in Response to Resistance Exercise Training Compared to an Isocaloric Western Diet. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.-L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef]
- Kuter, K.; Olech, Ł.; Głowacka, U. Prolonged Dysfunction of Astrocytes and Activation of Microglia Accelerate Degeneration of Dopaminergic Neurons in the Rat Substantia Nigra and Block Compensation of Early Motor Dysfunction Induced by 6-OHDA. Mol. Neurobiol. 2018, 55, 3049–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuter, K.; Olech, Ł.; Głowacka, U.; Paleczna, M. Astrocyte support is important for the compensatory potential of the nigrostriatal system neurons during early neurodegeneration. J. Neurochem. 2019, 148, 63–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuter, K.Z.; Olech, Ł.; Dencher, N.A. Increased energetic demand supported by mitochondrial electron transfer chain and astrocyte assistance is essential to maintain the compensatory ability of the dopaminergic neurons in an animal model of early Parkinson’s disease. Mitochondrion 2019, 47, 227–237. [Google Scholar] [CrossRef]
- Stott, S.R.; Barker, R.A. Time course of dopamine neuron loss and glial response in the 6-OHDA striatal mouse model of Parkinson’s disease. Eur. J. Neurosci. 2014, 39, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Kuter, K.; Kratochwil, M.; Berghauzen-Maciejewska, K.; Głowacka, U.; Sugawa, M.D.; Ossowska, K.; Dencher, N.A. Adaptation within mitochondrial oxidative phosphorylation supercomplexes and membrane viscosity during degeneration of dopaminergic neurons in an animal model of early Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 741–753. [Google Scholar] [CrossRef]
- Foster, J.D.; Pederson, B.A.; Nordlie, R.C. Glucose-6-phosphatase structure, regulation, and function: An update. Proc. Soc. Exp. Biol. Med. 1997, 215, 314–332. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L.C.; Chittó, A.L.; Müller, A.P.; Rocha, J.K.; Da Silva, M.C.; Quincozes-Santos, A.; Nardin, P.; Rotta, L.N.; Ziegler, D.R.; Gonçalves, C.-A.; et al. Ketogenic diet-fed rats have increased fat mass and phosphoenolpyruvate carboxykinase activity. Mol. Nutr. Food Res. 2008, 52, 1365–1371. [Google Scholar] [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Truong, L.; Allbutt, H.; Kassiou, M.; Henderson, J.M. Developing a preclinical model of Parkinson’s disease: A study of behaviour in rats with graded 6-OHDA lesions. Behav. Brain Res. 2006, 169. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, K.; Lenda, T.; Konieczny, J.; Czarnecka, A.; Lorenc-Koci, E. Depressive-like neurochemical and behavioral markers of Parkinson’s disease after 6-OHDA administered unilaterally to the rat medial forebrain bundle. Pharmacol. Rep. 2017, 69, 985–994. [Google Scholar] [CrossRef]
- Joniec-Maciejak, I.; Wawer, A.; Turzyńska, D.; Sobolewska, A.; Maciejak, P.; Szyndler, J.; Mirowska-Guzel, D.; Plaznik, A. Octanoic acid prevents reduction of striatal dopamine in the MPTP mouse model of Parkinson’s disease. Pharmacol. Rep. 2018, 70, 988–992. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.J.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Puri, B.K.; Maes, M.; Olive, L.; Berk, M.; Carvalho, A.F. The role of microglia in neuroprogressive disorders: Mechanisms and possible neurotherapeutic effects of induced ketosis. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Claverie, M.; Garrido-Gil, P.; Sebastian, W.S.; Izal-Azcárate, A.; Belzunegui, S.; Marcilla, I.; López, B.; Luquin, M.-R. Acute and chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administrations elicit similar microglial activation in the substantia nigra of monkeys. J. Neuropathol. Exp. Neurol. 2009, 68, 977–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Ren, H.; Wood, K.; Li, M.; Qiu, S.; Shi, F.-D.; Ma, C.; Liu, Q. Depletion of microglia augments the dopaminergic neurotoxicity of MPTP. FASEB J. 2018, 32, 3336–3345. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, Y.; Shinagawa, R.; Yamada, M.; Mori, T.; Tateishi, N.; Fujita, S. Long-lasting reactive changes observed in microglia in the striatal and substantia nigral of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2007, 1138, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral Gluconeogenesis and Diseases. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melø, T.M.; Nehlig, A.; Sonnewald, U. Neuronal–glial interactions in rats fed a ketogenic diet. Neurochem. Int. 2006, 48, 498–507. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M.; Berk, M.; Carvalho, A.F.; Puri, B.K. Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 2020, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghauzen-Maciejewska, K.; Kuter, K.; Kolasiewicz, W.; Głowacka, U.; Dziubina, A.; Ossowska, K.; Wardas, J. Pramipexole but not imipramine or fluoxetine reverses the “depressive-like” behaviour in a rat model of preclinical stages of Parkinson’s disease. Behav. Brain Res. 2014, 271, 343–353. [Google Scholar] [CrossRef]
- Kuter, K.; Kolasiewicz, W.; Gołembiowska, K.; Dziubina, A.; Schulze, G.; Berghauzen, K.; Wardas, J.; Ossowska, K. Partial lesion of the dopaminergic innervation of the ventral striatum induces “depressive-like” behavior of rats. Pharmacol. Rep. 2011, 63, 1383–1392. [Google Scholar] [PubMed]
- Bielohuby, M.; Menhofer, D.; Kirchner, H.; Stoehr, B.J.M.; Müller, T.D.; Stock, P.; Hempel, M.; Stemmer, K.; Pfluger, P.T.; Kienzle, E.; et al. Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Church, W.H.; Adams, R.E.; Wyss, L.S. Ketogenic diet alters dopaminergic activity in the mouse cortex. Neurosci. Lett. 2014, 571, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Barrio, J.R.; Huang, S.C.; Melega, W.P.; Yu, D.C.; Hoffman, J.M.; Schneider, J.S.; Satyamurthy, N.; Mazziotta, J.C.; Phelps, M.E. 6-[18F]fluoro-L-dopa probes dopamine turnover rates in central dopaminergic structures. J. Neurosci. Res. 1990, 27, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Sossi, V.; De La Fuente-Fernández, R.; Holden, J.E.; Doudet, D.J.; McKenzie, J.; Stoessl, A.J.; Ruth, T.J. Increase in dopamine turnover occurs early in Parkinson’s disease: Evidence from a new modeling approach to PET 18F-fluorodopa data. J. Cereb. Blood Flow Metab. 2002, 22, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.; Truckenbrod, L.; Federico, Q.; Campos, K.; Moon, B.; Ferekides, N.; Hoppe, M.; D’Agostino, D.; Burke, S. Metabolic switching is impaired by aging and facilitated by ketosis independent of glycogen. Aging 2020, 12, 7963–7984. [Google Scholar] [CrossRef] [PubMed]
- Al-Mudallal, A.S.; Lamanna, J.C.; Lust, W.D.; Harik, S.I. Diet-induced ketosis does not cause cerebral acidosis. Epilepsia 1996, 37, 258–261. [Google Scholar] [CrossRef]
- Kosinski, C.; Jornayvaz, F.R. Effects of Ketogenic Diets on Cardiovascular Risk Factors: Evidence from Animal and Human Studies. Nutrients 2017, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Veldhorst, M.A.; Westerterp-Plantenga, M.S.; Westerterp, K.R. Gluconeogenesis and energy expenditure after a high-protein, carbohydrate-free diet. Am. J. Clin. Nutr. 2009, 90, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsyad, A.; Idris, I.; Rasyid, A.A.; Usman, R.A.; Faradillah, K.R.; Latif, W.O.U.; Lubis, Z.I.; Aminuddin, A.; Yustisia, I.; Djabir, Y.Y. Long-Term Ketogenic Diet Induces Metabolic Acidosis, Anemia, and Oxidative Stress in Healthy Wistar Rats. J. Nutr. Metab. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-J.; Han, H.-S.; Kim, M.-J.; Koo, S.-H. Transcriptional regulators of hepatic gluconeogenesis. Arch. Pharmacal Res. 2013, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Bisschop, P.H.; Arias, A.M.P.; Ackermans, M.T.; Endert, E.; Pijl, H.; Kuipers, F.; Meijer, A.J.; Sauerwein, H.P.; Romijn, J.A. The effects of carbohydrate variation in isocaloric diets on glycogenolysis and gluconeogenesis in healthy men. J. Clin. Endocrinol. Metab. 2000, 85, 1963–1967. [Google Scholar] [CrossRef] [Green Version]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kim, D.S.; Kang, S.; Daily, J.W. A ketogenic diet impairs energy and glucose homeostasis by the attenuation of hypothalamic leptin signaling and hepatic insulin signaling in a rat model of non-obese type 2 diabetes. Exp. Biol. Med. 2011, 236, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.C.; Connolly, C.C.; Cherrington, A.D. Autoregulation of hepatic glucose production. Eur. J. Endocrinol. 1998, 138, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, R.W.; Garber, A.J. Phosphoenolpyruvate carboxykinase. I. Its role in gluconeogenesis. Am. J. Clin. Nutr. 1972, 25, 1010–1021. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, S.; Ferguson, S.; Wang, L.; Klepcyk, P.; Yun, J.S.; Friedman, J.E. Phosphoenolpyruvate carboxykinase overexpression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J. Biol. Chem. 2002, 277, 23301–23307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutfles, L.J.; Wilkins, H.M.; Koppel, S.J.; Weidling, I.W.; Selfridge, J.E.; Tan, E.; Thyfault, J.P.; Slawson, C.; Fenton, A.W.; Zhu, H.; et al. A bioenergetics systems evaluation of ketogenic diet liver effects. Appl. Physiol. Nutr. Metab. 2017, 42, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Bosma, K.J.; Rahim, M.; Oeser, J.K.; McGuinness, O.P.; Young, J.D.; O’Brien, R.M. G6PC2 confers protection against hypoglycemia upon ketogenic diet feeding and prolonged fasting. Mol. Metab. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Kading, J.; Finck, B.N.; DeBosch, B.J. Targeting hepatocyte carbohydrate transport to mimic fasting and calorie restriction. FEBS J. 2020, 288, 3784–3798. [Google Scholar] [CrossRef]
- Garnol, T.; Endlicher, R.; Kucera, O.; Drahota, Z.; Červinková, Z. Impairment of mitochondrial function of rat hepatocytes by high fat diet and oxidative stress. Physiol. Res. 2014, 63, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Diogo, C.V.; Mastrodonato, M.; de Bari, O.; Persichella, M.; Wang, D.Q.; Liquori, A.; Ferri, D.; Carratù, M.R.; Oliveira, P.J.; et al. A silybin-phospholipids complex counteracts rat fatty liver degeneration and mitochondrial oxidative changes. World J. Gastroenterol. 2013, 19, 3007–3017. [Google Scholar] [CrossRef]
- Strifler, G.; Tuboly, E.; Szél, E.; Kaszonyi, E.; Cao, C.; Kaszaki, J.; Meszaros, A.; Boros, M.; Hartmann, P. Inhaled Methane Limits the Mitochondrial Electron Transport Chain Dysfunction during Experimental Liver Ischemia-Reperfusion Injury. PLoS ONE 2016, 11, e0146363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, W.; Jelnes, P.; Rud, K.A.; Hansen, L.L.; Grunnet, N.; Quistorff, B. Progression of type 2 diabetes in GK rats affects muscle and liver mitochondria differently: Pronounced reduction of complex II flux is observed in liver only. Am. J. Physiol. Endocrinol. Metab. 2012, 303, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Oke, S.L.; Sohi, G.; Hardy, D.B. Perinatal protein restriction with postnatal catch-up growth leads to elevated p66Shc and mitochondrial dysfunction in the adult rat liver. Reproduction 2020, 159, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Kolasiewicz, W.; Kuter, K.; Berghauzen-Maciejewska, K.; Nowak, P.; Schulze, G.; Ossowska, K. 6-OHDA injections into A8–A9 dopaminergic neurons modelling early stages of Parkinson’s disease increase the harmaline-induced tremor in rats. Brain Res. 2012, 1477, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kuter, K.; Śmiałowska, M.; Wierońska, J.; Zięba, B.; Wardas, J.; Pietraszek, M.; Nowak, P.; Biedka, I.; Roczniak, W.; Konieczny, J.; et al. Toxic influence of subchronic paraquat administration on dopaminergic neurons in rats. Brain Res. 2007, 1155, 196–207. [Google Scholar] [CrossRef]
- Haan, C.; Behrmann, I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J. Immunol. Methods 2007, 318, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welinder, C.; Ekblad, L. Coomassie Staining as Loading Control in Western Blot Analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Beckett, T.L.; Studzinski, C.M.; Keller, J.N.; Murphy, M.P.; Niedowicz, D.M. A ketogenic diet improves motor performance but does not affect β-amyloid levels in a mouse model of Alzheimer’s Disease. Brain Res. 2013, 1505, 61–67. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Group | Path | Locomotion | Resting | Rearing | Rearing | Rearing | Rearing |
|---|---|---|---|---|---|---|---|---|
| Length | Time | Time | Time | Total | Supported | Free | ||
| (cm) | (% of Time) | (s) | (s) | (Number) | (Number) | (Number) | ||
| 3 DAYS | NS | 222.1 ± 11.7 | 13.2 ± 0.8 | 9558 ± 833 | 91.1 ± 4.4 | 10.0 ± 0.8 | 4.9 ± 0.5 | 5.1 ± 0.4 |
| NL | 111.6 * ± 15.8 | 4.9* ± 1.0 | 12,643 * ± 885 | 55.4 * ± 8.3 | 3.4 * ± 0.6 | 1.9 * ± 0.3 | 1.6 * ± 0.3 | |
| KS | 243.0 L ± 16.5 | 14.7 L ± 1.1 | 10,881 ± 731 | 61.9 * ± 3.9 | 8.5 L ± 0.9 | 4.8 L ± 0.5 | 3.6 *,L ± 0.4 | |
| KL | 63.8 *LK ± 13.2 | 2.6 *,K ± 0.9 | 16,395 *,L,K ± 1957 | 23.9 *,L,K ± 6.0 | 0.8 *,L,K ± 0.3 | 0.4 *,L,K ± 0.1 | 0.3 *,L,K ± 0.1 | |
| 5 DAYS | NS | 275.6 ± 17.6 | 16.2 ± 1.3 | 6334 ± 689 | 114.3 ± 8.9 | 12.5 ± 0.9 | 6.8 ± 0.7 | 5.6 ± 0.5 |
| NL | 157.0 * ± 33.6 | 7.8 * ± 2.4 | 11,627 * ± 1009 | 110.7 ± 24.3 | 4.8 * ± 1.5 | 2.5 * ± 0.9 | 2.3 * ± 0.6 | |
| KS | 346.7 *,L ± 35.1 | 20.8 *,L ± 2.2 | 6459 L ± 667 | 79.8 ± 8.1 | 10.6 L ± 1.6 | 6.4 L ± 1.0 | 3.7 *,L ± 0.4 | |
| KL | 116.5 *,K ± 19.7 | 4.5 *,K ± 1.0 | 13,814 *,K ± 1781 | 56.7 *,L,K ± 12.7 | 2.9 *,K ± 0.9 | 1.4 *,K ± 0.4 | 1.5 *,K ± 0.6 | |
| 13 DAYS | NS | 246.7 ± 13.8 | 13.4 ± 1.0 | 6871 ± 526 | 125.4 ± 5.4 | 10.9 ± 0.9 | 5.9 ± 0.6 | 5.0 ± 0.3 |
| NL | 213.4 ± 25.3 | 10.2 ± 1.8 | 6215 ± 692 | 168.1 * ± 17.1 | 10.7 ± 1.1 | 5.4 ± 0.8 | 4.3 ± 0.7 | |
| KS | 330.1 *,K ± 22.5 | 20.3 *,L ± 1.5 | 5285 ± 1044 | 101.7 L ± 9.0 | 10.7 ± 1.0 | 6.9 ± 0.8 | 3.6 *,L ± 0.3 | |
| KL | 248.8 K ± 36.6 | 12.32 K ± 2.8 | 7575 K ± 1194 | 119.8 L ± 11.5 | 7.2 *,L,K ± 1.2 | 4.4 K ± 0.7 | 2.3 *,L ± 0.6 | |
| 27 DAYS | NS | 294.2 ± 19.2 | 17.2 ± 1.3 | 4868 ± 617 | 130.6 ± 8.0 | 14.3 ± 0.8 | 8.1 ± 0.6 | 6.2 ± 0.3 |
| NL | 264.3 ± 31.7 | 13.7 ± 2.2 | 7705 * ± 1317 | 148.2 ± 11.4 | 12.2 ± 1.3 | 6.8 ± 0.8 | 5.4 ± 0.7 | |
| KS | 306.8 ± 23.6 | 18.4 ± 1.6 | 6616 ± 971 | 99.3 *,L ± 6.6 | 9.6 * ± 0.9 | 5.6 * ± 0.7 | 4.0 * ± 0.3 | |
| KL | 346.3 L ± 45.1 | 18.9 ± 2.9 | 8360 * ± 1313 | 112.6 L ± 11.7 | 8.5 *,L ± 1.7 | 4.9 * ± 0.9 | 3.6 *,L ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuter, K.Z.; Olech, Ł.; Głowacka, U.; Paleczna, M. Increased Beta-Hydroxybutyrate Level Is Not Sufficient for the Neuroprotective Effect of Long-Term Ketogenic Diet in an Animal Model of Early Parkinson’s Disease. Exploration of Brain and Liver Energy Metabolism Markers. Int. J. Mol. Sci. 2021, 22, 7556. https://doi.org/10.3390/ijms22147556
Kuter KZ, Olech Ł, Głowacka U, Paleczna M. Increased Beta-Hydroxybutyrate Level Is Not Sufficient for the Neuroprotective Effect of Long-Term Ketogenic Diet in an Animal Model of Early Parkinson’s Disease. Exploration of Brain and Liver Energy Metabolism Markers. International Journal of Molecular Sciences. 2021; 22(14):7556. https://doi.org/10.3390/ijms22147556
Chicago/Turabian StyleKuter, Katarzyna Z., Łukasz Olech, Urszula Głowacka, and Martyna Paleczna. 2021. "Increased Beta-Hydroxybutyrate Level Is Not Sufficient for the Neuroprotective Effect of Long-Term Ketogenic Diet in an Animal Model of Early Parkinson’s Disease. Exploration of Brain and Liver Energy Metabolism Markers" International Journal of Molecular Sciences 22, no. 14: 7556. https://doi.org/10.3390/ijms22147556
APA StyleKuter, K. Z., Olech, Ł., Głowacka, U., & Paleczna, M. (2021). Increased Beta-Hydroxybutyrate Level Is Not Sufficient for the Neuroprotective Effect of Long-Term Ketogenic Diet in an Animal Model of Early Parkinson’s Disease. Exploration of Brain and Liver Energy Metabolism Markers. International Journal of Molecular Sciences, 22(14), 7556. https://doi.org/10.3390/ijms22147556