
Chromatin Profiling Techniques: Exploring the Chromatin Environment and Its Contributions to Complex Traits


Abstract
1. Introduction
The Chromatin Environment

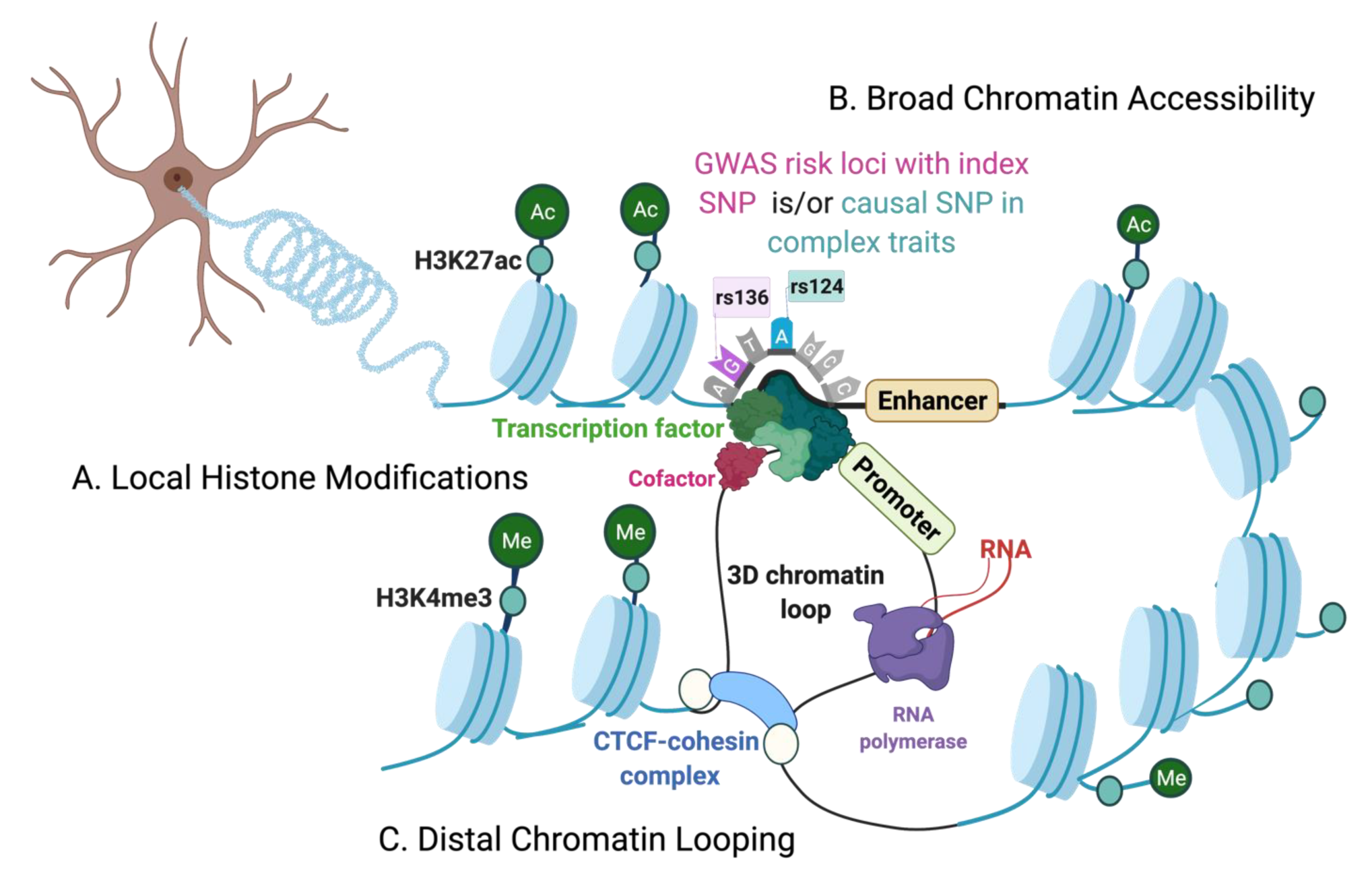{kind=link}
| Epigenomic Features | Techniques | Methods Overview | Benefits | Limitations | Single-Cell and Cell-Types |
|---|---|---|---|---|---|
| 1. Open chromatin regions. 2. Cis-regulatory elements. | DNase I hypersensitive sites sequencing (DNase-seq). [14] | DNase I digested fragments are extracted using biotin-streptavidin complex. | 1. High signal-to-noise ratio compared to FAIRE-seq. 2. No prior knowledge of locus-specific sequences, primers, or epitope tags is required. 3. Efficiently maps non-coding regions proximal to genes. | 1. DNase I sequence-specific cleavage biases may determine cleavage patterns at the predicted transcription factor (TF) binding sites or footprints. This complicates correctly assessing true transcription factor binding at open chromatin. [15] 2. Requires high number of cells (ideally >= 1 M cells) [14] and a high sequencing depth. 3. Maps relatively low distal regulatory sites compared to formaldehyde-assisted isolation of regulatory elements with sequencing (FAIRE-seq). [16] | Single-cell (sc)-DNase-seq. [17] |
| 1. Nucleosome positioning. 2. DNA-bound protein binding sites. | Micrococcal nuclease digestion of chromatin followed by sequencing (MNase-seq) [18], (alternative: nucleosome occupancy and methylome sequencing (NOME-seq). [19] | Cross-linking to covalently link proteins to the DNA, followed by micrococcal nuclease digestion to remove free DNA. | 1. MNase-seq can map DNA-protein binding for both histone and non-histone proteins. 2. Indirectly maps chromatin accessibility. 3. The digested fraction of accessible chromatin can be repurposed for chromatin immunoprecipitation-based assays (Native-ChIP). | 1. Requires a broad range of sequencing read-out (25 bps to 150 bps) to capture both sub-nucleosome and nucleosome fragments. [20] 2. High dependency on optimized MNase enzyme digestion for reproducibility between experiments. 3. MNase enzyme produces AT cleavage bias that needs bioinformatic corrections. 4. Requires large number of cellular input (ideally >= 1 M cells). | scMNase-seq, and scNOME-seq. [21,22,23,24] |
| 1. Open chromatin. 2. Cis-regulatory elements. 3. Nucleosome distribution. | Assay for transposase-accessible chromatin coupled to sequencing (ATAC-seq). [25] | Tn5 transposases-based cutting and tagging of open chromatin. | 1. Low input (ideally <= 50,000 cells) 2. Short and easy to use protocol. 3. Very high signal-to-noise ratio compared to other chromatin accessibility techniques. | 1. Tn5 sequence insertion bias can lead to mapping and/or TF footprinting biases and needs bioinformatic corrections. 2. Mitochondrial contamination of reads (although Omni-ATAC [26] is optimized for lower mitochondrial reads). | Flow cytometry-based approaches and single cell/nucleus ATAC-seq. [27,28,29,30,31,32] |
| 1. Protein-DNA interactions. 2. Histone post-translational modification. | Chromatin immunoprecipitation with sequencing (ChIP-seq). [33,34,35] | Formaldehyde crosslinked (X-ChIP) or micrococcal digested fragments (Native-ChIP) followed by immunoprecipitation. | 1. Gold standard to map genome-wide, direct DNA-protein interactions. 2. Single-nucleotide resolution (compared to ChIP-qPCR and ChIP-chip). 3. An ultra-low-input micrococcal nuclease-based native ChIP (ULI-NChIP) can profile genome-wide binding sites of histone proteins with as few as 1000 cells. [36] | 1. Cross-linking and sonication steps (X-ChIP) can lead to high background noise, requiring higher cellular input for optimal signal-to-noise ratio. [33] 2. Relies on the availability and quality of specific antibodies and can suffer from epitope masking due to cross-linking of fragments (X-ChIP). 3. Requires appropriate control experiments to minimize detection of false-positive protein-DNA binding sites. | sc-ChIP-seq [37] |
| 1. Protein-DNA interactions. 2. Histone post-translational modification. | ChIP with exonuclease (ChIP-exo) [38], Cleavage under targets & release using nuclease (CUT&RUN) [39], Cleavage under targets and tagmentation. (CUT&TAG) [40] | ChIP-exo: X-ChIP immunoprecipitated fragments followed by additional λ exonuclease digestion step. CUT&RUN: MNase tethered protein A, targeting specific antibody against the protein of interest. CUT&TAG: Tn5 transposase and protein A fusion protein, targeting antibody against the protein of interest. | 1.ChIP-exo: with an extra exonuclease treatment, it can remove unbound and non-specific DNA, providing higher signal-to-noise ratio over ChIP-seq. [38] 2. CUT&RUN: (i) Uses enzyme-tethering to avoid cross-linking and fragmentation of DNA that greatly reduces the background noise, and epitope masking, making it lower input over ChIP. (ii) It has been validated to map H3K27me3-marked heterochromatin regions. [39] (iii)Use of enzyme-tethering also maps local environment of binding sites, making it suitable to also detect long-range interactions of the protein. 3. CUT&TAG: (i) Requires the least number of cells compared to alternatives (ideally >= 100 cells) and can be performed at single-cell level. [40] (ii) It bypasses cross-linking (compared to ChIP) and library preparation step (compared to ChIP and CUT&RUN). (iii) More sensitive, easier workflow and cost-effective compared to CUT&RUN and alternatives | 1. ChIP-exo: High number of enzymatic steps in ChIP-exo makes it technically challenging and suffers from epitope masking, similar to ChIP. 2.CUT&RUN: (i) Calcium-activated MNase enzyme digestion of chromatin needs to be carefully optimized, to prevent over/under digestion of accessible chromatin. It also relies on antibody quality, like ChIP. (ii) Like X-ChIP, CUT&RUN cannot distinguish direct from indirect 3D contacts. [39] (iii) Requires higher number of cells relative to CUT&TAG (ideally >= 100,000 but can be performed with as low as 1000 cells). [39] 3. CUT&TAG: (i) A potential limitation is antibody-validation, since mapping certain protein-DNA interactions can be more efficient after cross-linking. (ii) Tn5 enzyme biases may confound detection of proteins at heterochromatin regions, since Tn5 preferentially tags accessible chromatin | CUT&TAG [40] |
| 3. Chromatin loops and 3D interactions. | Chromosome Conformation Capture 3C [41], 4C [42], 5C [43], and Hi-C. [44] | Formaldehyde cross-linking to covalently link physically interacting chromatin fragments. | 3C/4C/5C: these progressive modifications can map increasingly more chromatin conformations, i.e., one-to-one, one-to-many, and many-to-many epigenetic features, respectively. Hi-C (all-to-all): 1. An unbiased approach that maps genome-wide 3D chromatin conformations. 2. Long-range interactions several mega-base pairs away and high-resolution inter-chromosomal contacts can also be mapped. 3. Low cellular input over 3C/4C (ideally >= 1 M cells). Easy-Hi-C: a biotin-free strategy, more sensitive and requires relatively lower cell input over Hi-C (ideally >= 50,000 cells). [45] | 3C/4C/5C: 1. Maps to a limited resolution and genomic distances of interacting regions. 2. Need priori-defined regions of interests. 3. Cannot resolve long-range contacts by haplotypes (maternal/paternal) of the chromosomes. 4. Requires relatively higher number of cells (ideally >= 10M cells). Hi-C: (i) It cannot detect chromatin contacts with cell-type specificity and cannot detect functional relevance of the chromatin loops. (ii) Some proximity-ligation events can remain undetected due to low efficiency of biotin incorporation at ligation junctions. [45] | Flow cytometry-based approaches [46,47], sc-Hi-C-seq [48,49], sci-Hi-C-seq [50,51], Dip-C [52] |
| 4. Protein-bound 3D interactions | Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) [53], HiChIP [54], and Proximity ligation-assisted ChIP-seq (PLAC-seq). [55] | Formaldehyde cross-linking, followed by antibody-based immunoprecipitation of protein-bound chromatin interactions. | ChIA-PET, HiChIP & PLAC-seq: Can illustrate regulatory roles of 3D chromatin interactions. HiChIP & PLAC-seq: Higher signal-to-noise ratio and significantly lower cell input compared to ChIA-PET. | ChIA-PET: 1. Low sensitivity in detecting 3D interactions and can have false-positive reads by non-specific antibody binding. 2. Requires very high number of cellular input (ideally >= 100 M cells) [54,56] and high sequencing depth. 3. Ligation of DNA linkers to chromatin fragments can also lead to self-ligation of linkers and false-positive read-outs. ChIA-PET, HiChIP, and PLAC-seq: They all require a priori of target protein of interest and need bioinformatic correction for biases introduced by: ChIP procedure, different fragment lengths, and restriction enzymes cut-site biases. HiChIP and PLAC-seq also require high cell-number (ideally >= 1 M cells). | Flow cytometry approach [55,57], and multiplex chromatin interaction analysis via droplet-based and barcode-linked sequencing (ChIA-Drop) [58] |
2. Chromatin Accessibility Techniques
2.1. DNase I Hypersensitive Sites Sequencing (DNase-seq)
2.2. Formaldehyde-Assisted Isolation of Regulatory Elements with Sequencing (FAIRE-seq)
2.3. Micrococcal Nuclease Digestion of Chromatin Followed by Sequencing (MNase-seq)
2.4. Assay for Transposase-Accessible Chromatin (ATAC-seq)
3. Chromatin-Bound Proteins and Histone Modifications
3.1. Chromatin Immunoprecipitation with Sequencing (ChIP-seq)
3.2. ChIP-seq Alternatives
3.2.1. DNA Adenine Methyltransferase (DAM)-Identification (DamID)
3.2.2. Cleavage under Targets and Release Using Nuclease (CUT&RUN)
4. 3D Chromatin Interactions: Techniques and Applications
4.1. Chromosome Conformation Capture
4.1.1. 3C: One-to-One Mapping
4.1.2. 4C: One-to-Many Mapping
4.1.3. 5C: Many-to-Many Mapping
4.1.4. Hi-C: All-to-All Mapping
4.2. Protein-Centric 3D Interactions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association studies |
| LD | Linkage disequilibrium |
| SNPs | Single nucleotide polymorphisms |
| INDELs | Insertion/deletion polymorphisms |
| ENCODE | Encyclopedia of DNA Elements |
| TFs | Transcription factors |
| TSSs | Transcription start sites |
| CTCF | CCCTC-binding factor |
| TADs | Topologically associated domains |
| NPCs | Neural progenitor cells |
| FAC/NS | Fluorescence assisted cell/nuclei sorting |
| RNAPII | RNA polymerases II |
| DHSs | DNase I hypersensitive sites |
| DNase-seq | DNase I hypersensitive sites sequencing |
| MNase-seq | Micrococcal nuclease digestion of chromatin followed by sequencing |
| FAIRE-seq | Formaldehyde-assisted isolation of regulatory elements with sequencing |
| ATAC-seq | Assay for transposase-accessible chromatin coupled to sequencing |
| ChIP-seq | Chromatin immunoprecipitation with sequencing |
| CUT&RUN | Cleavage under targets and release using nuclease |
| DamID | DNA adenine methyltransferase (DAM)-identification |
| ChIA-PET | Chromatin interaction analysis by paired-end tag sequencing |
| PLAC-seq | Proximity ligation-assisted ChIP-Seq |
References
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.G.; Farnham, P.J. Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenet. Chromatin 2015, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; St Clair, D.; Weinberger, D.R.; Wendland, J.R.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- O’Connor, L.J.; Schoech, A.P.; Hormozdiari, F.; Gazal, S.; Patterson, N.; Price, A.L. Extreme polygenicity of complex traits is explained by negative selection. Am. J. Hum. Genet. 2019, 105, 456–476. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Milosavljevic, A.; Ren, B.; Stamatoyannopoulos, J.A.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Lutz, P.-E.; Tanti, A.; Gasecka, A.; Barnett-Burns, S.; Kim, J.J.; Zhou, Y.; Chen, G.G.; Wakid, M.; Shaw, M.; Almeida, D.; et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: Convergent epigenetic, transcriptional, and morphological evidence. AJP 2017, 174, 1185–1194. [Google Scholar] [CrossRef]
- Labonté, B.; Suderman, M.; Maussion, G.; Navaro, L.; Yerko, V.; Mahar, I.; Bureau, A.; Mechawar, N.; Szyf, M.; Meaney, M.J.; et al. Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry 2012, 69, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Patrick, E.; Taga, M.; Ergun, A.; Ng, B.; Casazza, W.; Cimpean, M.; Yung, C.; Schneider, J.A.; Bennett, D.A.; Gaiteri, C.; et al. Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. PLoS Comput. Biol. 2020, 16, e1008120. [Google Scholar] [CrossRef]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005, 12, 46–53. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- DDD Study; Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G.; Santoro, S.L.; Shuss, C.; Ziegler, A.; et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 2019, 21, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Crawford, G.E. DNase-Seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.-H.; Guertin, M.J.; Baek, S.; Hager, G.L. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol. Cell 2014, 56, 275–285. [Google Scholar] [CrossRef]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ahmad, K.; Henikoff, S. Transcription and remodeling produce asymmetrically unwrapped nucleosomal intermediates. Mol. Cell 2017, 68, 1038–1053.e4. [Google Scholar] [CrossRef]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef]
- Henikoff, J.G.; Belsky, J.A.; Krassovsky, K.; MacAlpine, D.M.; Henikoff, S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. USA 2011, 108, 18318–18323. [Google Scholar] [CrossRef]
- Gao, W.; Lai, B.; Ni, B.; Zhao, K. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scmnase-seq. Nat. Protoc. 2020, 15, 68–85. [Google Scholar] [CrossRef]
- Lai, B.; Gao, W.; Cui, K.; Xie, W.; Tang, Q.; Jin, W.; Hu, G.; Ni, B.; Zhao, K. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature 2018, 562, 281–285. [Google Scholar] [CrossRef]
- Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 2017, 6, e23203. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.-A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. ScNMT-Seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 2018, 9, 781. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-Seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Fullard, J.F.; Hauberg, M.E.; Bendl, J.; Egervari, G.; Cirnaru, M.-D.; Reach, S.M.; Motl, J.; Ehrlich, M.E.; Hurd, Y.L.; Roussos, P. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018, 28, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Trevino, A.E.; Sinnott-Armstrong, N.; Andersen, J.; Yoon, S.-J.; Huber, N.; Pritchard, J.K.; Chang, H.Y.; Greenleaf, W.J.; Pașca, S.P. Chromatin accessibility dynamics in a model of human forebrain development. Science 2020, 367, eaay1645. [Google Scholar] [CrossRef]
- Hauberg, M.E.; Creus-Muncunill, J.; Bendl, J.; Kozlenkov, A.; Zeng, B.; Corwin, C.; Chowdhury, S.; Kranz, H.; Hurd, Y.L.; Wegner, M.; et al. Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat. Commun. 2020, 11, 5581. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Ziffra, R.S.; Kim, C.N.; Wilfert, A.; Turner, T.N.; Haeussler, M.; Casella, A.M.; Przytycki, P.F.; Kreimer, A.; Pollard, K.S.; Ament, S.A.; et al. Single cell epigenomic atlas of the developing human brain and organoids. Dev. Biol. 2019. preprint. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-Seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Brind’Amour, J.; Liu, S.; Hudson, M.; Chen, C.; Karimi, M.M.; Lorincz, M.C. An Ultra-Low-Input Native ChIP-Seq Protocol for Genome-Wide Profiling of Rare Cell Populations. Nat Commun. 2015, 6, 6033. [Google Scholar] [CrossRef] [PubMed]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-Seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.S.; Pugh, B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-Chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, X.; Huang, W.-K.; Giusti-Rodríguez, P.; Cui, J.; Zhang, S.; Xu, W.; Wen, Z.; Ma, S.; Rosen, J.D.; et al. Robust Hi-C maps of enhancer-promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol. Cell 2020, 79, 521–534.e15. [Google Scholar] [CrossRef] [PubMed]
- Espeso-Gil, S.; Halene, T.; Bendl, J.; Kassim, B.; Hutta, G.B.; Iskhakova, M.; Shokrian, N.; Auluck, P.; Javidfar, B.; Rajarajan, P.; et al. A chromosomal connectome for psychiatric and metabolic risk variants in adult dopaminergic neurons. Genome Med. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; le Dily, F.; Garcia, F.; Salvà-Castro, J.; Gómez, H.L.; Julià, E.; Moutinho, C.; Cigliano, R.A.; et al. Three-dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 2019, 28, 352–367.e9. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Lee, C.; Disteche, C.M.; Noble, W.S.; Shendure, J.; Duan, Z. Sci-Hi-C: A single-Cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 2020, 170, 61–68. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef]
- Tan, L.; Xing, D.; Chang, C.-H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Fang, R.; Yu, M.; Li, G.; Chee, S.; Liu, T.; Schmitt, A.D.; Ren, B. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-Seq. Cell Res. 2016, 26, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Song, M.; Pebworth, M.-P.; Yang, X.; Abnousi, A.; Fan, C.; Wen, J.; Rosen, J.D.; Choudhary, M.N.K.; Cui, X.; Jones, I.R.; et al. Cell-type-specific 3D epigenomes in the developing human cortex. Nature 2020, 587, 644–649. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, S.Z.; Capurso, D.; Kim, M.; Maurya, R.; Lee, B.; Piecuch, E.; Gong, L.; Zhu, J.J.; Li, Z.; et al. Multiplex chromatin interactions with single-molecule precision. Nature 2019, 566, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, D.A.; Chan, U.; Chen, L.-F.; West, A.E. chromatin regulation of neuronal maturation and plasticity. Trends Neurosci. 2018, 41, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.E.; Davis, S.; Scacheri, P.C.; Renaud, G.; Halawi, M.J.; Erdos, M.R.; Green, R.; Meltzer, P.S.; Wolfsberg, T.G.; Collins, F.S. DNase-Chip: A high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 2006, 3, 503–509. [Google Scholar] [CrossRef]
- Dorschner, M.O.; Hawrylycz, M.; Humbert, R.; Wallace, J.C.; Shafer, A.; Kawamoto, J.; Mack, J.; Hall, R.; Goldy, J.; Sabo, P.J.; et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 2004, 1, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Funk, C.C.; Casella, A.M.; Jung, S.; Richards, M.A.; Rodriguez, A.; Shannon, P.; Donovan-Maiye, R.; Heavner, B.; Chard, K.; Xiao, Y.; et al. Atlas of transcription factor binding sites from ENCODE DNase hypersensitivity data across 27 tissue types. Cell Rep. 2020, 32, 108029. [Google Scholar] [CrossRef]
- Shibata, Y.; Sheffield, N.C.; Fedrigo, O.; Babbitt, C.C.; Wortham, M.; Tewari, A.K.; London, D.; Song, L.; Lee, B.-K.; Iyer, V.R.; et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012, 8, e1002789. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, X.; Yu, H.; Li, J.; Jiang, Z.; Chen, B.; Lu, Y.; Wang, W.; Han, C.; Ouyang, Y.; et al. Evolution and comprehensive analysis of DNaseI hypersensitive sites in regulatory regions of primate brain-related genes. Front. Genet. 2019, 10, 152. [Google Scholar] [CrossRef]
- ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; The RACI Consortium; Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.-K.; Sheffield, N.C.; Graf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767. [Google Scholar] [CrossRef]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.-W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity 2015, 42, 186–198. [Google Scholar] [CrossRef]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-Seq and FAIRE-Seq open chromatin profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef]
- Voong, L.N.; Xi, L.; Wang, J.-P.; Wang, X. Genome-wide mapping of the nucleosome landscape by micrococcal nuclease and chemical mapping. Trends Genet. 2017, 33, 495–507. [Google Scholar] [CrossRef]
- Ozsolak, F.; Song, J.S.; Liu, X.S.; Fisher, D.E. High-throughput mapping of the chromatin structure of human promoters. Nat. Biotechnol. 2007, 25, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, P.G.; Pedersen, B.A.; Taylor, J.F.; Khattab, O.S.; Chen, Y.-H.; Chen, Y.; Jacobsen, S.E.; Wang, P.H. Nucleosome organization in human embryonic stem cells. PLoS ONE 2015, 10, e0136314. [Google Scholar] [CrossRef]
- Teif, V.B.; Vainshtein, Y.; Caudron-Herger, M.; Mallm, J.-P.; Marth, C.; Höfer, T.; Rippe, K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012, 19, 1185–1192. [Google Scholar] [CrossRef]
- Kundaje, A.; Kyriazopoulou-Panagiotopoulou, S.; Libbrecht, M.; Smith, C.L.; Raha, D.; Winters, E.E.; Johnson, S.M.; Snyder, M.; Batzoglou, S.; Sidow, A. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 2012, 22, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.C.; Chereji, R.V.; Lee, P.R.; Fields, R.D.; Clark, D.J. Differential nucleosome spacing in neurons and glia. Neurosci. Lett. 2020, 714, 134559. [Google Scholar] [CrossRef]
- Berkowitz, E.M.; Sanborn, A.C.; Vaughan, D.W. Chromatin structure in neuronal and neuroglial cell nuclei as a function of age. J. Neurochem. 1983, 41, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Druliner, B.R.; Vera, D.; Johnson, R.; Ruan, X.; Apone, L.M.; Dimalanta, E.T.; Stewart, F.J.; Boardman, L.; Dennis, J.H. Comprehensive nucleosome mapping of the human genome in cancer progression. Oncotarget 2015, 7, 13429–13445. [Google Scholar] [CrossRef]
- Chutake, Y.K.; Costello, W.N.; Lam, C.; Bidichandani, S.I. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in friedreich ataxia. J. Biol. Chem. 2014, 289, 15194–15202. [Google Scholar] [CrossRef]
- Sun, H.; Damez-Werno, D.M.; Scobie, K.N.; Shao, N.-Y.; Dias, C.; Rabkin, J.; Koo, J.W.; Korb, E.; Bagot, R.C.; Ahn, F.H.; et al. ACF chromatin-remodeling complex mediates stress-induced depressive-like behavior. Nat. Med. 2015, 21, 1146–1153. [Google Scholar] [CrossRef]
- Goodman, J.V.; Bonni, A. Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68. [Google Scholar] [CrossRef]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Luscombe, N.M. Nucleosome positioning stability is a significant modulator of germline mutation rate variation across the human genome. Genomics 2018. preprint. [Google Scholar] [CrossRef]
- Bryois, J.; Garrett, M.E.; Song, L.; Safi, A.; Giusti-Rodriguez, P.; Johnson, G.D.; Shieh, A.W.; Buil, A.; Fullard, J.F.; Roussos, P.; et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 2018, 9, 3121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.E.; Preissl, S.; Hou, X.; Zhang, Z.; Zhang, K.; Fang, R.; Qiu, Y.; Poirion, O.; Li, B.; Liu, H.; et al. An atlas of gene regulatory elements in adult mouse cerebrum. Neuroscience 2020. preprint. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, R.M.; DeRosa, B.A.; Thornton, C.A.; Sayar, Z.; Torkenczy, K.A.; Fields, A.J.; Wright, K.M.; Nan, X.; Ramji, R.; Steemers, F.J.; et al. Improved single-cell ATAC-Seq reveals chromatin dynamics of in vitro corticogenesis. Genomics 2019. preprint. [Google Scholar] [CrossRef]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef]
- Guertin, M.J.; Lis, J.T. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr. Opin. Genet. Dev. 2013, 23, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.P.; Hill, M.J.; Kavanagh, D.H.; Tansey, K.E.; Waite, A.J.; Blake, D.J. The psychiatric risk gene transcription Factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr. Bull. 2018, 44, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.C.; Javidfar, B.; Pothula, V.; Ibi, D.; Shen, E.Y.; Peter, C.J.; Bicks, L.K.; Fehr, T.; Jiang, Y.; Brennand, K.J.; et al. MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice. Mol. Psychiatry 2018, 23, 123–132. [Google Scholar] [CrossRef]
- Shulha, H.P.; Crisci, J.L.; Reshetov, D.; Tushir, J.S.; Cheung, I.; Bharadwaj, R.; Chou, H.-J.; Houston, I.B.; Peter, C.J.; Mitchell, A.C.; et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012, 10, e1001427. [Google Scholar] [CrossRef]
- Shulha, H.P.; Cheung, I.; Guo, Y.; Akbarian, S.; Weng, Z. Coordinated cell type–specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013, 9, e1003433. [Google Scholar] [CrossRef] [PubMed]
- Cheung, I.; Shulha, H.P.; Jiang, Y.; Matevossian, A.; Wang, J.; Weng, Z.; Akbarian, S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8824–8829. [Google Scholar] [CrossRef]
- Sun, W.; Poschmann, J.; del Rosario, R.C.-H.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 2016, 167, 1385–1397.e11. [Google Scholar] [CrossRef]
- Shulha, H.P. Epigenetic signatures of autism: Trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 2012, 69, 314. [Google Scholar] [CrossRef]
- Notwell, J.H.; Heavner, W.E.; Darbandi, S.F.; Katzman, S.; McKenna, W.L.; Ortiz-Londono, C.F.; Tastad, D.; Eckler, M.J.; Rubenstein, J.L.R.; McConnell, S.K.; et al. TBR1 regulates autism risk genes in the developing neocortex. Genome Res. 2016, 26, 1013–1022. [Google Scholar] [CrossRef]
- Kasinathan, S.; Orsi, G.A.; Zentner, G.E.; Ahmad, K.; Henikoff, S. High-Resolution Mapping of Transcription Factor Binding Sites on Native Chromatin. Nat Methods 2014, 11, 203–209. [Google Scholar] [CrossRef]
- Van Steensel, B.; Delrow, J.; Henikoff, S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat. Genet. 2001, 27, 304–308. [Google Scholar] [CrossRef]
- Tosti, L.; Ashmore, J.; Tan, B.S.N.; Carbone, B.; Mistri, T.K.; Wilson, V.; Tomlinson, S.R.; Kaji, K. Mapping transcription factor occupancy using minimal numbers of cells in vitro and in vivo. Genome Res. 2018, 28, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.A.; van den Ameele, J.; Cheetham, S.W.; Yakob, R.; Brand, A.H.; Nord, A.S. Novel CHD8 genomic targets identified in fetal mouse brain by in vivo targeted DamID. Genomics 2021. preprint. [Google Scholar] [CrossRef]
- Mitchell, A.C.; Javidfar, B.; Bicks, L.K.; Neve, R.; Garbett, K.; Lander, S.S.; Mirnics, K.; Morishita, H.; Wood, M.A.; Jiang, Y.; et al. Longitudinal assessment of neuronal 3D genomes in mouse prefrontal cortex. Nat. Commun. 2016, 7, 12743. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef]
- Rooijers, K.; Markodimitraki, C.M.; Rang, F.J.; de Vries, S.S.; Chialastri, A.; de Luca, K.L.; Mooijman, D.; Dey, S.S.; Kind, J. Simultaneous quantification of protein—DNA contacts and transcriptomes in single cells. Nat. Biotechnol. 2019, 37, 766–772. [Google Scholar] [CrossRef]
- Aughey, G.N.; Southall, T.D. Dam It’s Good! DamID profiling of protein-DNA interactions: Dam It’s Good! WIREs Dev. Biol. 2016, 5, 25–37. [Google Scholar] [CrossRef]
- Stroud, H.; Yang, M.G.; Tsitohay, Y.N.; Davis, C.P.; Sherman, M.A.; Hrvatin, S.; Ling, E.; Greenberg, M.E. An activity-mediated transition in transcription in early postnatal neurons. Neuron 2020, 107, 874–890.e8. [Google Scholar] [CrossRef] [PubMed]
- Gegenhuber, B.; Wu, M.V.; Bronstein, R.; Tollkuhn, J. Regulation of neural gene expression by estrogen receptor alpha. Neuroscience 2020. [Google Scholar] [CrossRef]
- Ruzicka, W.B.; Mohammadi, S.; Davila-Velderrain, J.; Subburaju, S.; Tso, D.R.; Hourihan, M.; Kellis, M. Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience. Psychiatry Clin. Psychol. 2020. preprint. [Google Scholar] [CrossRef]
- Borden, J.; Manuelidis, L. Movement of the x chromosome in epilepsy. Science 1988, 242, 1687–1691. [Google Scholar] [CrossRef]
- Kadauke, S.; Blobel, G.A. Chromatin loops in gene regulation. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 17–25. [Google Scholar] [CrossRef]
- The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Gusev, F.E.; Reshetov, D.A.; Mitchell, A.C.; Andreeva, T.V.; Dincer, A.; Grigorenko, A.P.; Fedonin, G.; Halene, T.; Aliseychik, M.; Filippova, E.; et al. Chromatin profiling of cortical neurons identifies individual epigenetic signatures in schizophrenia. Transl. Psychiatry 2019, 9, 256. [Google Scholar] [CrossRef]
- Mitchell, A.C.; Bharadwaj, R.; Whittle, C.; Krueger, W.; Mirnics, K.; Hurd, Y.; Rasmussen, T.; Akbarian, S. The genome in 3D: A new frontier in human brain research. Biol. Psychiatry 2014, 75, 961–969. [Google Scholar] [CrossRef]
- McAllister, A.K. Major histocompatibility Complex I in brain development and schizophrenia. Biol. Psychiatry 2014, 75, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.-F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Jiang, Y.; Mao, W.; Jakovcevski, M.; Dincer, A.; Krueger, W.; Garbett, K.; Whittle, C.; Tushir, J.S.; Liu, J.; et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J. Neurosci. 2013, 33, 11839–11851. [Google Scholar] [CrossRef]
- Zhao, Z.; Tavoosidana, G.; Sjölinder, M.; Göndör, A.; Mariano, P.; Wang, S.; Kanduri, C.; Lezcano, M.; Singh Sandhu, K.; Singh, U.; et al. Circular Chromosome Conformation Capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 2006, 38, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Splinter, E.; de Wit, E.; Nora, E.P.; Klous, P.; van de Werken, H.J.G.; Zhu, Y.; Kaaij, L.J.T.; van IJcken, W.; Gribnau, J.; Heard, E.; et al. The inactive X Chromosome adopts a unique three-dimensional conformation that is dependent on xist RNA. Genes Dev. 2011, 25, 1371–1383. [Google Scholar] [CrossRef]
- 2p15 Consortium; 16p11.2 Consortium; Loviglio, M.N.; Leleu, M.; Männik, K.; Passeggeri, M.; Giannuzzi, G.; van der Werf, I.; Jacquemont, S.; Reymond, A.; et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol. Psychiatry 2017, 22, 836–849. [Google Scholar] [CrossRef]
- Zeitz, M.J.; Lerner, P.P.; Ay, F.; van Nostrand, E.; Heidmann, J.D.; Noble, W.S.; Hoffman, A.R. Implications of COMT long-range interactions on the phenotypic variability of 22q11.2 deletion syndrome. Nucleus 2013, 4, 487–493. [Google Scholar] [CrossRef]
- Eckart, N.; Song, Q.; Yang, R.; Wang, R.; Zhu, H.; McCallion, A.S.; Avramopoulos, D. Functional characterization of schizophrenia-associated variation in CACNA1C. PLoS ONE 2016, 11, e0157086. [Google Scholar] [CrossRef] [PubMed]
- Dostie, J.; Dekker, J. Mapping networks of physical interactions between genomic elements using 5C technology. Nat. Protoc. 2007, 2, 988–1002. [Google Scholar] [CrossRef]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Beagan, J.A.; Pastuzyn, E.D.; Fernandez, L.R.; Guo, M.H.; Feng, K.; Titus, K.R.; Chandrashekar, H.; Shepherd, J.D.; Phillips-Cremins, J.E. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci. 2020, 23, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Beagan, J.A.; Duong, M.T.; Titus, K.R.; Zhou, L.; Cao, Z.; Ma, J.; Lachanski, C.V.; Gillis, D.R.; Phillips-Cremins, J.E. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017, 27, 1139–1152. [Google Scholar] [CrossRef]
- Sams, D.S.; Nardone, S.; Getselter, D.; Raz, D.; Tal, M.; Rayi, P.R.; Kaphzan, H.; Hakim, O.; Elliott, E. Neuronal CTCF is necessary for basal and experience-dependent gene regulation, memory formation, and genomic structure of BDNF and Arc. Cell Rep. 2016, 17, 2418–2430. [Google Scholar] [CrossRef]
- Huo, Y.; Li, S.; Liu, J.; Li, X.; Luo, X.-J. Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat. Commun. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; la Marca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362, eaat8464. [Google Scholar] [CrossRef]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef]
- Whalen, S.; Pollard, K.S. Most regulatory interactions are not. in linkage disequilibrium. Genomics 2018. preprint. [Google Scholar] [CrossRef]
- Giusti-Rodriguez, P.M.; Sullivan, P.F. Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. Genetics 2018. preprint. [Google Scholar] [CrossRef]
- Tan, L.; Ma, W.; Wu, H.; Zheng, Y.; Xing, D.; Chen, R.; Li, X.; Daley, N.; Deisseroth, K.; Xie, X.S. Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development. Cell 2021, 184, 741–758.e17. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef]
- Grubert, F.; Srivas, R.; Spacek, D.V.; Kasowski, M.; Ruiz-Velasco, M.; Sinnott-Armstrong, N.; Greenside, P.; Narasimha, A.; Liu, Q.; Geller, B.; et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature 2020, 583, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Arloth, J.; Bogdan, R.; Weber, P.; Frishman, G.; Menke, A.; Wagner, K.V.; Balsevich, G.; Schmidt, M.V.; Karbalai, N.; Neale, B.; et al. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron 2015, 86, 1189–1202. [Google Scholar] [CrossRef]
- Nott, A.; Holtman, I.R.; Coufal, N.G.; Schlachetzki, J.C.M.; Yu, M.; Hu, R.; Han, C.Z.; Pena, M.; Xiao, J.; Wu, Y.; et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019, 366, 1134–1139. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chawla, A.; Nagy, C.; Turecki, G. Chromatin Profiling Techniques: Exploring the Chromatin Environment and Its Contributions to Complex Traits. Int. J. Mol. Sci. 2021, 22, 7612. https://doi.org/10.3390/ijms22147612
Chawla A, Nagy C, Turecki G. Chromatin Profiling Techniques: Exploring the Chromatin Environment and Its Contributions to Complex Traits. International Journal of Molecular Sciences. 2021; 22(14):7612. https://doi.org/10.3390/ijms22147612
Chicago/Turabian StyleChawla, Anjali, Corina Nagy, and Gustavo Turecki. 2021. "Chromatin Profiling Techniques: Exploring the Chromatin Environment and Its Contributions to Complex Traits" International Journal of Molecular Sciences 22, no. 14: 7612. https://doi.org/10.3390/ijms22147612
APA StyleChawla, A., Nagy, C., & Turecki, G. (2021). Chromatin Profiling Techniques: Exploring the Chromatin Environment and Its Contributions to Complex Traits. International Journal of Molecular Sciences, 22(14), 7612. https://doi.org/10.3390/ijms22147612