
The Role of TRPM2 in Endothelial Function and Dysfunction




{kind=link}
{kind=link}
Abstract
:1. Introduction
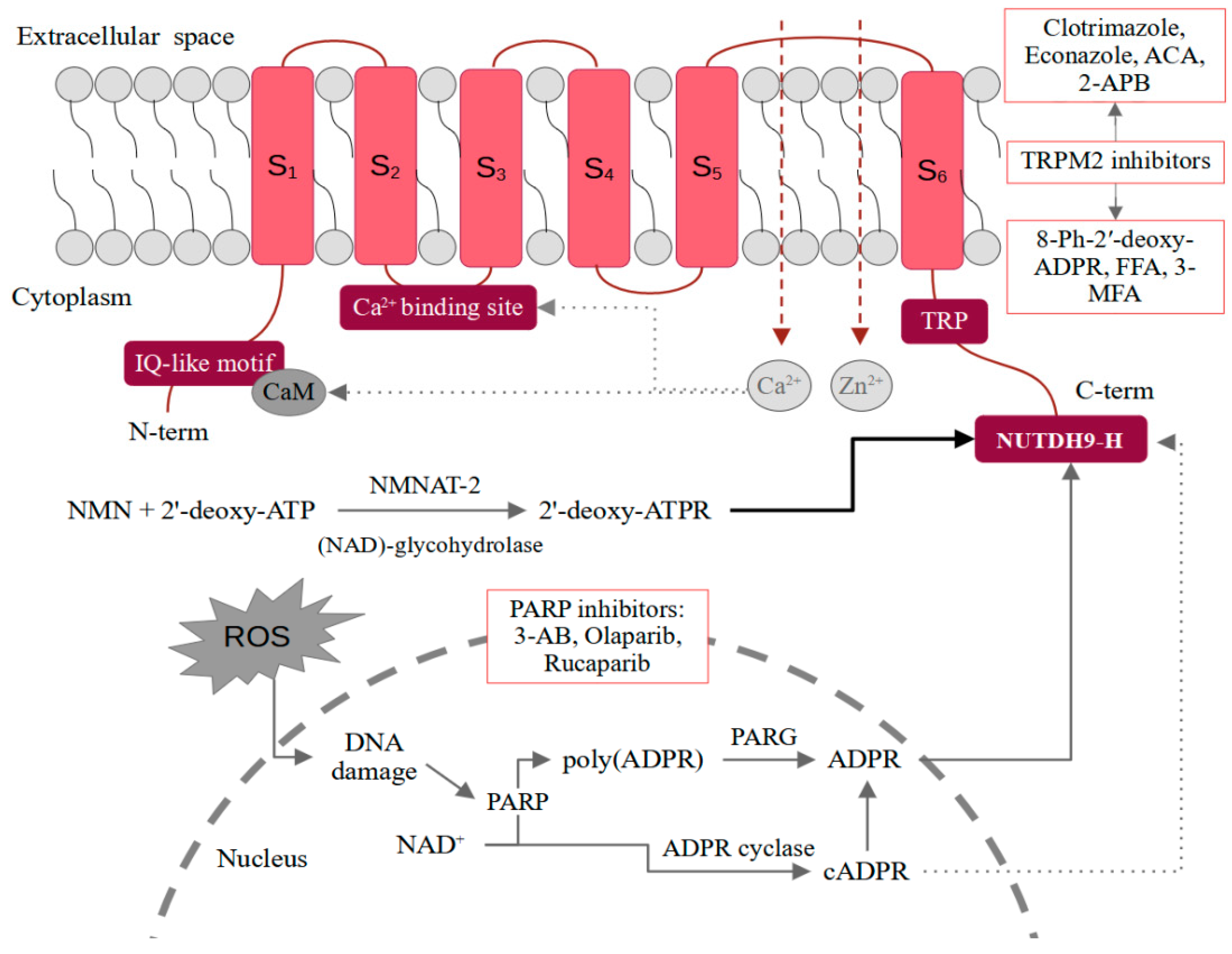
2. TRPM2 Structure and Activation Mechanisms
3. TRPM2 in Endothelial Permeability
4. TRPM2 in Endothelial Cell Death
5. TRPM2 in Endothelial Cell Migration and Angiogenesis
6. TRPM2 Inhibitors in Clinical Application and Further Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 Channel in Mediating H2O2 -Induced Ca2+ Entry and Endothelial Hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A Novel TRPM2 Isoform Inhibits Calcium Influx and Susceptibility to Cell Death. J. Biol. Chem. 2003, 278, 16222–16229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jüngling, E.; Zitt, C.; Lückhoff, A. Activation of the Cation Channel Long Transient Receptor Potential Channel 2 (LTRPC2) by Hydrogen Peroxide. J. Biol. Chem. 2002, 277, 23150–23156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegert, R.; Bauche, A.; Pérez, A.-M.W.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Zhang, W.; Conrad, K.; Mostoller, K.; Cheung, J.Y.; Peterson, B.Z.; Miller, B.A. Regulation of the Transient Receptor Potential Channel TRPM2 by the Ca2+ Sensor Calmodulin. J. Biol. Chem. 2006, 281, 9076–9085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yu, P.; Lin, H.; Jin, Z.; Zhao, S.; Zhang, Y.; Xu, Q.; Liu, Z.; Yang, W.; Zhang, L. The Discovery of Novel ACA Derivatives as Specific TRPM2 Inhibitors that Reduce Ischemic Injury Both In Vitro and In Vivo. J. Med. Chem. 2021, 64, 3976–3996. [Google Scholar] [CrossRef]
- Miyanohara, J.; Kakae, M.; Nagayasu, K.; Nakagawa, T.; Mori, Y.; Arai, K.; Shirakawa, H.; Kaneko, S. TRPM2 Channel Aggravates CNS Inflammation and Cognitive Impairment via Activation of Microglia in Chronic Cerebral Hypoperfusion. J. Neurosci. 2018, 38, 3520–3533. [Google Scholar] [CrossRef]
- Zeng, X.; Sikka, S.C.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.B.; E Pottle, J.; Taylor, J.T.; Li, M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2009, 13, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yau, H.-Y.; Wong, W.-Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H2O2-Induced Cell Apoptosis in Endothelial Cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, J.; Ko, W.-H.; Kwan, Y.-W.; Jiang, L.; Sun, L.; Yao, X. TRPM2 promotes autophagic degradation in vascular smooth muscle cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Kühn, F.J.P.; Lückhoff, A. Sites of the NUDT9-H Domain Critical for ADP-ribose Activation of the Cation Channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) – Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Fliegert, R.; Watt, J.M.; Schöbel, A.; Rozewitz, M.D.; Moreau, C.; Kirchberger, T.; Thomas, M.P.; Sick, W.; Araujo, A.C.; Harneit, A.; et al. Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349. Biochem. J. 2017, 474, 2159–2175. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.-M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.-H.; Yu, X.-F.; Ma, C.; Yang, F.; Yang, W. Effects of calcium-binding sites in the S2–S3 loop on human and Nematostella vectensis TRPM2 channel gating processes. J. Zhejiang Univ. Sci. B 2019, 20, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Riekehr, W.M.; Guse, A.H. Does Cyclic ADP-Ribose (cADPR) Activate the Non-selective Cation Channel TRPM2? Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695.e4. [Google Scholar] [CrossRef] [Green Version]
- Tóth, B.; Iordanov, I.; Csanády, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J. Gen. Physiol. 2015, 145, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Roth, B.; Lü, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nat. Cell Biol. 2018, 562, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nat. Cell Biol. 2016, 536, 460–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.; Redmond, E.M. Vascular endothelium–Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Volk, T.; Hensel, M.; Kox, W.J. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: Role of hydrogen peroxide. Mol. Cell. Biochem. 1997, 171, 11–21. [Google Scholar] [CrossRef]
- Dreher, D.; Junod, A.F. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J. Cell. Physiol. 1995, 162, 147–153. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Ahmmed, G.U.; Malik, A.B. TRPM2 Channel Regulates Endothelial Barrier Function. Chem. Biol. Pteridines Folates 2009, 661, 155–167. [Google Scholar] [CrossRef]
- Sandoval, R.; Malik, A.B.; Minshall, R.D.; Kouklis, P.; Ellis, C.A.; Tiruppathi, C. Ca2+ signalling and PKCα activate increased endothelial permeability by disassembly of VE—Cadherin junctions. J. Physiol. 2001, 533, 433–445. [Google Scholar] [CrossRef]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil Activation of Endothelial Cell-Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef]
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2012, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative Interaction oftrpMelastatin Channel Transient Receptor Potential (TRPM2) With Its Splice Variant TRPM2 Short Variant Is Essential for Endothelial Cell Apoptosis. Circ. Res. 2014, 114, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Osmanlıoğlu, H. Ömer; Yıldırım, M.K.; Akyuva, Y.; Yıldızhan, K.; Nazıroğlu, M. Morphine Induces Apoptosis, Inflammation, and Mitochondrial Oxidative Stress via Activation of TRPM2 Channel and Nitric Oxide Signaling Pathways in the Hippocampus. Mol. Neurobiol. 2020, 57, 3376–3389. [Google Scholar] [CrossRef]
- Zhang, W.; Hirschler-Laszkiewicz, I.; Tong, Q.; Conrad, K.; Sun, S.-C.; Penn, L.; Barber, D.L.; Stahl, R.; Carey, D.J.; Cheung, J.Y.; et al. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am. J. Physiol. Cell Physiol. 2006, 290, C1146–C1159. [Google Scholar] [CrossRef]
- Gasser, A.; Glassmeier, G.; Fliegert, R.; Langhorst, M.F.; Meinke, S.; Hein, D.; Krüger, S.; Weber, K.; Heiner, I.; Oppenheimer, N.; et al. Activation of T Cell Calcium Influx by the Second Messenger ADP-ribose. J. Biol. Chem. 2006, 281, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Lin-Hua, J.; Sivaprasadarao, A. High glucose–induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef]
- Jiang, Q.; Gao, Y.; Wang, C.; Tao, R.; Wu, Y.; Zhan, K.; Liao, M.; Lu, N.; Lu, Y.; Wilcox, C.S.; et al. Nitration of TRPM2 as a Molecular Switch Induces Autophagy During Brain Pericyte Injury. Antioxid. Redox Signal. 2017, 27, 1297–1316. [Google Scholar] [CrossRef] [PubMed]
- Izdebska, M.; Zielińska, W.; Hałas-Wiśniewska, M.; Grzanka, A. Involvement of Actin in Autophagy and Autophagy-Dependent Multidrug Resistance in Cancer. Cancers 2019, 11, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, K.; Franco, C.; Philippides, A.; Blanco, R.; Dierkes, M.; Gebala, V.; Stanchi, F.; Jones, M.; Aspalter, I.M.; Cagna, G.; et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat. Cell Biol. 2014, 16, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca 2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2020, 10, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.-P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel Role of Reactive Oxygen Species–Activated trp Melastatin Channel-2 in Mediating Angiogenesis and Postischemic Neovascularization. Arter. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Quillinan, N.; Yang, Y.-F.; Nakayama, S.; Cheng, J.; Kelley, M.; Herson, P. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 2012, 530, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Bilecik, T.; Karateke, F.; Elkan, H.; Gokce, H.; Bilecik, T.; Karateke, F.; Elkan, H.; Gokce, H. The effects of TRPM2, TRPM6, TRPM7 and TRPM8 gene expression in hepatic ischemia reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3088–3095. [Google Scholar]
- Zhang, H.; Zhao, S.; Yu, J.; Yang, W.; Liu, Z.; Zhang, L. Medicinal chemistry perspective of TRPM2 channel inhibitors: Where we are and where we might be heading? Drug Discov. Today 2020, 25, 2326–2334. [Google Scholar] [CrossRef]
- Luo, X.; Lihe, Z.; Zhan, K.; Yang, W.; Zhang, L.; Wang, K.; Kaiyu, Z.; Zhang, L. Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues. Chem. Biol. Drug Des. 2018, 91, 552–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourgeaud, L.; Dvorak, C.; Faouzi, M.; Starkus, J.; Sahdeo, S.; Wang, Q.; Lord, B.; Coate, H.; Taylor, N.; He, Y.; et al. Pharmacology of JNJ-28583113: A novel TRPM2 antagonist. Eur. J. Pharmacol. 2019, 853, 299–307. [Google Scholar] [CrossRef]
- Akyuva, Y.; Nazıroğlu, M. Resveratrol attenuates hypoxia-induced neuronal cell death, inflammation and mitochondrial oxidative stress by modulation of TRPM2 channel. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Özkaya, D.; Nazıroğlu, M. Curcumin diminishes cisplatin-induced apoptosis and mitochondrial oxidative stress through inhibition of TRPM2 channel signaling pathway in mouse optic nerve. J. Recept. Signal Transduct. 2020, 40, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, F.; Zhang, J.; Wang, T.; Wei, X.; Wu, J.; Feng, Y.; Dai, Z.; Wu, Q. Neuroprotective Effect of Resveratrol on Ischemia/Reperfusion Injury in Rats Through TRPC6/CREB Pathways. J. Mol. Neurosci. 2013, 50, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Lückhoff, A. Effects of antioxidants on calcium influx through TRPM2 channels in transfected cells activated by hydrogen peroxide. J. Neurol. Sci. 2008, 270, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Radovits, T.; Zotkina, J.; Lin, L.-N.; Bömicke, T.; Arif, R.; Gerö, D.; Horvath, E.M.; Karck, M.; Szabó, C.; Szabó, G. Poly(ADP-Ribose) Polymerase Inhibition Improves Endothelial Dysfunction Induced by Hypochlorite. Exp. Biol. Med. 2007, 232, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Yabluchanskiy, A.; Csipo, T.; Fulop, G.; Kiss, T.; Balasubramanian, P.; DelFavero, J.; Ahire, C.; Ungvari, A.; Nyúl-Tóth, Ádám; et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. GeroScience 2019, 41, 533–542. [Google Scholar] [CrossRef]
- Zhang, G.-H.; Chao, M.; Hui, L.-H.; Xu, D.L.; Cai, W.-L.; Zheng, J.; Gao, M.; Zhang, M.-X.; Wang, J.; Lu, Q.-H. Poly(ADP-ribose)Polymerase 1 Inhibition Protects Against Age-Dependent Endothelial Dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1266–1274. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zielińska, W.; Zabrzyński, J.; Gagat, M.; Grzanka, A. The Role of TRPM2 in Endothelial Function and Dysfunction. Int. J. Mol. Sci. 2021, 22, 7635. https://doi.org/10.3390/ijms22147635
Zielińska W, Zabrzyński J, Gagat M, Grzanka A. The Role of TRPM2 in Endothelial Function and Dysfunction. International Journal of Molecular Sciences. 2021; 22(14):7635. https://doi.org/10.3390/ijms22147635
Chicago/Turabian StyleZielińska, Wioletta, Jan Zabrzyński, Maciej Gagat, and Alina Grzanka. 2021. "The Role of TRPM2 in Endothelial Function and Dysfunction" International Journal of Molecular Sciences 22, no. 14: 7635. https://doi.org/10.3390/ijms22147635
APA StyleZielińska, W., Zabrzyński, J., Gagat, M., & Grzanka, A. (2021). The Role of TRPM2 in Endothelial Function and Dysfunction. International Journal of Molecular Sciences, 22(14), 7635. https://doi.org/10.3390/ijms22147635