
Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond



Abstract
:1. Introduction
- Acute onset of an illness (minutes to hours) with involvement of the skin (urticaria, pruritus, flushing) and/or mucosa (angioedema/swelling of lips, tongue, larynx)Plus at Least One of the Following:
- Respiratory involvement (dyspnea, wheezing, stridor, reduced peak flows or hypoxemia);
- Reduced blood pressure and associated symptoms of end-organ dysfunction (syncope, fecal or urinary incontinence, hypotonia/collapse);
- Severe gastrointestinal symptoms (severe crampy abdominal pain, severe diarrhea, repetitive vomiting).
- Acute onset of hypotension * or bronchospasm or laryngeal involvement (stridor, voice change, odynophagia) after exposure to a known or highly probable allergen for the patient even in the absence of skin involvement.
2. Materials and Methods
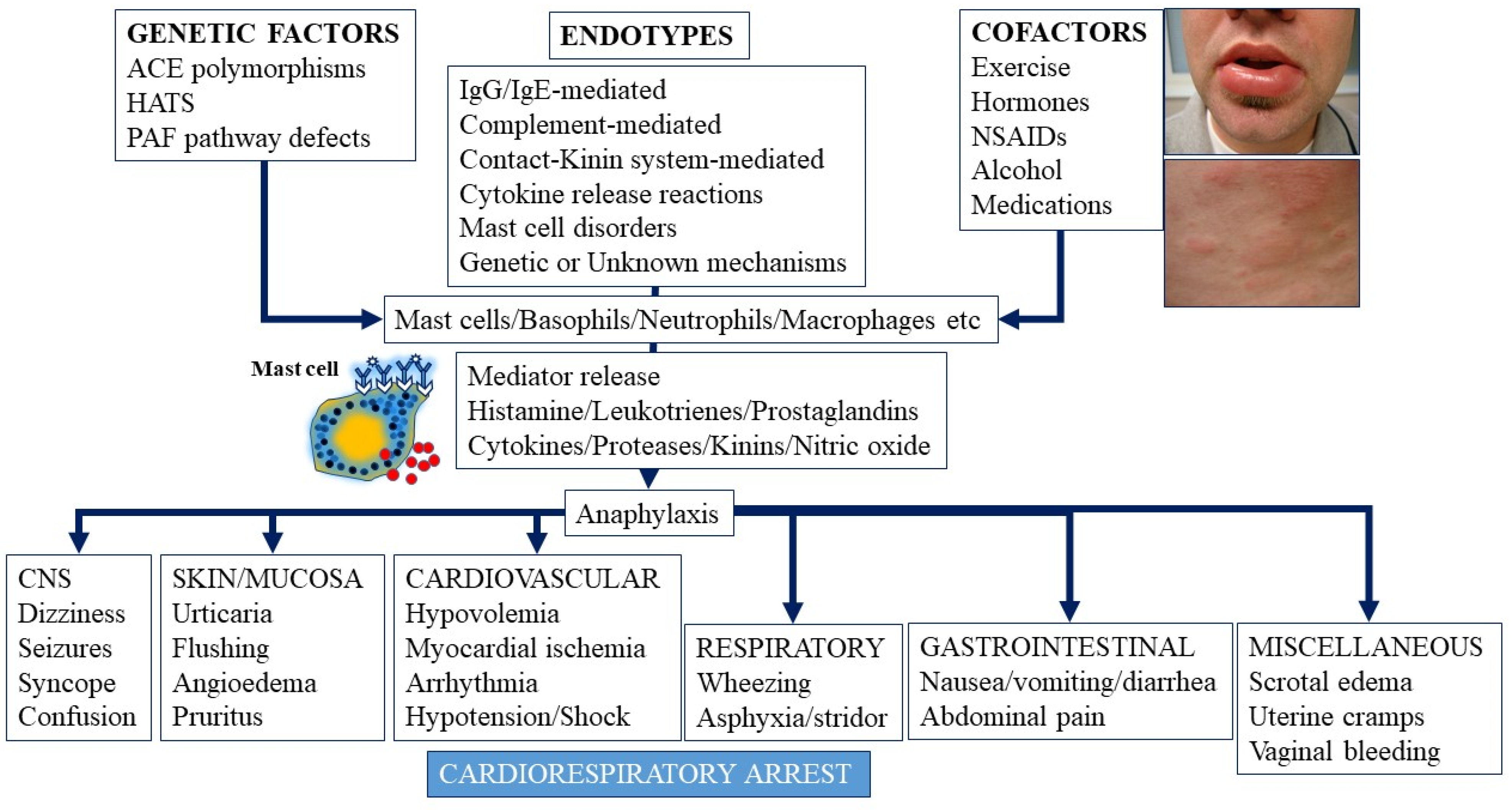
3. Molecular Mechanisms in Anaphylaxis: Endotypes and Phenotypes
3.1. Immunoglobulin E-Dependent Anaphylaxis
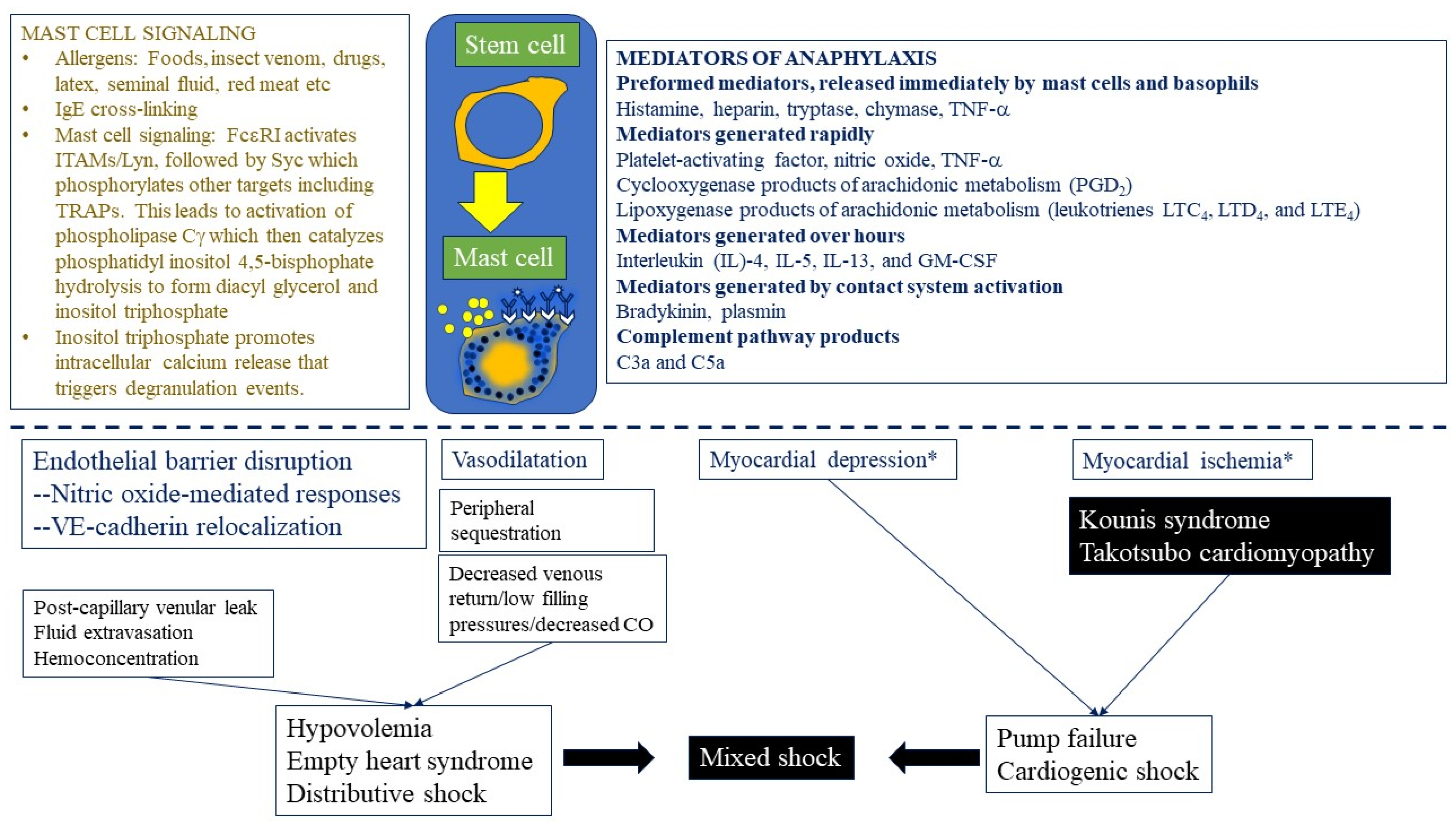
3.1.1. Human Mast Cells and Basophils: Overview, Phylogeny, and Differentiation
3.1.2. Mast Cells: Activation and IgE-mediated Signaling
3.1.3. Mast Cell Degranulation
3.1.4. Pivotal Mediators Expressed by Mast Cells and Basophils
3.1.5. Role of Mast Cell Mediators in Anaphylaxis
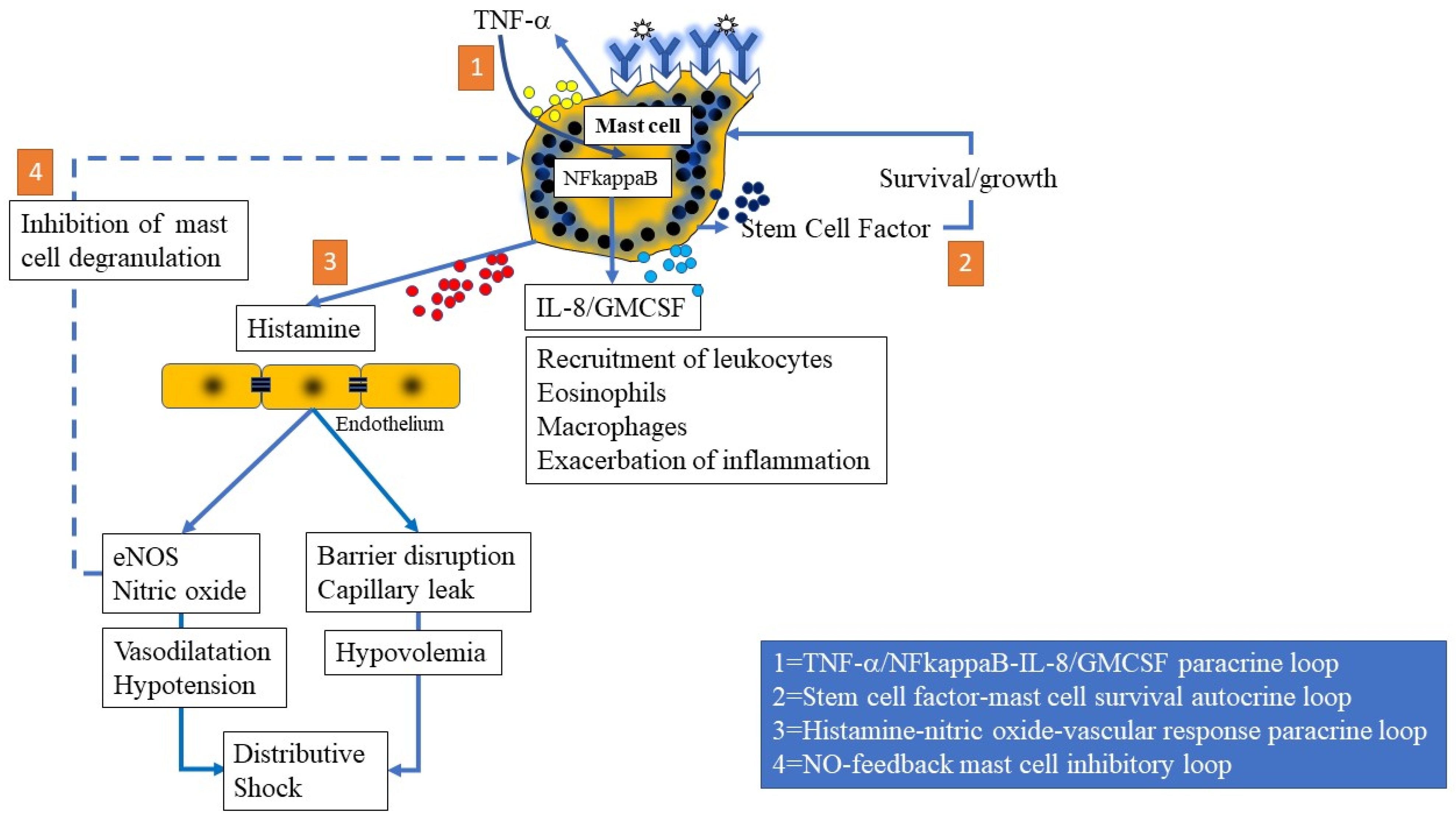
3.1.6. Mast Cell-Mediated Paracrine, Endocrine, and Autocrine Loops
3.1.7. The Classical IgE-histamine Pathway versus the IgG-FcγR-PAF Pathway
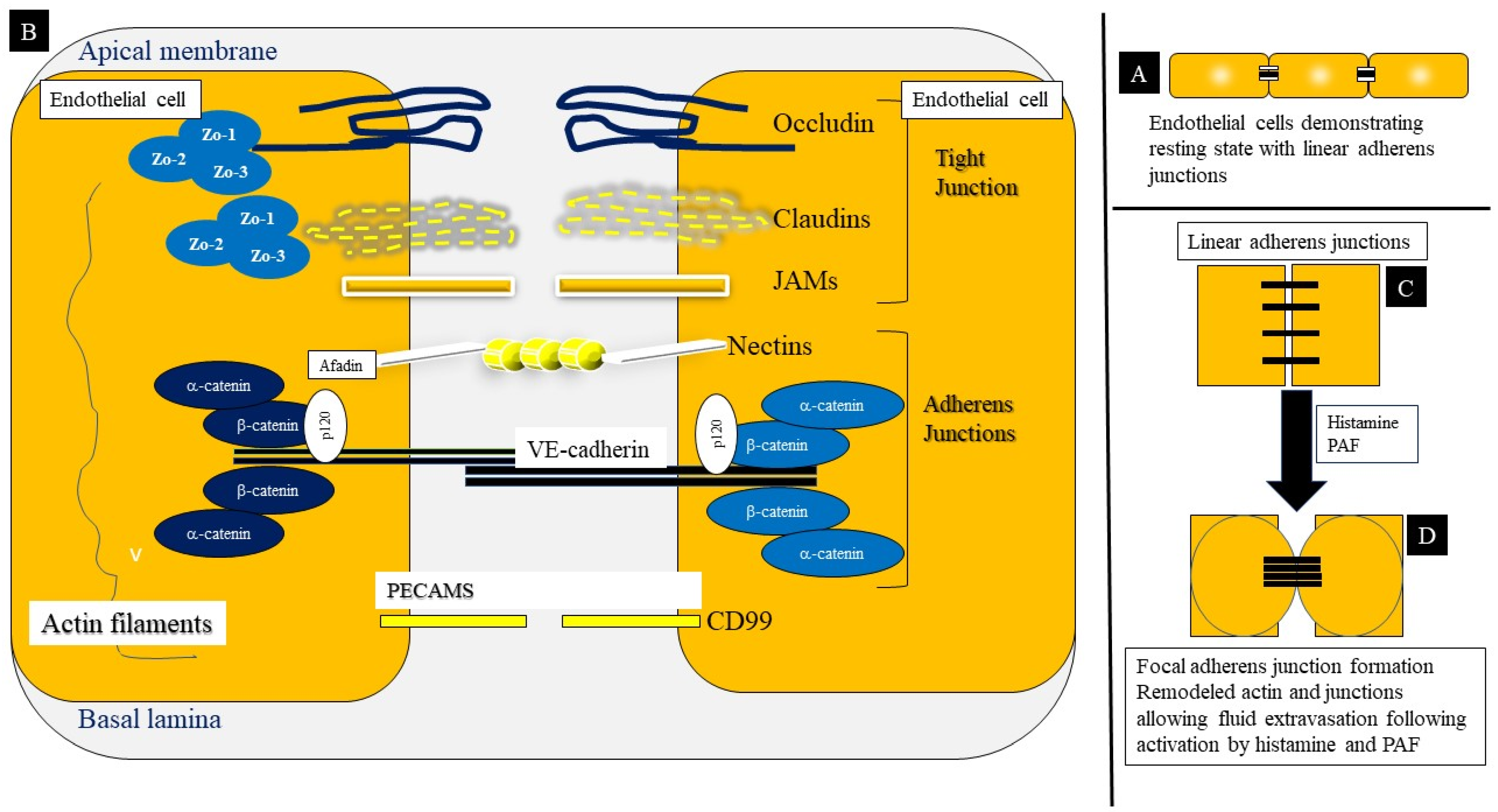
3.1.8. Endothelial Cell–Cell Adhesions: Structure and Function
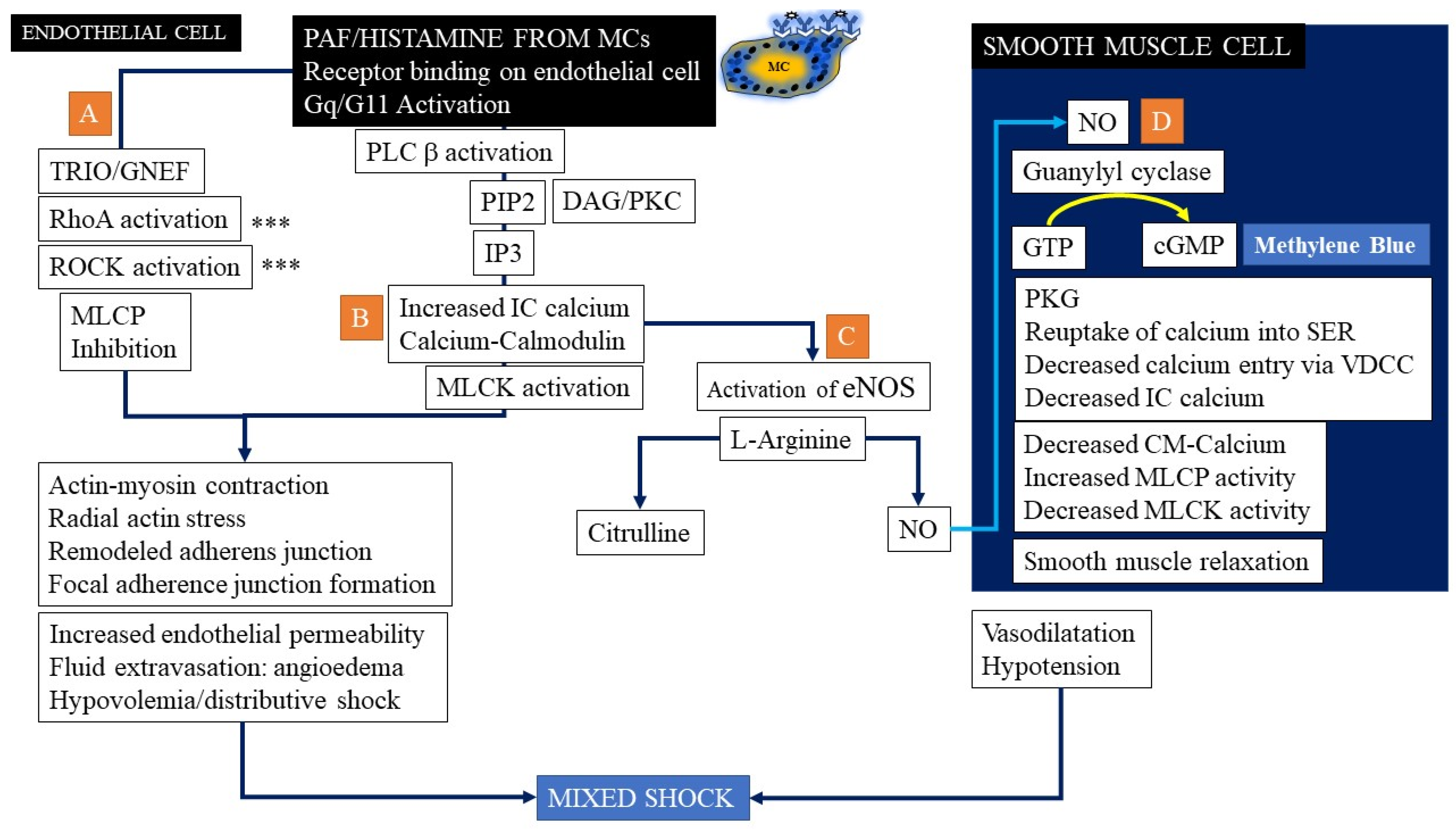
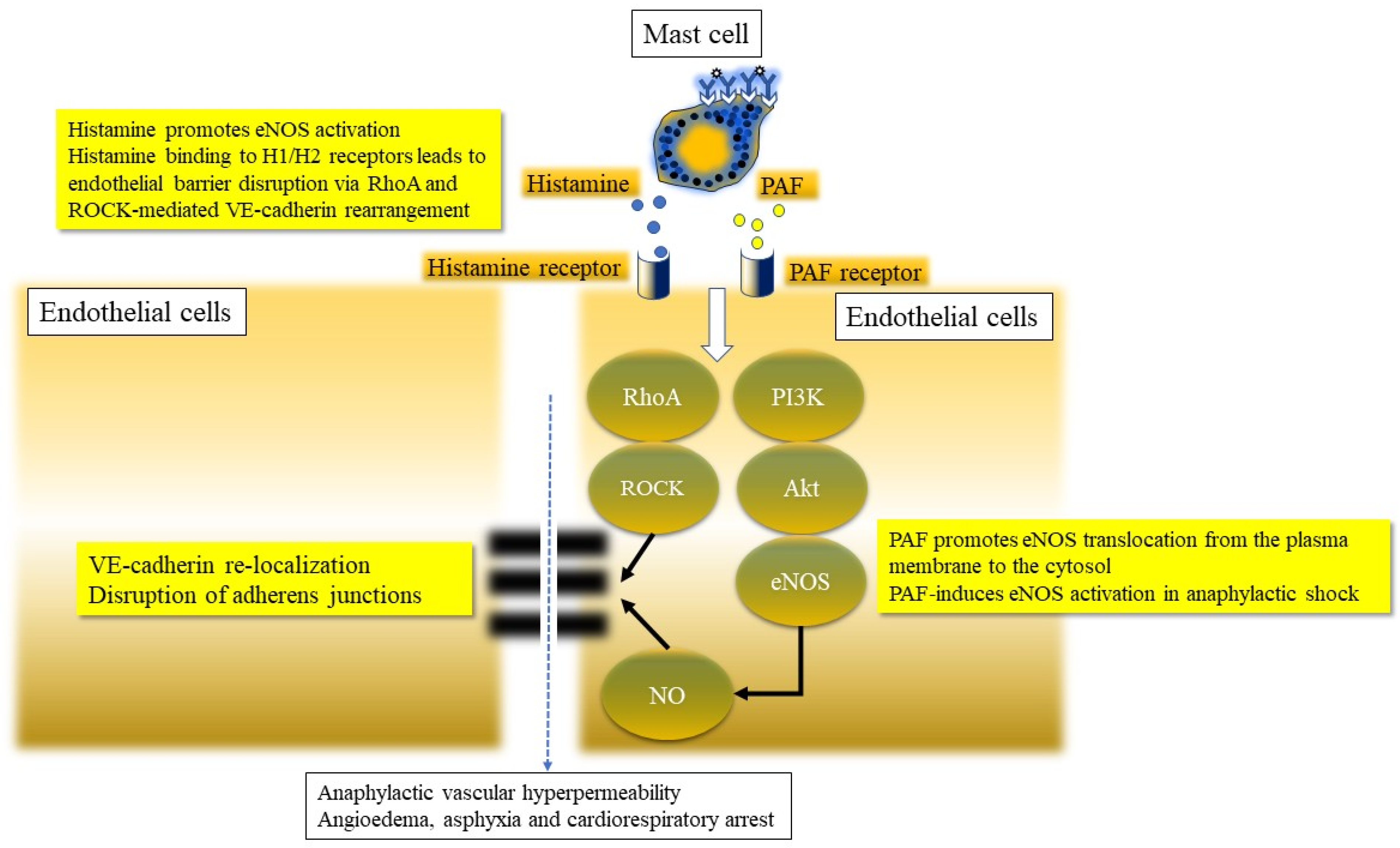
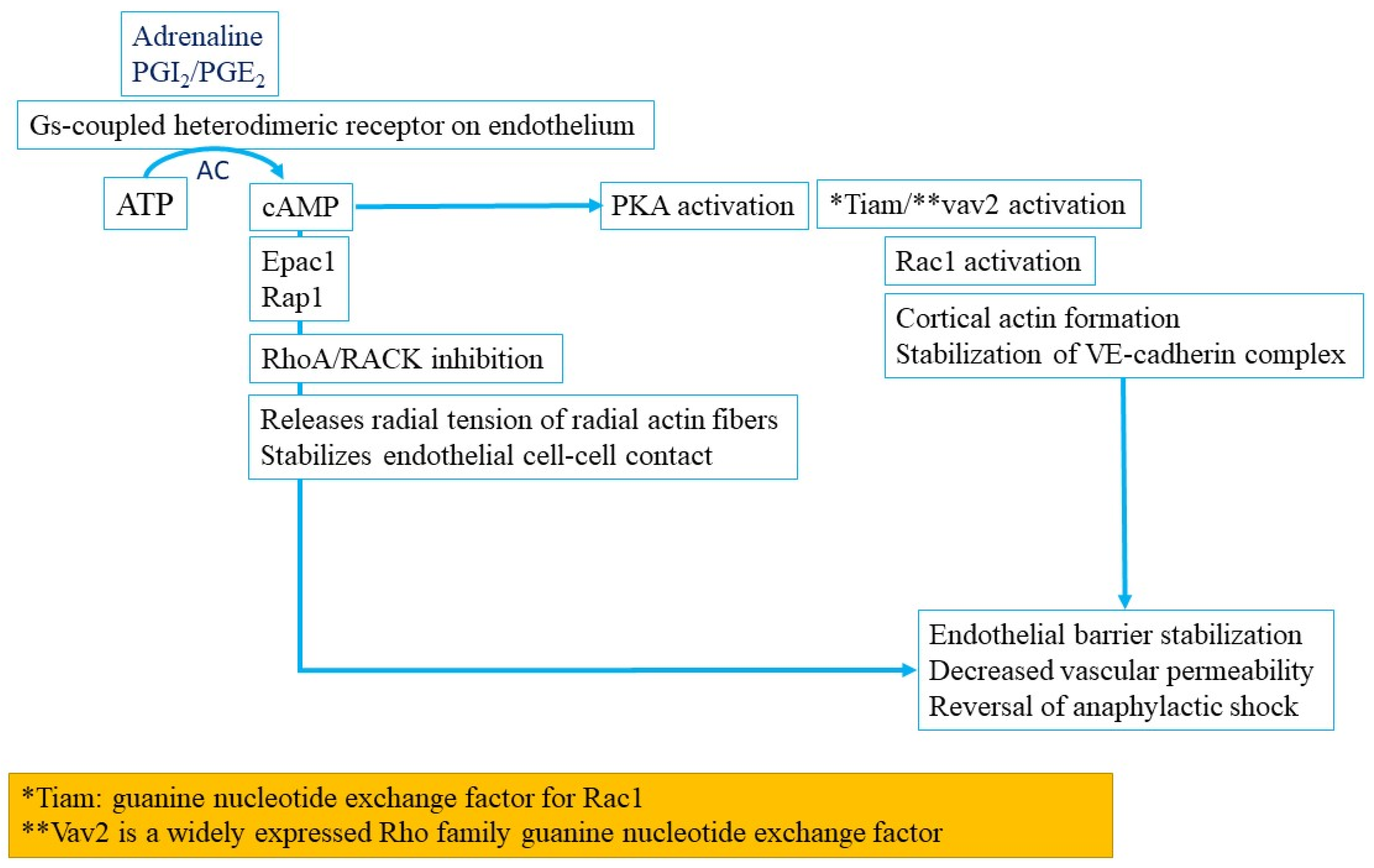
3.1.9. Endothelial Barrier Dysfunction in Anaphylaxis
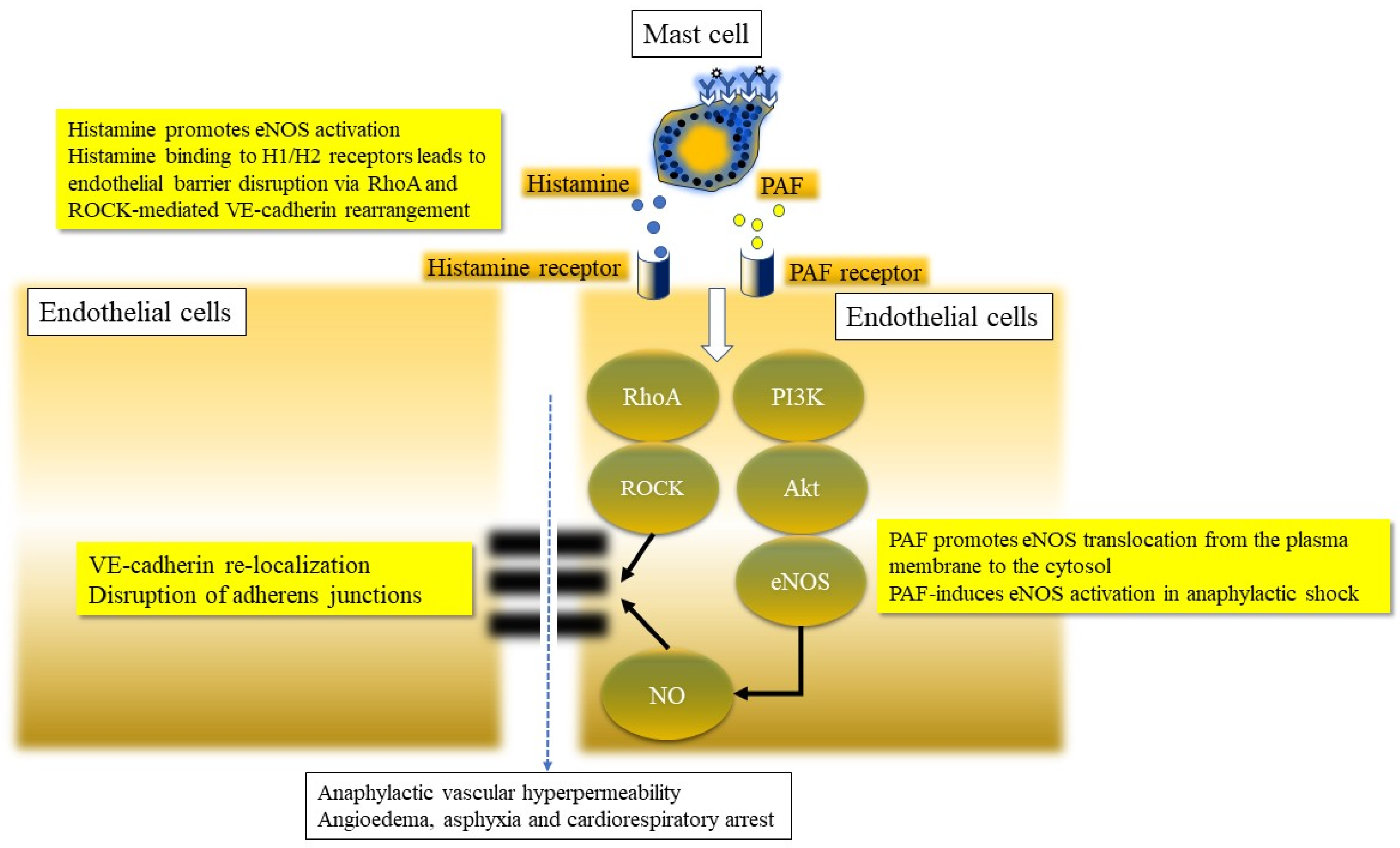
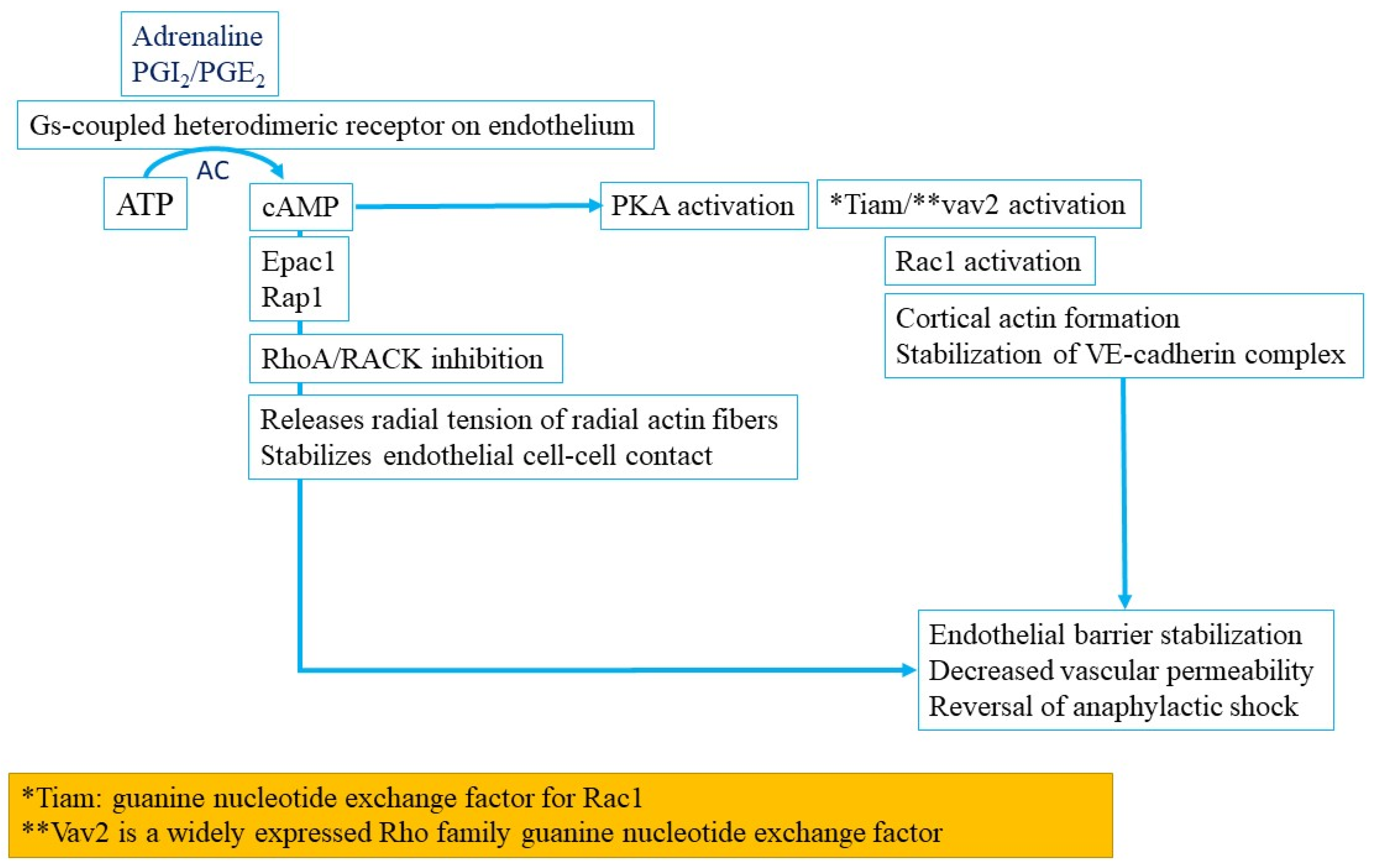
3.1.10. Nitric Oxide Pathway Activation in Anaphylaxis
3.2. IgG-Mediated Anaphylaxis
3.3. Cytokine Release and Cytokine Storm Reactions
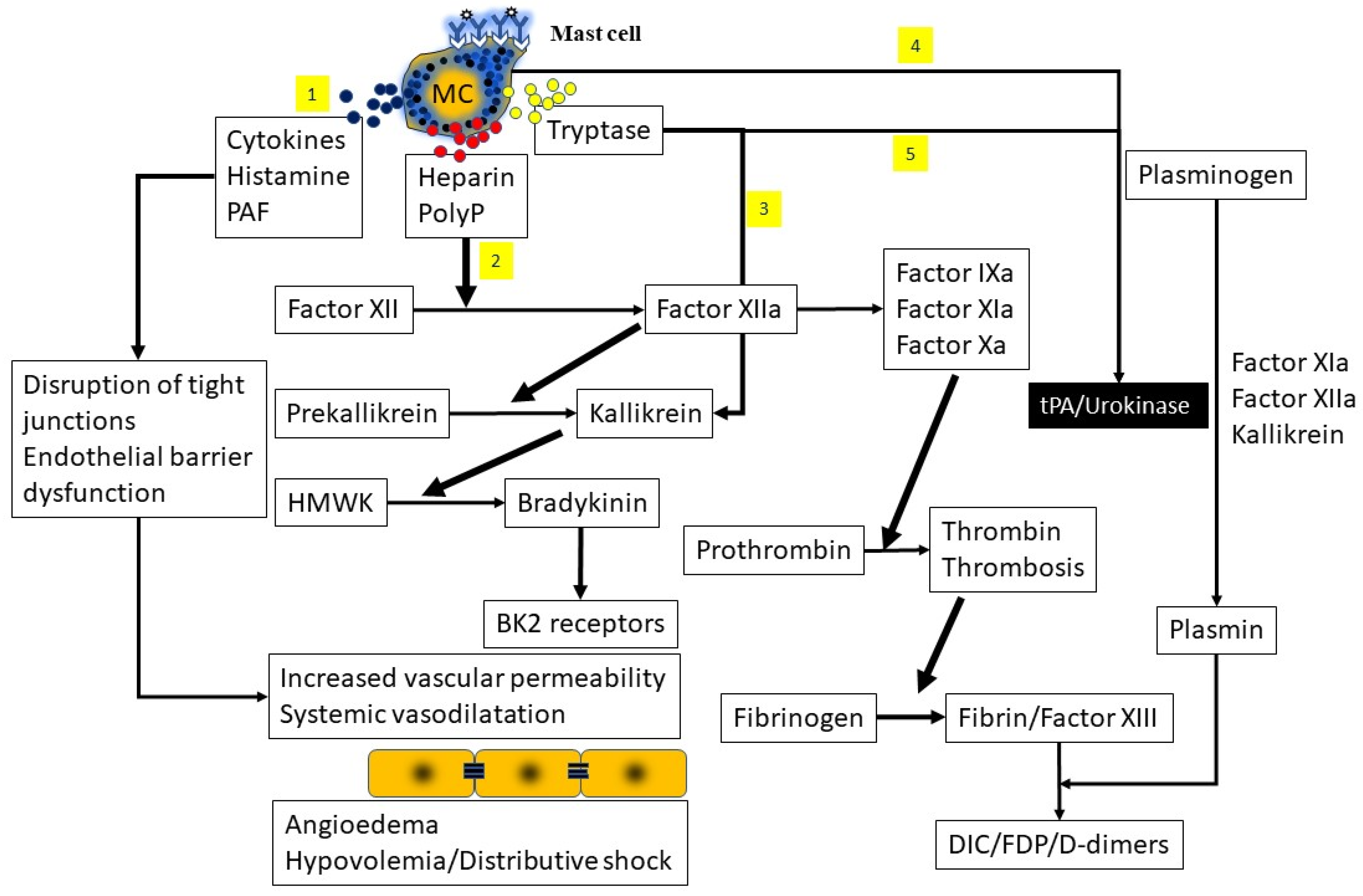
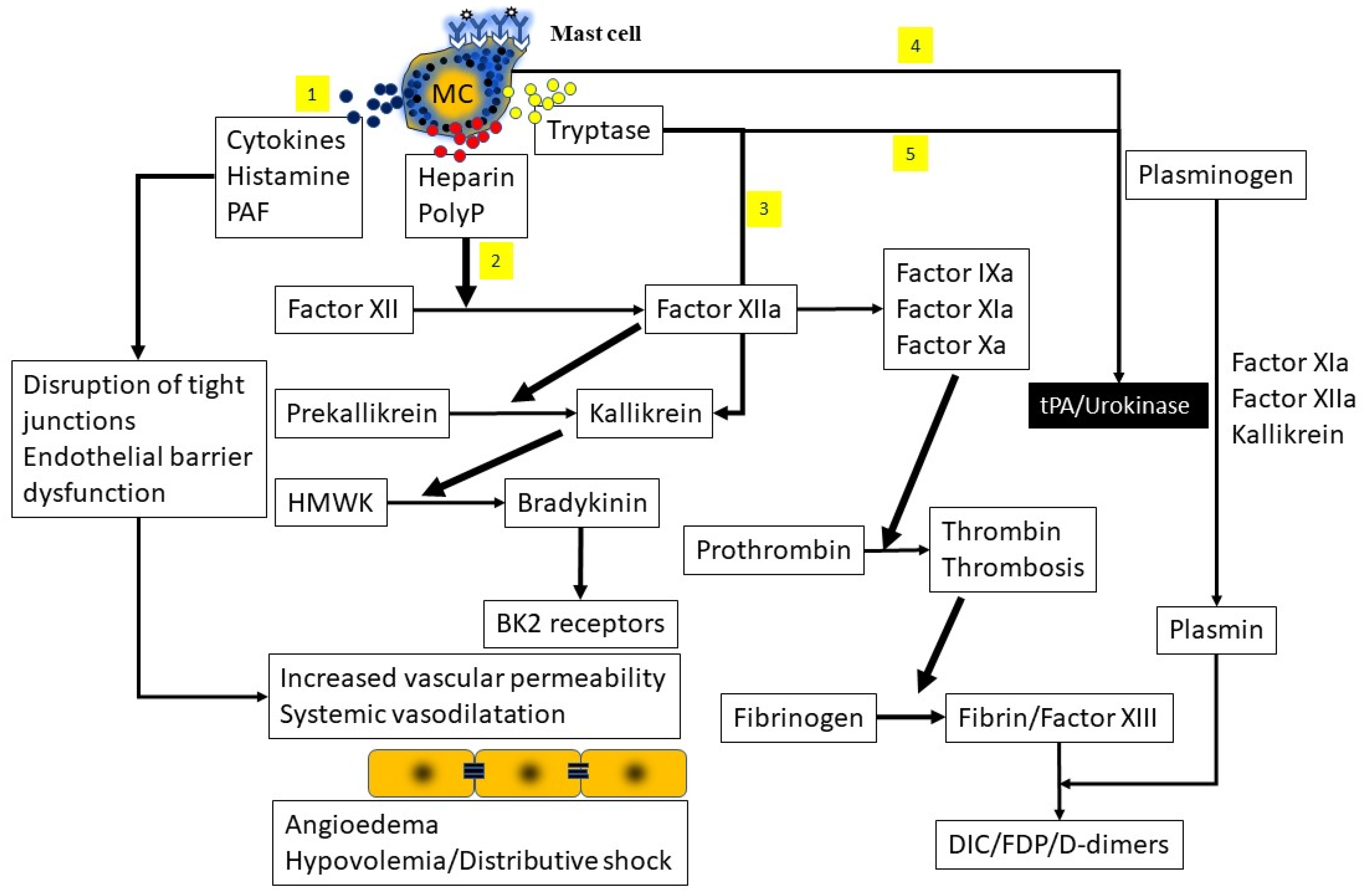
3.4. Contact System Activation
3.5. Complement-Mediated Reactions
3.6. Direct Mast Cell Activation
3.7. Hereditary Alpha-Tryptasemia (HAT) and Other Genetic Disorders
3.8. Idiopathic/Unknown Mechanisms
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- LoVerde, D.; Iweala, O.I.; Eginli, A.; Krishnaswamy, G. Anaphylaxis. Chest 2018, 153, 528–543. [Google Scholar] [CrossRef]
- Krishnaswamy, G. Critical Care Management of the Patient With Anaphylaxis: A Concise Definitive Review. Crit. Care Med. 2021, 49, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Cardona, V.; Ansotegui, I.J.; Ebisawa, M.; El-Gamal, Y.; Rivas, M.F.; Fineman, S.; Geller, M.; Gonzalez-Estrada, A.; Greenberger, P.A.; Borges, M.S.; et al. World Allergy Organization Anaphylaxis Guidance 2020. World Allergy Organ. J. 2020, 13. [Google Scholar] [CrossRef]
- Sclar, D.A.; Lieberman, P.L. Anaphylaxis: Underdiagnosed, Underreported, and Undertreated. Am. J. Med. 2014, 127, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Sampson, H.A.; Muñoz-Furlong, A.; Campbell, R.L.; Adkinson, N.F.; Bock, S.A.; Branum, A.; Brown, S.; Camargo, C.A.; Cydulka, R.; Galli, S.J.; et al. Second symposium on the definition and management of anaphylaxis: Summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J. Allergy Clin. Immunol. 2006, 117, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.; Campbell, D.E.; Motosue, M.S.; Campbell, R.L. Global Trends in Anaphylaxis Epidemiology and Clinical Implications. J. Allergy Clin. Immunol. Pract. 2020, 8, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Saleh, H.; Embry, S.; Nauli, A.; Atyia, S.; Krishnaswamy, G. Anaphylactic Reactions to Oligosaccharides in Red Meat: A Syndrome in Evolution. Clin. Mol. Allergy 2012, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.L.; Krishnaswamy, G. Autoimmune progesterone dermatitis and its manifestation as anaphylaxis: A case report and literature review. Ann. Allergy Asthma Immunol. 2003, 90, 469–477. [Google Scholar] [CrossRef]
- Foer, D.; Buchheit, K.M.; Gargiulo, A.R.; Lynch, D.M.; Castells, M.; Wickner, P.G. Progestogen Hypersensitivity in 24 Cases: Diagnosis, Management, and Proposed Renaming and Classification. J. Allergy Clin. Immunol. Pract. 2016, 4, 723–729. [Google Scholar] [CrossRef]
- Motosue, M.S.; Bellolio, M.F.; Van Houten, H.K.; Shah, N.D.; Li, J.T.; Campbell, R.L. Outcomes of Emergency Department Anaphylaxis Visits from 2005 to 2014. J. Allergy Clin. Immunol. Pract. 2018, 6, 1002–1009. [Google Scholar] [CrossRef]
- Tejedor-Alonso, M.A.; Moro-Moro, M.; Múgica-García, M.V. Epidemiology of Anaphylaxis: Contributions from the Last 10 Years. J. Investig. Allergol. Clin. Immunol. 2015, 25, 163–175. [Google Scholar]
- Moneret-Vautrin, D.A.; Morisset, M.; Flabbee, J.; Beaudouin, E.; Kanny, G. Epidemiology of life-threatening and lethal anaphylaxis: A review. Allergy 2005, 60, 443–451. [Google Scholar] [CrossRef]
- Miller, C.; Guha, B.; Krishnaswamy, G. Exercise-Induced Anaphylaxis: A Serious but Preventable Disorder. Phys. Sportsmed. 2008, 36, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Castells, M. Diagnosis and management of anaphylaxis in precision medicine. J. Allergy Clin. Immunol. 2017, 140, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Labella, M.; Garcia-Neuer, M.; Castells, M. Application of precision medicine to the treatment of anaphylaxis. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 190–197. [Google Scholar] [CrossRef]
- Jimenez-Rodriguez, T.W.; Garcia-Neuer, M.; Alenazy, L.A.; Castells, M. Anaphylaxis in the 21st century: Phenotypes, endotypes, and biomarkers. J. Asthma Allergy 2018, 11, 121–142. [Google Scholar] [CrossRef] [Green Version]
- Agache, I.; Akdis, C.A. Precision medicine and phenotypes, endotypes, genotypes, regiotypes, and theratypes of allergic diseases. J. Clin. Investig. 2019, 129, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Cunill, A.S.; Guilarte, M.; Cardona, V. Phenotypes, endotypes and biomarkers in anaphylaxis: Current insights. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 370–376. [Google Scholar] [CrossRef]
- Fenny, N.; Grammer, L.C. Idiopathic Anaphylaxis. Immunol. Allergy Clin. N. Am. 2015, 35, 349–362. [Google Scholar] [CrossRef]
- Greenberger, P.A.; Lieberman, P. Idiopathic Anaphylaxis. J. Allergy Clin. Immunol. Pract. 2014, 2, 243–250. [Google Scholar] [CrossRef]
- Kuhlen, J.L.; Virkud, Y.V. Pathogenesis, newly recognized etiologies, and management of idiopathic anaphylaxis. Discov. Med. 2015, 19, 137–144. [Google Scholar] [PubMed]
- Ebo, D.G.; Clarke, R.C.; Mertes, P.-M.; Platt, P.R.; Sabato, V.; Sadleir, P. Molecular mechanisms and pathophysiology of perioperative hypersensitivity and anaphylaxis: A narrative review. Br. J. Anaesth. 2019, 123, e38–e49. [Google Scholar] [CrossRef] [PubMed]
- Komi, D.E.A.; Wöhrl, S.; Bielory, L. Mast Cell Biology at Molecular Level: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 58, 342–365. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Ajitawi, O.; Chi, D.S. The Human Mast Cell: An Overview. Methods Mol. Biol. 2006, 315, 013–034. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Kelly, J.; Johnson, D.; Youngberg, W.; Stone, S.; Huang, K.; Bieber, J.; Shi, S.D. The human mast cell: Functions in physiology and disease. Front. Biosci. 2001, 6, d1109–d1127. [Google Scholar] [CrossRef] [Green Version]
- Finkelman, F.D.; Khodoun, M.V.; Strait, R. Human IgE-independent systemic anaphylaxis. J. Allergy Clin. Immunol. 2016, 137, 1674–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Gilfillan, A.M.; Austin, S.J.; Metcalfe, D.D. Mast Cell Biology: Introduction and Overview. Adv. Exp. Med. Biol. 2011, 716, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Vyas, H.; Krishnaswamy, G.; Chi, D.S. Paul Ehrlich’s. Mast Cells 2005, 315, 003–012. [Google Scholar] [CrossRef]
- Ghably, J.; Saleh, H.; Vyas, H.; Peiris, E.; Misra, N.; Krishnaswamy, G. Paul Ehrlich’s Mastzellen: A Historical Perspective of Relevant Developments in Mast Cell Biology. Adv. Struct. Saf. Stud. 2014, 1220, 3–10. [Google Scholar] [CrossRef]
- Reber, L.; Hernandez, J.D.; Galli, S.J. The pathophysiology of anaphylaxis. J. Allergy Clin. Immunol. 2017, 140, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Karasuyama, H.; Mukai, K.; Obata-Ninomiya, K.; Tsujimura, Y.; Wada, T. Nonredundant Roles of Basophils in Immunity. Annu. Rev. Immunol. 2011, 29, 45–69. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The development of human mast cells. An historical reappraisal. Exp. Cell Res. 2016, 342, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, G.; Lakshman, T.; Miller, A.; Srikanth, S.; Hall, K.; Huang, S.-K.; Suttles, J.; Smith, J.; Stout, R. Multifunctional Cytokine Expression by Human Mast Cells: Regulation by T Cell Membrane Contact and Glucocorticoids. J. Interf. Cytokine Res. 1997, 17, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Watkins, C.E.; Bokor, W.B.; Leicht, S.; Youngberg, G.; Krishnaswamy, G. Telangiectasia Macularis Eruptiva Perstans: More than skin deep. Dermatol. Rep. 2011, 3, e12. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.C.; Metcalfe, D.D.; Komarow, H.D. Mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Anderson, D.F.; Bradding, P.; Coward, W.R.; Baddeley, S.M.; MacLeod, J.D.A.; McGill, J.I.; Church, M.K.; Holgate, S.T.; Roche, W.R. Human mast cells express stem cell factor. J. Pathol. 1998, 186, 59–66. [Google Scholar] [CrossRef]
- Coward, W.R.; Okayama, Y.; Sagara, H.; Wilson, S.J.; Holgate, S.T.; Church, M.K. NF-κB and TNF-α: A Positive Autocrine Loop in Human Lung Mast Cells? J. Immunol. 2002, 169, 5287–5293. [Google Scholar] [CrossRef] [Green Version]
- Satitsuksanoa, P.; Van De Veen, W.; Akdis, M. B cells in food allergy. J. Allergy Clin. Immunol. 2021, 147, 49–51. [Google Scholar] [CrossRef]
- Sallis, B.F.; Fiebiger, E. A Shocking Type of Communication. Immunity 2018, 49, 999–1001. [Google Scholar] [CrossRef] [Green Version]
- Kraft, S.; Kinet, J.-P.P. New developments in FcεRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, H.C. Fifty years later: Emerging functions of IgE antibodies in host defense, immune regulation, and allergic diseases. J. Allergy Clin. Immunol. 2016, 137, 1631–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, M.K.; Pao, G.J.; Holgate, S.T. Characterization of histamine secretion from mechanically dispersed human lung mast cells: Effects of anti-IgE, calcium ionophore A23187, compound 48/80, and basic polypeptides. J. Immunol. 1982, 129, 2116–2121. [Google Scholar] [PubMed]
- Shakoory, B.; Fitzgerald, S.M.; Lee, S.A.; Chi, D.S.; Krishnaswamy, G. The Role of Human Mast Cell-Derived Cytokines in Eosinophil Biology. J. Interf. Cytokine Res. 2004, 24, 271–281. [Google Scholar] [CrossRef]
- Kunder, C.; John, A.S.; Abraham, S.N. Mast cell modulation of the vascular and lymphatic endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [Green Version]
- White, M.V. The role of histamine in allergic diseases. J. Allergy Clin. Immunol. 1990, 86, 599–605. [Google Scholar] [CrossRef]
- Ono, E.; Taniguchi, M.; Mita, H.; Fukutomi, Y.; Higashi, N.; Miyazaki, E.; Kumamoto, T.; Akiyama, K. Increased production of cysteinyl leukotrienes and prostaglandin D2 during human anaphylaxis. Clin. Exp. Allergy 2009, 39, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, Y.; Maekawa, A.; Penrose, J.F.; Austen, K.F.; Lam, B.K. Attenuated Zymosan-induced Peritoneal Vascular Permeability and IgE-dependent Passive Cutaneous Anaphylaxis in Mice Lacking Leukotriene C4 Synthase. J. Biol. Chem. 2001, 276, 22608–22613. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, A.; Austen, K.F.; Kanaoka, Y. Targeted Gene Disruption Reveals the Role of Cysteinyl Leukotriene 1 Receptor in the Enhanced Vascular Permeability of Mice Undergoing Acute Inflammatory Responses. J. Biol. Chem. 2002, 277, 20820–20824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Machado, F.R.; Assem, E.S.; Ezeamuzie, C.I. Cardiac anaphylaxis: The role of different mediators, Part I: Histamine. Allergol. Immunopathol. 1985, 13, 259–272. [Google Scholar]
- Gill, P.; Jindal, N.L.; Jagdis, A.; Vadas, P. Platelets in the immune response: Revisiting platelet-activating factor in anaphylaxis. J. Allergy Clin. Immunol. 2015, 135, 1424–1432. [Google Scholar] [CrossRef] [Green Version]
- Vadas, P.; Gold, M.; Perelman, B.; Liss, G.M.; Lack, G.; Blyth, T.; Simons, F.E.R.; Simons, K.J.; Cass, D.; Yeung, J. Platelet-Activating Factor, PAF Acetylhydrolase, and Severe Anaphylaxis. N. Engl. J. Med. 2008, 358, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.G.A.; Stone, S.F.; Fatovich, D.M.; Burrows, S.A.; Holdgate, A.; Celenza, A.; Coulson, A.; Hartnett, L.; Nagree, Y.; Cotterell, C.; et al. Anaphylaxis: Clinical patterns, mediator release, and severity. J. Allergy Clin. Immunol. 2013, 132, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.C.; Wilding, T.; Buka, R.J.; Baretto, R.L.; Huissoon, A.P.; Krishna, M.T. Biomarkers in Human Anaphylaxis: A Critical Appraisal of Current Evidence and Perspectives. Front. Immunol. 2019, 10, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadas, P.; Perelman, B.; Liss, G. Platelet-activating factor, histamine, and tryptase levels in human anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 144–149. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Bonadonna, P.; Hartmann, K.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Siebenhaar, F.; Sperr, W.R.; Elberink, J.N.O.; et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 1125–1133.e1. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var---Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef]
- Tanimoto, A.; Wang, K.-Y.; Murata, Y.; Kimura, S.; Nomaguchi, M.; Nakata, S.; Tsutsui, M.; Sasaguri, Y. Histamine Upregulates the Expression of Inducible Nitric Oxide Synthase in Human Intimal Smooth Muscle Cells via Histamine H1 Receptor and NF-κB Signaling Pathway. Arter. Thromb. Vasc. Biol. 2007, 27, 1556–1561. [Google Scholar] [CrossRef] [Green Version]
- Mayhan, W.G. Nitric oxide accounts for histamine-induced increases in macromolecular extravasation. Am. J. Physiol. Circ. Physiol. 1994, 266, H2369–H2373. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide: A regulator of mast cell activation and mast cell-mediated inflammation. Clin. Exp. Immunol. 2002, 129, 4–10. [Google Scholar] [CrossRef]
- Cauwels, A.; Janssen, B.; Buys, E.; Sips, P.; Brouckaert, P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J. Clin. Investig. 2006, 116, 2244–2251. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, C.J.; Michel, T. What’s in a name? eNOS and anaphylactic shock. J. Clin. Investig. 2006, 116, 2075–2078. [Google Scholar] [CrossRef]
- Evora, P.R.; Simon, M.R. Role of nitric oxide production in anaphylaxis and its relevance for the treatment of anaphylactic hypotension with methylene blue. Ann. Allergy Asthma Immunol. 2007, 99, 306–313. [Google Scholar] [CrossRef]
- Nakamura, T.; Murata, T. Regulation of vascular permeability in anaphylaxis. Br. J. Pharmacol. 2018, 175, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, I.; Dombrowicz, D.; Martin, T.R.; Ravetch, J.V.; Kinet, J.P.; Galli, S.J. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Investig. 1997, 99, 901–914. [Google Scholar] [CrossRef]
- Dombrowicz, D.; Flamand, V.; Miyajima, I.; Ravetch, J.V.; Galli, S.J.; Kinet, J.P. Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. J. Clin. Investig. 1997, 99, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, C.; Ridley, A. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef]
- Van Buul, J.; Timmerman, I. Small Rho GTPase-mediated actin dynamics at endothelial adherens junctions. Small GTPases 2016, 7, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Ashina, K.; Tsubosaka, Y.; Nakamura, T.; Omori, K.; Kobayashi, K.; Hori, M.; Ozaki, H.; Murata, T. Histamine Induces Vascular Hyperpermeability by Increasing Blood Flow and Endothelial Barrier Disruption In Vivo. PLoS ONE 2015, 10, e0132367. [Google Scholar] [CrossRef]
- Korhonen, H.; Fisslthaler, B.; Moers, A.; Wirth, A.; Habermehl, D.; Wieland, T.; Schütz, G.; Wettschureck, N.; Fleming, I.; Offermanns, S. Anaphylactic shock depends on endothelial Gq/G11. J. Exp. Med. 2009, 206, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef] [Green Version]
- Kugelmann, D.; Rotkopf, L.T.; Radeva, M.Y.; Garcia-Ponce, A.; Walter, E.; Waschke, J. Histamine causes endothelial barrier disruption via Ca2+-mediated RhoA activation and tension at adherens junctions. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Mayhan, W.G. Role of nitric oxide in histamine-induced increases in permeability of the blood–brain barrier. Brain Res. 1996, 743, 70–76. [Google Scholar] [CrossRef]
- Klinger, J.R.; Kadowitz, P.J. The Nitric Oxide Pathway in Pulmonary Vascular Disease. Am. J. Cardiol. 2017, 120, S71–S79. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F. The cGMP system: Components and function. Biol. Chem. 2020, 401, 447–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mónica, F.; Bian, K.; Murad, F. The Endothelium-Dependent Nitric Oxide–cGMP Pathway. Adv. Pharmacol. 2016, 77, 1–27. [Google Scholar] [CrossRef]
- Jang, D.H.; Nelson, L.S.; Hoffman, R.S. Methylene Blue for Distributive Shock: A Potential New Use of an Old Antidote. J. Med. Toxicol. 2013, 9, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, K.W.; Ruiz-Garcia, M.; Patel, N.; Boyle, R.J.; Turner, P.J. Reaction phenotypes in IgE-mediated food allergy and anaphylaxis. Ann. Allergy Asthma Immunol. 2020, 124, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Saiz, R. Drug-induced IgG-neutrophil-mediated anaphylaxis in humans: Uncovered. Allergy 2019, 75, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Kow, A.S.F.; Chik, A.; Soo, K.-M.; Khoo, L.W.; Abas, F.; Tham, C.L. Identification of Soluble Mediators in IgG-Mediated Anaphylaxis via Fcγ Receptor: A Meta-Analysis. Front. Immunol. 2019, 10, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, M.M.; DeStefano, F. Vaccine-associated hypersensitivity. J. Allergy Clin. Immunol. 2018, 141, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Bretterklieber, A.; Beham-Schmid, C.; Sturm, G.J.; Berghold, A.; Brezinschek, R.; Aberer, W.; Aberer, E. Anaphylaxis with clonal mast cells in normal looking skin–A new entity? Allergy 2015, 70, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, F.; De Chaisemartin, L.; Granger, V.; Gouel, A.; Gillis, C.M.; Zhu, Q.; Dib, F.; Nicaise-Roland, P.; Ganneau, C.; Hurtado-Nedelec, M.; et al. An IgG-induced neutrophil activation pathway contributes to human drug-induced anaphylaxis. Sci. Transl. Med. 2019, 11, eaat1479. [Google Scholar] [CrossRef] [PubMed]
- Zinderman, C.E.; Landow, L.; Wise, R.P. Anaphylactoid reactions to Dextran 40 and 70: Reports to the United States Food and Drug Administration, 1969 to 2004. J. Vasc. Surg. 2006, 43, 1004–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, W.; Späth, P.; Zühlsdorf, M.; Dalichau, H.; Kirchhoff, P.G.; Kuppe, H.; Preiss, D.U.; Mayer, G. Anaphylactic Reactions to Aprotinin Reexposure in Cardiac Surgery. Anesthesiology 2001, 95, 64–71. [Google Scholar] [CrossRef]
- Williams, S.J.; Gupta, S. Anaphylaxis to IVIG. Arch. Immunol. Ther. Exp. 2016, 65, 11–19. [Google Scholar] [CrossRef]
- Jonsson, F.; Mancardi, D.A.; Kita, Y.; Karasuyama, H.; Iannascoli, B.; Van Rooijen, N.; Shimizu, T.; Daeron, M.; Bruhns, P. Mouse and human neutrophils induce anaphylaxis. J. Clin. Investig. 2011, 121, 1484–1496. [Google Scholar] [CrossRef] [Green Version]
- Lukan, N. “Cytokine storm”, not only in COVID-19 patients. Mini-review. Immunol. Lett. 2020, 228, 38–44. [Google Scholar] [CrossRef]
- Khan, D.A. Hypersensitivity and immunologic reactions to biologics: Opportunities for the allergist. Ann. Allergy Asthma Immunol. 2016, 117, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Macy, E. Immune-Related Adverse Drug Reactions and Immunologically Mediated Drug Hypersensitivity. Immunol. Allergy Clin. N. Am. 2020, 40, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Freyer, C.W.; Porter, D.L. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic malignancies. J. Allergy Clin. Immunol. 2020, 146, 940–948. [Google Scholar] [CrossRef]
- Guilarte, M.; Sala-Cunill, A.; Luengo, O.; Labrador-Horrillo, M.; Cardona, V. The Mast Cell, Contact, and Coagulation System Connection in Anaphylaxis. Front. Immunol. 2017, 8, 846. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.P. Preventing anaphylaxis fatalities: Should we target bradykinin? J. Allergy Clin. Immunol. 2020, 145, 1365–1366. [Google Scholar] [CrossRef] [Green Version]
- Bender, L.; Weidmann, H.; Rose-John, S.; Renné, T.; Long, A.T. Factor XII-Driven Inflammatory Reactions with Implications for Anaphylaxis. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnee, T.; Reddigari, S.R.; Shibayama, Y.; Kaplan, A.P.; Silverberg, M. Mast cell derived heparin activates the contact system: A link to kinin generation in allergic reactions. Clin. Exp. Allergy 1997, 27, 653–663. [Google Scholar] [CrossRef]
- Noga, O.; Brunnée, T.; Schäper, C.; Kunkel, G. Heparin, derived from the mast cells of human lungs is responsible for the generation of kinins in allergic reactions due to the activation of the contact system. Int. Arch. Allergy Immunol. 1999, 120, 310–316. [Google Scholar] [CrossRef]
- E Warkentin, T.; Greinacher, A. Heparin-induced anaphylactic and anaphylactoid reactions: Two distinct but overlapping syndromes. Expert Opin. Drug Saf. 2009, 8, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, K.D.; Coyne, D.P.; Sanford, D.M.; Huddelson, J. The heparin recall of 2008. Perfus. 2012, 28, 61–65. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated Heparin Associated with Adverse Clinical Events and Activation of the Contact System. N. Eng. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunill, A.S.; Björkqvist, J.; Senter, R.; Guilarte, M.; Cardona, V.; Labrador-Horrillo, M.; Nickel, K.F.; Butler, L.; Luengo, O.; Kumar, P.; et al. Plasma contact system activation drives anaphylaxis in severe mast cell–mediated allergic reactions. J. Allergy Clin. Immunol. 2015, 135, 1031–1043.e6. [Google Scholar] [CrossRef] [PubMed]
- Inal, M.T.; Memis, D.; Top, H.; Bahar, M.; Celik, E.; Sıkar, E.Y.; Kement, B.; Sikar, E.Y. Late-Onset Pulmonary Edema and Disseminated Intravascular Coagulation Due to Latex Anaphylaxis. Aesthet. Plast. Surg. 2009, 34, 394–396. [Google Scholar] [CrossRef]
- Choi, I.H.; Ha, T.Y.; Lee, D.G.; Park, J.S.; Lee, J.H.; Park, Y.M.; Lee, H.K. Occurrence of disseminated Intravascular coagulation (DIC) in active systemic anaphylaxis: Role of platelet-activating factor. Clin. Exp. Immunol. 2008, 100, 390–394. [Google Scholar] [CrossRef]
- Lombardini, C.; Helia, R.-E.; Boehlen, F.; Merlani, P. “Heparinization” and hyperfibrinogenolysis by wasp sting. Am. J. Emerg. Med. 2009, 27, 1176.e1–1176.e3. [Google Scholar] [CrossRef]
- Truong, H.; Browning, R. Anaphylaxis-induced hyperfibrinolysis in pregnancy. Int. J. Obstet. Anesth. 2015, 24, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Asero, R.; Tedeschi, A.; Lazzari, R.; Marzano, A.V. Inflammation and Coagulation in Urticaria and Angioedema. Curr. Vasc. Pharmacol. 2012, 10, 653–658. [Google Scholar] [CrossRef]
- Yanase, Y.; Takahagi, S.; Hide, M. Chronic spontaneous urticaria and the extrinsic coagulation system. Allergol. Int. 2018, 67, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Kolkhir, P.; Asero, R.; Pogorelov, D.; Olisova, O.; Kochergin, N.; Cugno, M. Chronic urticaria and coagulation: Pathophysiological and clinical aspects. Allergy 2014, 69, 683–691. [Google Scholar] [CrossRef]
- Jung, J.W.; Jeon, E.J.; Kim, J.W.; Choi, J.C.; Shin, J.W.; Kim, J.Y.; Park, I.W.; Choi, B.W. A Fatal Case of Intravascular Coagulation After Bee Sting Acupuncture. Allergy Asthma Immunol. Res. 2012, 4, 107–109. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef]
- Cano, R.M.; Picado, C.; Valero, A.; Bartra, J. Mechanisms of Anaphylaxis Beyond IgE. J. Investig. Allergol. Clin. Immunol. 2016, 26, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Akin, C. Mast Cell Activation Syndromes Presenting as Anaphylaxis. Immunol. Allergy Clin. N. Am. 2015, 35, 277–285. [Google Scholar] [CrossRef]
- Bonadonna, P.; Bonifacio, M.; Lombardo, C.; Zanotti, R. Hymenoptera Allergy and Mast Cell Activation Syndromes. Curr. Allergy Asthma Rep. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Nedoszytko, B.; Bonadonna, P.; Hartmann, K.; Niedoszytko, M.; Brockow, K.; Siebenhaar, F.; Triggiani, M.; Arock, M.; et al. Diagnosis, Classification and Management of Mast Cell Activation Syndromes (MCAS) in the Era of Personalized Medicine. Int. J. Mol. Sci. 2020, 21, 9030. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.J. Hereditary Alpha Tryptasemia. Immunol. Allergy Clin. N. Am. 2018, 38, 483–495. [Google Scholar] [CrossRef]
- Robey, R.C.; Wilcock, A.; Bonin, H.; Beaman, G.; Myers, B.; Grattan, C.; Briggs, T.A.; Arkwright, P.D. Hereditary Alpha-Tryptasemia: UK Prevalence and Variability in Disease Expression. J. Allergy Clin. Immunol. Pract. 2020, 8, 3549–3556. [Google Scholar] [CrossRef]
- Giannetti, M.P.; Weller, E.; Bormans, C.; Novak, P.; Hamilton, M.J.; Castells, M. Hereditary alpha-tryptasemia in 101 patients with mast cell activation–related symptomatology including anaphylaxis. Ann. Allergy Asthma Immunol. 2021, 126, 655–660. [Google Scholar] [CrossRef]
- Vadas, P. The platelet-activating factor pathway in food allergy and anaphylaxis. Ann. Allergy Asthma Immunol. 2016, 117, 455–457. [Google Scholar] [CrossRef]
- Ribó, P.; Guo, Y.; Aranda, J.; Ainsua-Enrich, E.; Navinés-Ferrer, A.; Guerrero, M.; Pascal, M.; de la Cruz, C.; Orozco, M.; Muñoz-Cano, R.; et al. Mutation in KARS: A novel mechanism for severe anaphylaxis. J. Allergy Clin. Immunol. 2021, 147, 1855–1864. [Google Scholar] [CrossRef] [PubMed]

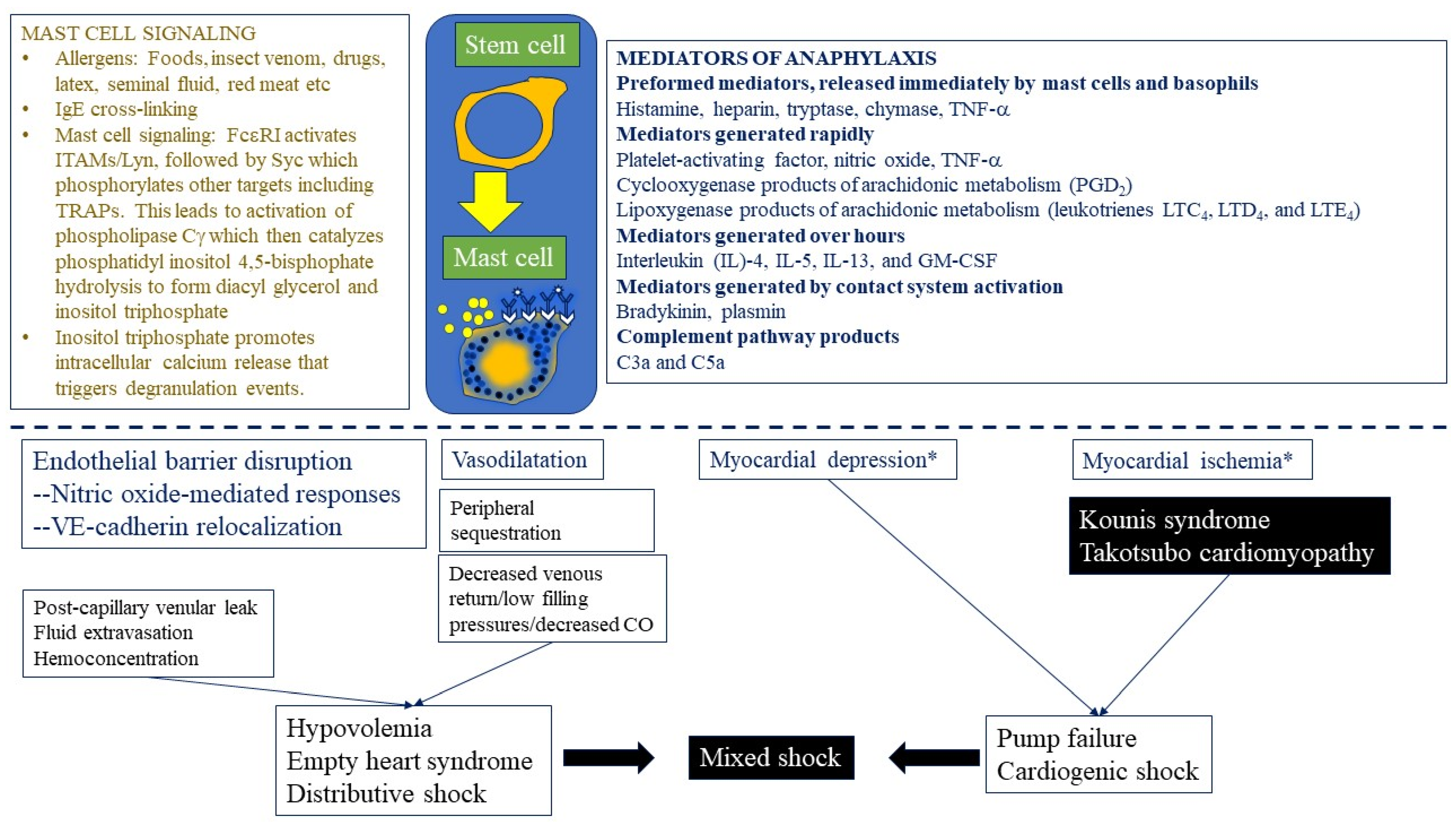
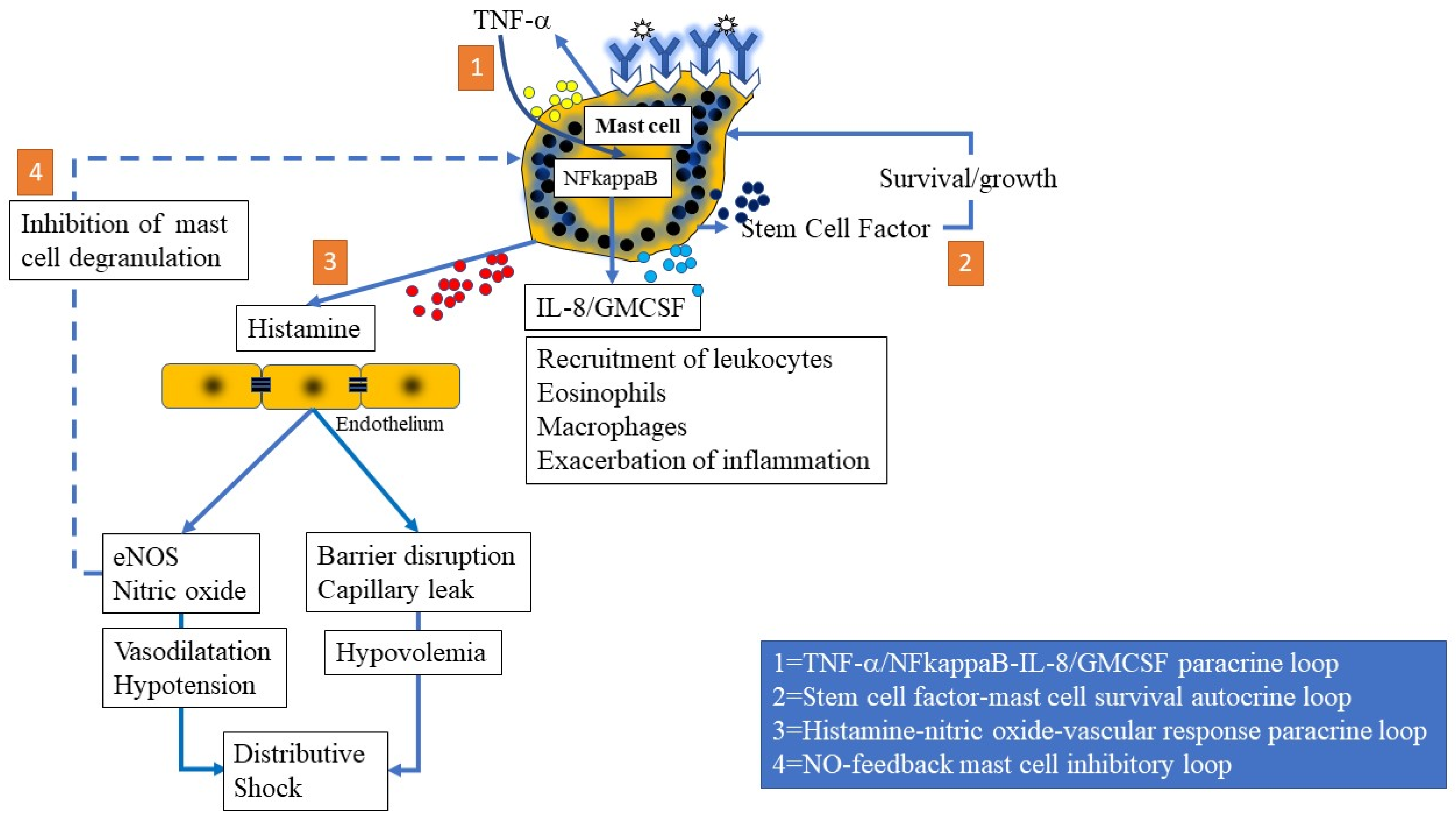
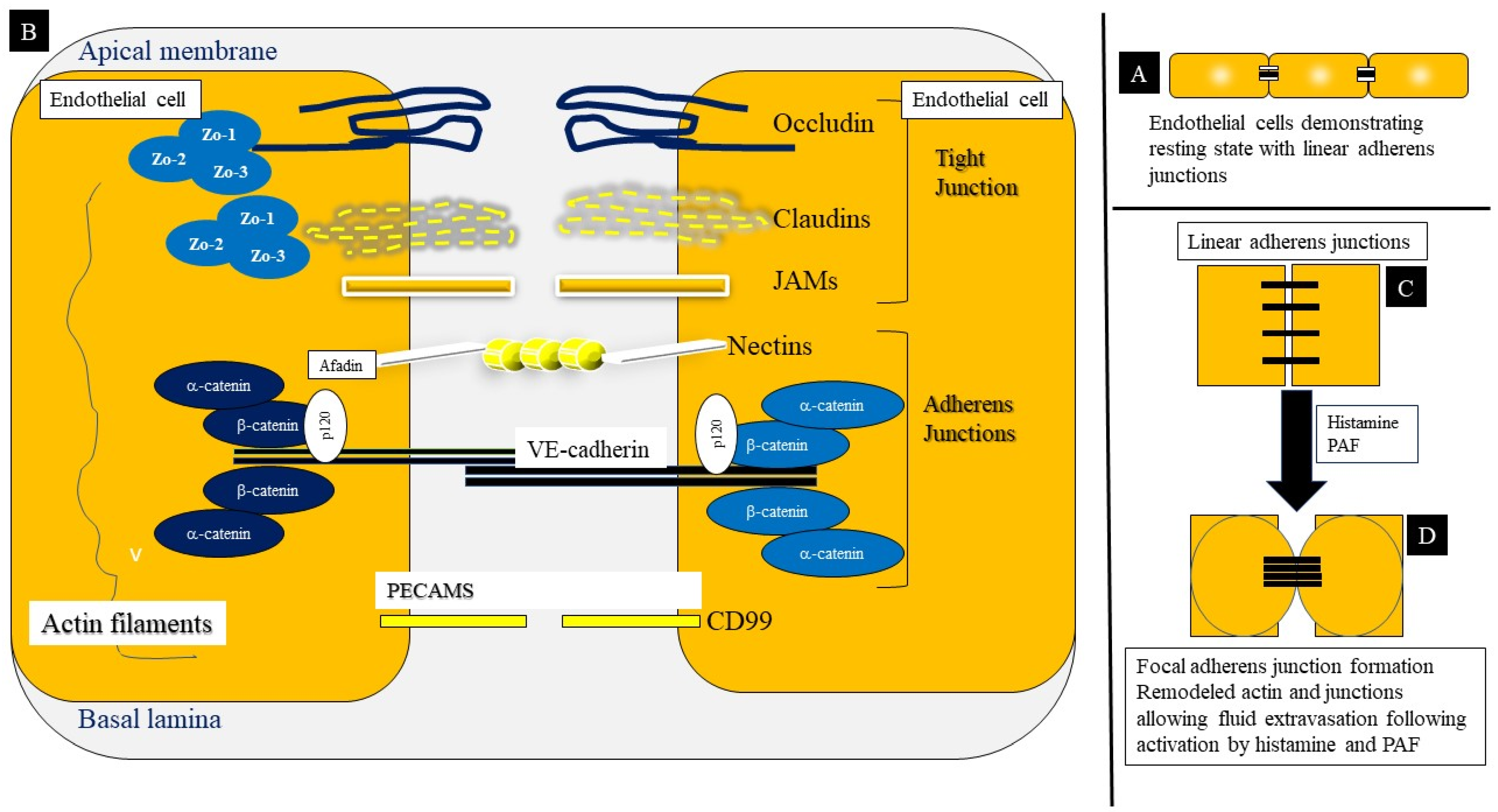
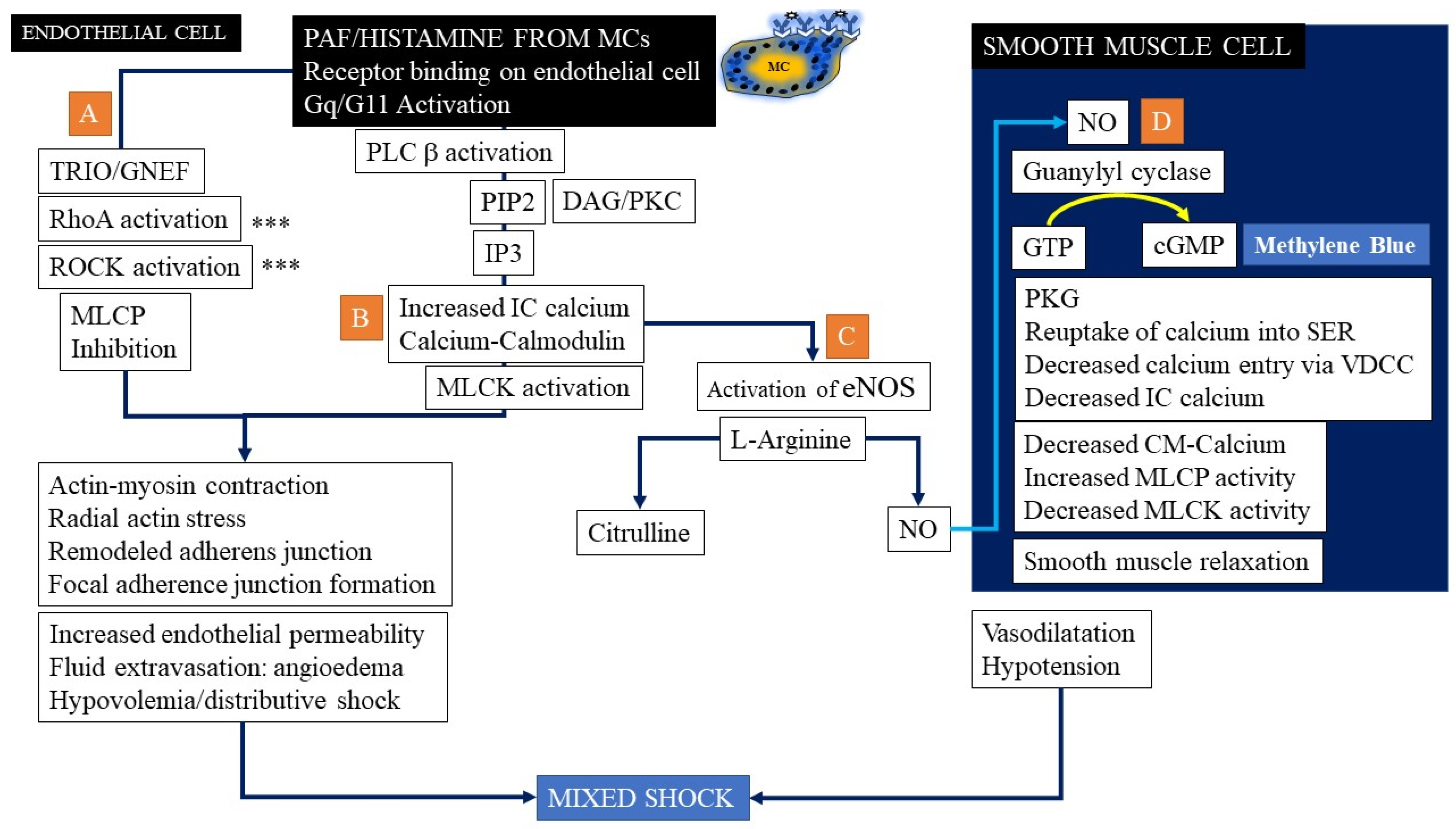

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endotype/Mediators | Phenotype/Symptoms | Triggers | Examples |
|---|---|---|---|
| Immune-mediated | |||
| IgE-dependent reactions Mast cells, histamine, tryptase, platelet activating factor, nitric oxide, leukotrienes, proteases (chymase), prostaglandins, TNF-α | Urticaria, angioedema, vomiting, diarrhea, hypotension | Foods | Peanut, wheat, soy, milk, egg, shellfish |
| Drugs | Antibiotics, anesthetics, chemotherapy drugs (platins, taxanes), biologics | ||
| Alpha-Gal | Mammalian meat | ||
| Hormones | Progesterone | ||
| Hymenoptera venom | Wasps, Hornets | ||
| IgG-mediated reactions IgG immune complexes, serotonin, PAF synthesis, thromboxane A2 | Wheezing, hypovolemia, hypotension, angioedema, diarrhea | IVIG | IgG/IgE anti-IgA antibodies (Common variable immune deficiency, IgA deficiency) |
| Blood transfusion | IgA deficiency | ||
| Immune compounds | Complement/immune complexes | ||
| Monoclonal antibodies | Rituximab | ||
| Cytokine release and cytokine storm reactions (T-cell, TNF-α, IL-1β, IL-6, and MCP-1) | Flushing, nausea, chills, fever, hypoxemia, hypotension. | Chemotherapy drugs (cytokine release) | |
| Monoclonal antibodies (cytokine storm) | |||
| Contact system activation Factor XII-kallikrein-bradykinin activation, bradykinin receptors, endothelium | Angioedema, wheezing, hypotension | Drugs | Heparin and oversulfated chondroitin sulfate |
| Complement-mediated reactions Anaphylatoxins-Factors C5a/C3a, mast cell complement receptors, inflammatory pathway activation and cellular recruitment | Drugs | Protamine (heparin antidote) | |
| Membrane | Hemodialysis | ||
| Toxins | Vespid Toxin (also IgE mediated) | ||
| Vehicle | Polyethylene glycol | ||
| Non-immune mediated (Idiopathic) | |||
| Direct mast cell/basophil activation Tryptase, cytokine, histamine release from mast cells/basophils | Angioedema, flushing, wheezing, hypotension | Aspirin and Nonsteroidal anti-inflammatory drugs | |
| Opiates | |||
| Physical | Exercise, sunlight, temperature changes | ||
| Media | Radiocontrast dyes | ||
| Antibiotics and anesthetics | Fluoroquinolones, vancomycin, tubocurarine, atracuronium | ||
| Primary Mast Cell Disorders | Systemic mastocytosis | Exercise, drugs, hymenoptera venom | |
| Mast cell activation syndrome | Food, stress, alcohol | ||
| Hereditary Alpha-Tryptasemia syndrome Autosomal dominant syndrome | Diarrhea, myalgia, fatigue, urticaria, pruritus and anaphylaxis | Increased copy numbers of the gene encoding alpha-tryptase (TPSAB1) | Molecular testing confirms the diagnosis Droplet digital PCR testing of buccal samples |
| Idiopathic anaphylaxis | Urticaria, angioedema, hypotension, syncope, anaphylaxis | Exclude mast cell disorders, mammalian meat allergy and Hereditary alpha-tryptasemia | Diagnosis of exclusion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, S.M.T.; Rupprecht, C.P.; Haque, A.; Pattanaik, D.; Yusin, J.; Krishnaswamy, G. Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. Int. J. Mol. Sci. 2021, 22, 7785. https://doi.org/10.3390/ijms22157785
Nguyen SMT, Rupprecht CP, Haque A, Pattanaik D, Yusin J, Krishnaswamy G. Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. International Journal of Molecular Sciences. 2021; 22(15):7785. https://doi.org/10.3390/ijms22157785
Chicago/Turabian StyleNguyen, Samantha Minh Thy, Chase Preston Rupprecht, Aaisha Haque, Debendra Pattanaik, Joseph Yusin, and Guha Krishnaswamy. 2021. "Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond" International Journal of Molecular Sciences 22, no. 15: 7785. https://doi.org/10.3390/ijms22157785
APA StyleNguyen, S. M. T., Rupprecht, C. P., Haque, A., Pattanaik, D., Yusin, J., & Krishnaswamy, G. (2021). Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. International Journal of Molecular Sciences, 22(15), 7785. https://doi.org/10.3390/ijms22157785