
Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies

 ,
,  , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Docking Compound Library with HcProRS Ligand-Bound Structure
2.2. Confirmation of the Hits from Virtual Screening by a Thermal Shift Assay
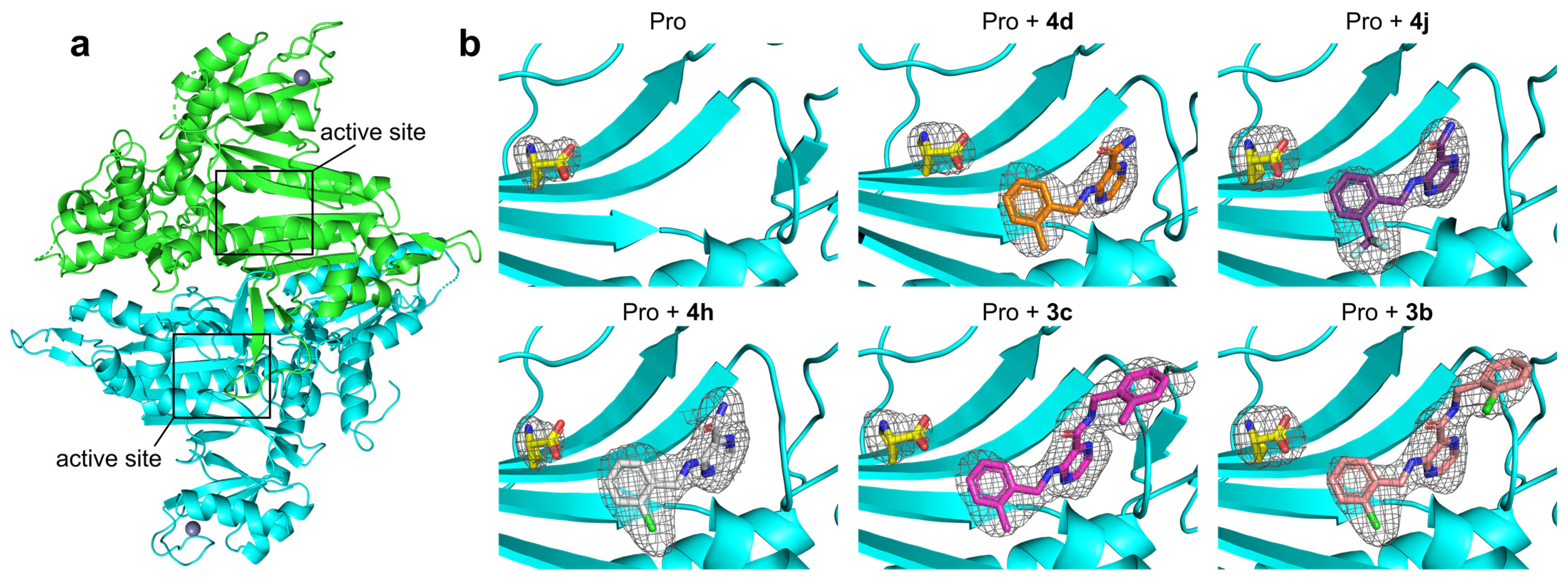
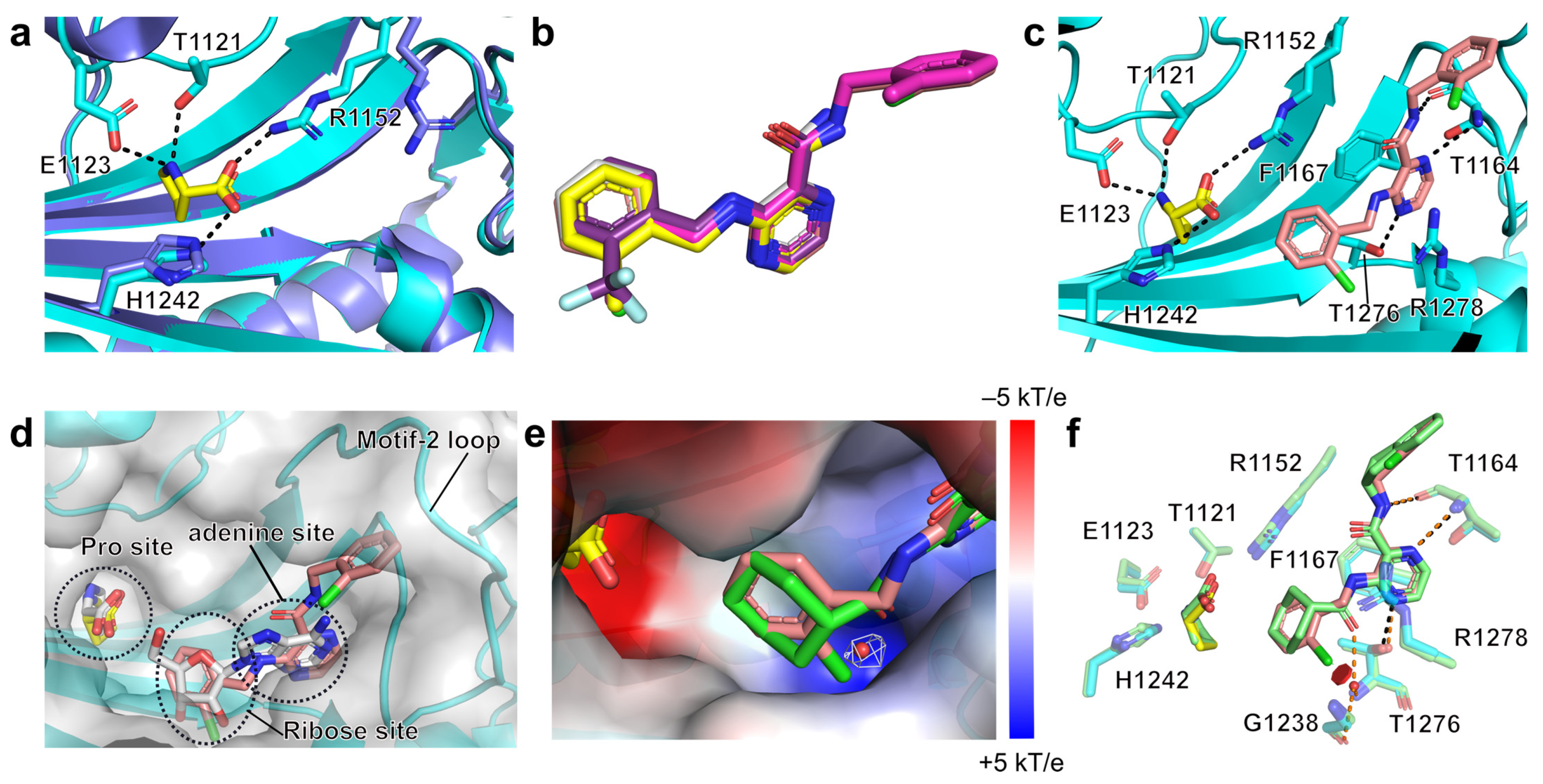
2.3. Structural Studies of the Binding Mechanism of the Compounds with HcProRS
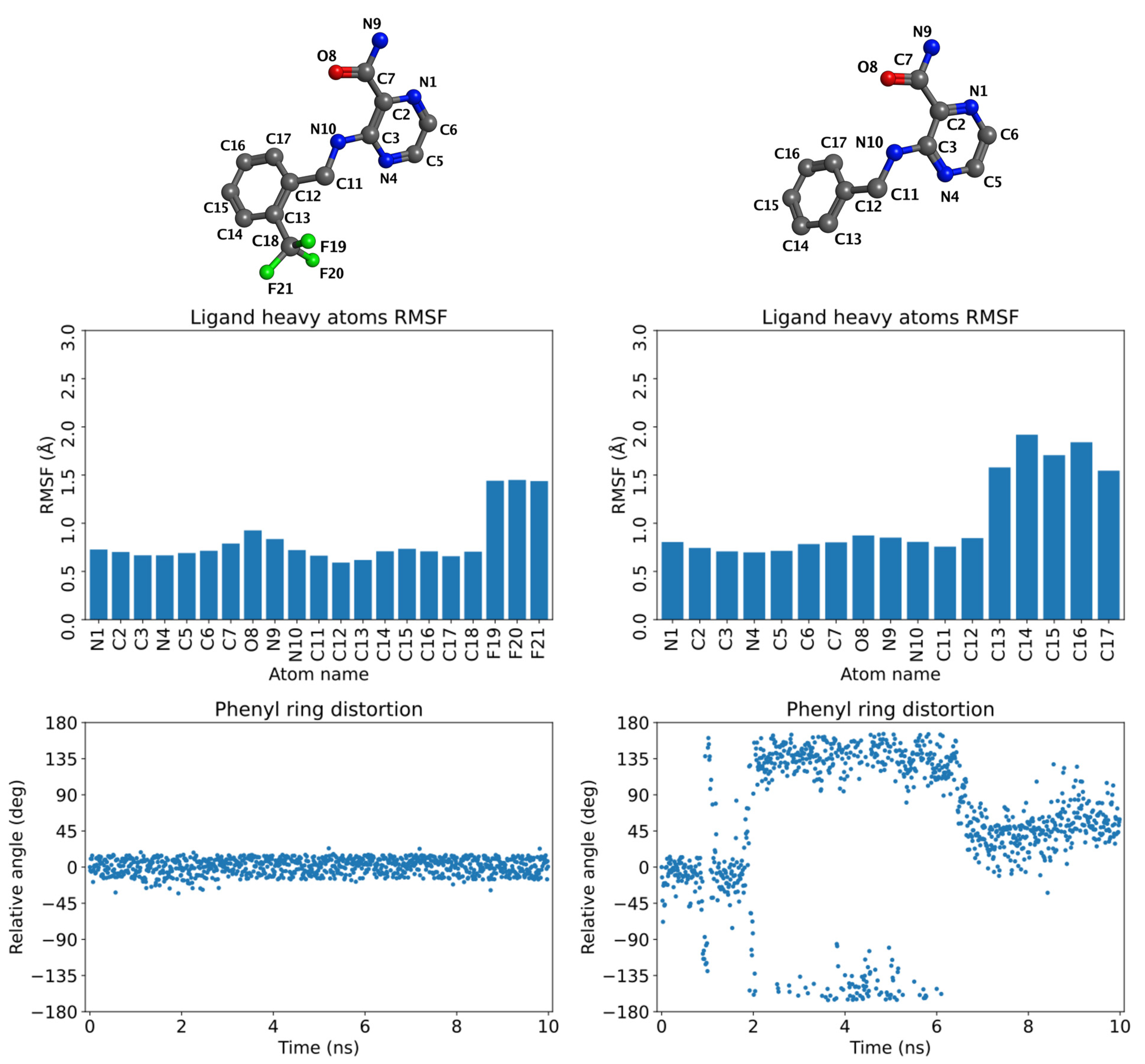
2.4. Molecular Dynamics (MD) Simulations
2.5. Potential Selectivity of Title Compounds towards HcProRS over Other Class II aaRS Members
3. Discussion
4. Materials and Methods
4.1. Protein Preparation
4.2. Crystallization of HcProRS Complexes
4.3. Data Collection and Structure Determination
4.4. Thermal Shift Assay
4.5. In Silico Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ibba, M.; Soll, D. Aminoacyl-TRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Antonellis, A.; Green, E.D. The Role of Aminoacyl-TRNA Synthetases in Genetic Diseases. Annu. Rev. Genomics Hum. Genet. 2008, 9, 87–107. [Google Scholar] [CrossRef]
- Kim, S.; You, S.; Hwang, D. Aminoacyl-TRNA Synthetases and Tumorigenesis: More than Housekeeping. Nat. Rev. Cancer 2011, 11, 708–718. [Google Scholar] [CrossRef]
- Yao, P.; Fox, P.L. Aminoacyl-TRNA Synthetases in Medicine and Disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef]
- Datt, M.; Sharma, A. Evolutionary and Structural Annotation of Disease-Associated Mutations in Human Aminoacyl-TRNA Synthetases. BMC Genomics 2014, 15, 1063. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Schuman, R.; Antonellis, A. Emerging Mechanisms of Aminoacyl-TRNA Synthetase Mutations in Recessive and Dominant Human Disease. Hum. Mol. Genet. 2017, 26, R114–R127. [Google Scholar] [CrossRef]
- Guo, M.; Schimmel, P. Essential Non-Translational Functions of TRNA Synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Bae, S.; Song, M. Recent Development of Aminoacyl-TRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules 2020, 10, 1625. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S.; Mullen, P. Progress and Challenges in Aminoacyl-TRNA Synthetase-Based Therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, V.; Yogavel, M.; Oshima, Y.; Kikuchi, H.; Touquet, B.; Hakimi, M.-A.; Sharma, A. Structure of Prolyl-TRNA Synthetase-Halofuginone Complex Provides Basis for Development of Drugs against Malaria and Toxoplasmosis. Structure 2015, 23, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pines, M.; Nagler, A. Halofuginone: A Novel Antifibrotic Therapy. Gen. Pharmacol. 1998, 30, 445–450. [Google Scholar] [CrossRef]
- Gavish, Z.; Pinthus, J.H.; Barak, V.; Ramon, J.; Nagler, A.; Eshhar, Z.; Pines, M. Growth Inhibition of Prostate Cancer Xenografts by Halofuginone. Prostate 2002, 51, 73–83. [Google Scholar] [CrossRef]
- Elkin, M.; Ariel, I.; Miao, H.Q.; Nagler, A.; Pines, M.; de-Groot, N.; Hochberg, A.; Vlodavsky, I. Inhibition of Bladder Carcinoma Angiogenesis, Stromal Support, and Tumor Growth by Halofuginone. Cancer Res. 1999, 59, 4111–4118. [Google Scholar] [PubMed]
- Leiba, M.; Jakubikova, J.; Klippel, S.; Mitsiades, C.S.; Hideshima, T.; Tai, Y.-T.; Leiba, A.; Pines, M.; Richardson, P.G.; Nagler, A.; et al. Halofuginone Inhibits Multiple Myeloma Growth in Vitro and in Vivo and Enhances Cytotoxicity of Conventional and Novel Agents. Br. J. Haematol 2012, 157, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zeng, Q.; Gettayacamin, M.; Tungtaeng, A.; Wannaying, S.; Lim, A.; Hansukjariya, P.; Okunji, C.O.; Zhu, S.; Fang, D. Antimalarial Activities and Therapeutic Properties of Febrifugine Analogs. Antimicrob. Agents Chemother. 2005, 49, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
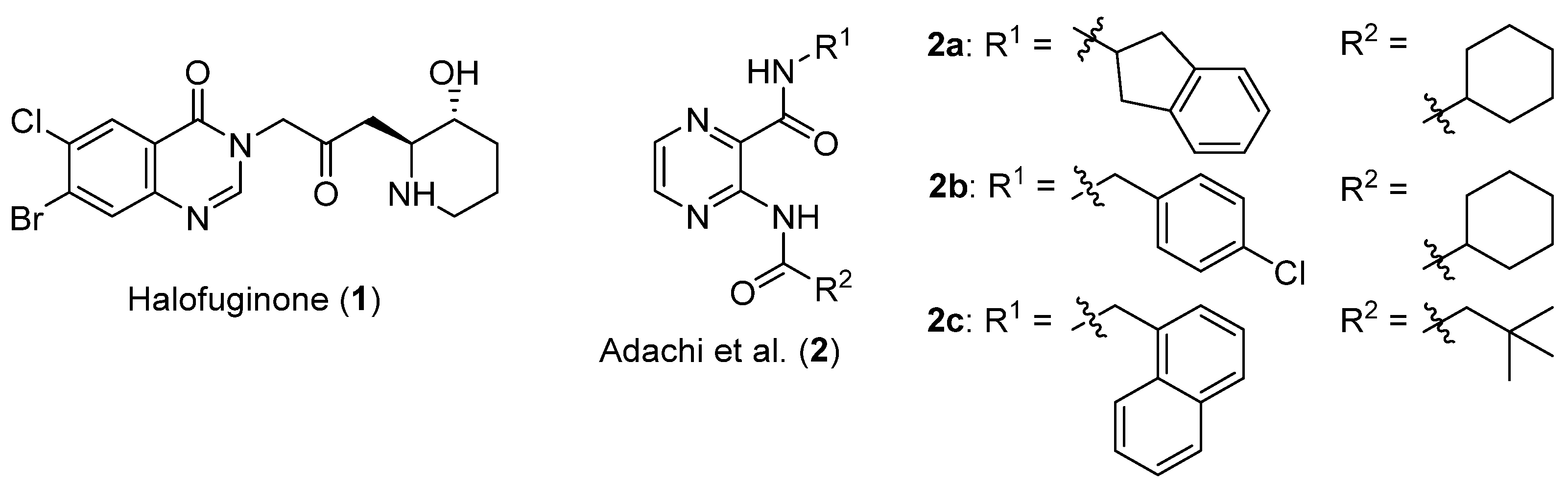
- Adachi, R.; Okada, K.; Skene, R.; Ogawa, K.; Miwa, M.; Tsuchinaga, K.; Ohkubo, S.; Henta, T.; Kawamoto, T. Discovery of a Novel Prolyl-TRNA Synthetase Inhibitor and Elucidation of Its Binding Mode to the ATP Site in Complex with l-Proline. Biochem. Biophys. Res. Commun. 2017, 488, 393–399. [Google Scholar] [CrossRef]
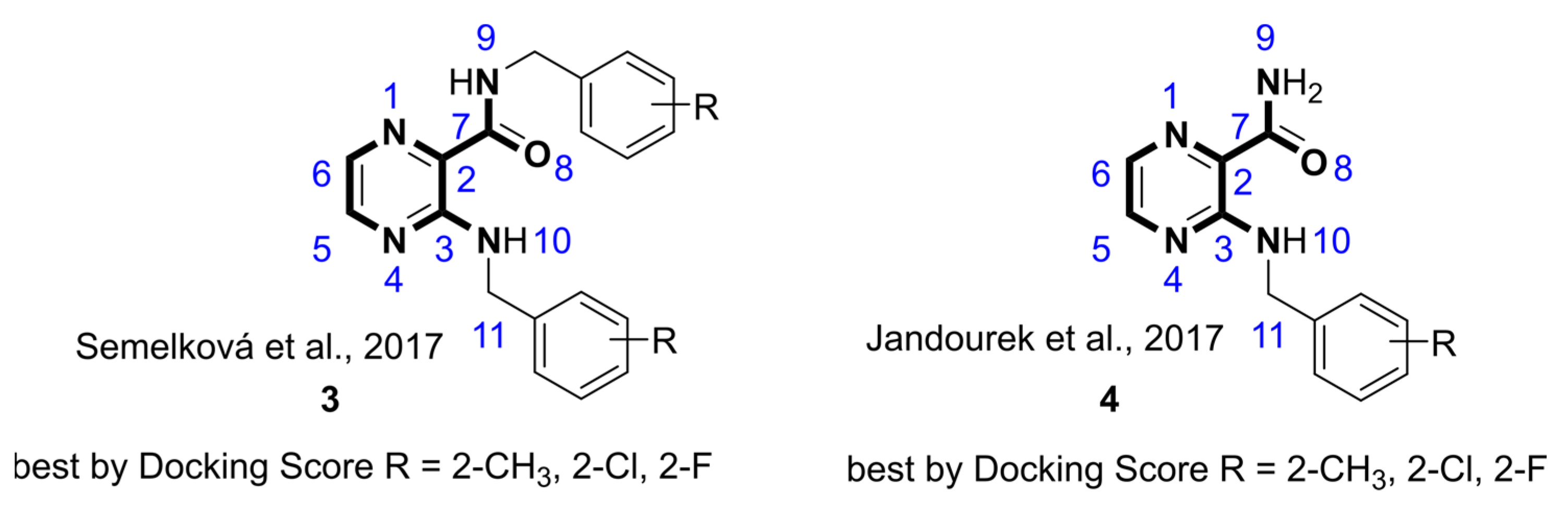
- Jandourek, O.; Tauchman, M.; Paterova, P.; Konecna, K.; Navratilova, L.; Kubicek, V.; Holas, O.; Zitko, J.; Dolezal, M. Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-Carboxamide and Their Antimicrobial Evaluation. Molecules 2017, 22, 223. [Google Scholar] [CrossRef] [Green Version]
- Semelková, L.; Janďourek, O.; Konečná, K.; Paterová, P.; Navrátilová, L.; Trejtnar, F.; Kubíček, V.; Kuneš, J.; Doležal, M.; Zitko, J. 3-Substituted N-Benzylpyrazine-2-Carboxamide Derivatives: Synthesis, Antimycobacterial and Antibacterial Evaluation. Molecules 2017, 22, 495. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.P.-D.S.; Pavan, F.R.; Leite, C.Q.F.; Felli, V.M.A. Synthesis and Evaluation of a Pyrazinoic Acid Prodrug in Mycobacterium Tuberculosis. Saudi Pharm. J. 2014, 22, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Zimhony, O.; Vilchèze, C.; Arai, M.; Welch, J.T.; Jacobs, W.R. Pyrazinoic Acid and Its N-Propyl Ester Inhibit Fatty Acid Synthase Type I in Replicating Tubercle Bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef] [Green Version]
- Lima, C.H.S.; Henriques, M.G.M.O.; Candéa, A.L.P.; Lourenço, M.C.S.; Bezerra, F.A.F.M.; Ferreira, M.L.; Kaiser, C.R.; de Souza, M.V.N. Synthesis and Antimycobacterial Evaluation of N′-(E)-Heteroaromaticpyrazine-2-Carbohydrazide Derivatives. Med. Chem. 2011, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Servusova-Vanaskova, B.; Jandourek, O.; Paterova, P.; Kordulakova, J.; Plevakova, M.; Kubicek, V.; Kucera, R.; Garaj, V.; Naesens, L.; Kunes, J.; et al. Alkylamino Derivatives of N-Benzylpyrazine-2-Carboxamide: Synthesis and Antimycobacterial Evaluation. Med. Chem. Commun. 2015, 6, 1311–1317. [Google Scholar] [CrossRef]
- Kranz, J.K.; Schalk-Hihi, C. Protein Thermal Shifts to Identify Low Molecular Weight Fragments. Methods Enzymol. 2011, 493, 277–298. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder Maps: Improving OMIT Maps by Excluding Bulk Solvent. Acta Crystallogr. D Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Carter, C.W. Cognition, Mechanism, and Evolutionary Relationships in Aminoacyl-TRNA Synthetases. Annu. Rev. Biochem. 1993, 62, 715–748. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Delarue, M.; Moras, D. The Aminoacyl-TRNA Synthetase Family: Modules at Work. Bioessays 1993, 15, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.; Musier-Forsyth, K.; Martinis, S.A. Aminoacyl-TRNA Synthetases in Biology and Disease: New Evidence for Structural and Functional Diversity in an Ancient Family of Enzymes. RNA 1997, 3, 954–960. [Google Scholar] [PubMed]
- Ribas de Pouplana, L.; Schimmel, P. Two Classes of TRNA Synthetases Suggested by Sterically Compatible Dockings on TRNA Acceptor Stem. Cell 2001, 104, 191–193. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Han, H.; Wang, J.; Chen, K.; Chen, X.; Guo, M. Structural Basis for Specific Inhibition of TRNA Synthetase by an ATP Competitive Inhibitor. Chem. Biol. 2015, 22, 734–744. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhou, H.; Vo, M.-N.; Shi, Y.; Nawaz, M.H.; Vargas-Rodriguez, O.; Diedrich, J.K.; Yates, J.R.; Kishi, S.; Musier-Forsyth, K.; et al. Double Mimicry Evades TRNA Synthetase Editing by Toxic Vegetable-Sourced Non-Proteinogenic Amino Acid. Nat. Commun. 2017, 8, 2281. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Van Aerschot, A. Aminoacyl-TRNA Synthetase Inhibitors as Potential Antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef]
- Pang, L.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-TRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int. J. Mol. Sci. 2021, 22, 1750. [Google Scholar] [CrossRef]
- Kwon, N.H.; Fox, P.L.; Kim, S. Aminoacyl-TRNA Synthetases as Therapeutic Targets. Nat. Rev. Drug Discov. 2019, 18, 629–650. [Google Scholar] [CrossRef] [PubMed]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for Aminoacyl-TRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Kuno, M.; Adachi, R.; Sato, Y.; Hattori, H.; Matsuda, A.; Okuzono, Y.; Igaki, K.; Tominari, Y.; Takagi, T.; et al. Discovery and Pharmacological Characterization of a New Class of Prolyl-TRNA Synthetase Inhibitor for Anti-Fibrosis Therapy. PLoS ONE 2017, 12, e0186587. [Google Scholar] [CrossRef] [Green Version]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone Inhibits TH17 Cell Differentiation by Activating the Amino Acid Starvation Response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.D.; Rice, D.P.; Ribacke, U.; Silterra, J.; Deik, A.A.; Moss, E.L.; Broadbent, K.M.; Neafsey, D.E.; Desai, M.M.; Clish, C.B.; et al. A Genomic and Evolutionary Approach Reveals Non-Genetic Drug Resistance in Malaria. Genome Biol. 2014, 15, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershenobich, D.; Fierro, F.J.; Rojkind, M. The Relationship between the Free Pool of Proline and Collagen Content in Human Liver Cirrhosis. J. Clin. Investig. 1970, 49, 2246–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation Independent Cloning Vectors for Expression of SUMO Fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
| Code | Structure/R | Without L-Pro | With 1 mM L-Pro | Docking 5 | |||||
| Tm 1 (°C) | ∆Tm 2 (°C) | Tm 3 (°C) | ∆ Tm 4 (°C) | EC50 (µM) | S | RMSD (Å) | Pose | ||
| HcProRS | / | 56.15 | / | 60.20 | / | / | / | / | / |
| POA |  | 55.52 | −0.63 | 59.06 | −1.14 | ND | ND | ND | ND |
| PZA | 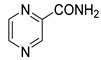 | 56.06 | −0.09 | 59.04 | −1.16 | ND | ND | ND | ND |
| 3-NH2-POA |  | 55.87 | −0.28 | 59.50 | −0.70 | ND | ND | ND | ND |
| 3a | 4-F | 55.25 | −0.90 | 58.21 | −1.99 | ND | −8.672 | 0.540 | 1 |
| 3b | 2-Cl | 56.05 | −0.10 | 64.92 | +4.72 | 3.77 ± 0.85 | −9.240 | 0.425 | 1 |
| 3c | 2-CH3 | 55.25 | −0.90 | 62.72 | +2.52 | 7.34 ± 1.66 | −9.283 | 0.443 | 1 |
| 4a | H | 55.80 | −0.35 | 59.36 | −0.84 | ND | −6.603 | 0.308 | 1 |
| 4b | 3-Cl | 55.38 | −0.77 | 60.42 | +0.22 | ND | −6.666 | 0.964 | 2 |
| 4c | 3,4-diCl | 54.57 | −1.58 | 57.47 | −2.73 | ND | −6.959 | 2.765 | no |
| 4d | 2-CH3 | 55.91 | −0.24 | 63.33 | +3.13 | 91.11 ± 18.39 | −6.949 | 0.247 | 2 |
| 4e | 4-Cl | 55.06 | −1.09 | 58.70 | −1.5 | ND | −6.824 | 2.507 | no |
| 4f | 4-OCH3 | 56.34 | +0.19 | 58.33 | −1.87 | ND | −6.990 | 2.615 | no |
| 4g | 4-CH3 | 55.76 | −0.39 | 59.09 | −1.11 | ND | −7.052 | 2.477 | no |
| 4h | 2-Cl | 57.04 | +0.89 | 62.47 | +2.27 | 13.63 ± 3.27 | −6.651 | 0.926 | 2 |
| 4i | 2-F | 55.46 | −0.69 | 60.30 | +0.10 | ND | −6.777 | 0.299 | 1 |
| 4j | 2-CF3 | 58.13 | +1.98 | 67.12 | +6.92 | 15.28 ± 1.04 | −7.133 | 0.706 | 1 |
| 4k | 2,4-(OCH3)2 | 56.08 | −0.07 | 58.48 | −1.72 | ND | −7.160 | 0.438 | 2 |
| 2a6 | 63.15 | +7.00 | 73.35 | +13.15 | 3.74 ± 0.67 | −9.767 | 0.190 | 1 | |
| PDB Code | HcProRS:Pro | HcProRS:Pro:4d | HcProRS:Pro:4h | HcProRS:Pro:4j | HcProRS:Pro:3b | HcProRS:Pro:3c |
|---|---|---|---|---|---|---|
| 7OSY | 7OSZ | 7OT0 | 7OT2 | 7OT3 | 7OT1 | |
| Data collection | ||||||
| Resolution range (Å) | 39.3–2.23 (2.31–2.23) | 65.41–2.46 (2.548–2.46) | 63.09–2.32 (2.403–2.32) | 54.91–2.48 (2.569–2.48) | 52.36–2.53 (2.62–2.53) | 62.51–2.711 (2.808–2.711) |
| Space group | P 1 21 1 | P 1 21 1 | P 1 21 1 | P 1 21 1 | P 1 21 1 | P 1 21 1 |
| Unit cell | 69.9 88.4 83.8 90 110.2 90 | 69.7 86.7 83.1 90 110.1 90 | 71.2 93.5 87.5 90 108.6 90 | 72.0 92.8 87.9 90 109.0 90 | 71.9 92.4 87.3 90 108.9 90 | 70.3 89.0 84.8 90 110.3 90 |
| Unique reflections | 47,843 (4596) | 33,698 (3333) | 45,788 (4523) | 38,563 (3849) | 35,859 (3530) | 26,094 (2642) |
| Multiplicity | 4.8 (4.9) | 3.7 (3.8) | 3.8 (3.8) | 6.7 (7.2) | 6.9 (7.0) | 3.7 (3.9) |
| Completeness (%) | 98.05 (99.07) | 99.48 (99.52) | 96.97 (96.14) | 99.01 (99.35) | 99.05 (98.85) | 97.69 (99.36) |
| Mean I/σ (I) | 11.67 (1.99) | 7.03 (1.46) | 8.50 (1.52) | 14.44 (2.02) | 18.10 (1.97) | 8.26 (2.24) |
| Wilson B factor (Å2) | 46.34 | 50.69 | 55.22 | 67.28 | 80.11 | 49.02 |
| Rmerge | 0.07098 (0.8346) | 0.1066 (0.9065) | 0.08095 (1.054) | 0.06697 (1.023) | 0.04557 (0.8425) | 0.1284 (0.83) |
| Rmeas | 0.07969 (0.9342) | 0.1253 (1.055) | 0.09489 (1.232) | 0.0727 (1.103) | 0.04929 (0.9104) | 0.1504 (0.9602) |
| Rpim | 0.03575 (0.415) | 0.06494 (0.5341) | 0.04872 (0.6289) | 0.02793 (0.4093) | 0.01858 (0.3414) | 0.07727 (0.4768) |
| CC1/2 | 0.999 (0.713) | 0.996 (0.711) | 0.996 (0.696) | 0.999 (0.84) | 0.999 (0.781) | 0.992 (0.702) |
| Refinement | ||||||
| Reflections used for Rfree | 1940 (209) | 1724 (183) | 2190 (196) | 1865 (186) | 1768 (173) | 1230 (130) |
| Rwork | 0.2090 (0.2637) | 0.2200 (0.3128) | 0.1966 (0.3146) | 0.1936 (0.2928) | 0.2180 (0.3048) | 0.2140 (0.2871) |
| Rfree | 0.2601 (0.3210) | 0.2777 (0.3869) | 0.2503 (0.3632) | 0.2548 (0.3860) | 0.2757 (0.3872) | 0.2833 (0.3630) |
| Number of non-H atoms | 7653 | 7646 | 7847 | 7764 | 7581 | 7631 |
| Macromolecules | 7502 | 7505 | 7691 | 7655 | 7481 | 7486 |
| Ligands | 16(Pro) | 16(Pro)/36(4d) | 16(Pro)/36(4h) | 16(Pro)/42(4j) | 16(Pro)/52(3b) | 16(Pro)/52(3c) |
| Solvent | 127 | 80 | 100 | 47 | 26 | 69 |
| RMS bonds (Å) | 0.003 | 0.003 | 0.005 | 0.006 | 0.003 | 0.003 |
| RMS angles (°) | 0.91 | 0.91 | 0.98 | 1.09 | 0.92 | 0.90 |
| Ramachandran favored (%) | 97.28 | 97.08 | 98.06 | 97.34 | 97.21 | 96.33 |
| Ramachandran allowed (%) | 2.62 | 2.61 | 1.94 | 2.66 | 2.68 | 3.67 |
| Average B-factor (Å2) | 63.57 | 62.84 | 71.61 | 85.06 | 115.2 | 52.18 |
| Protein | 63.8 | 62.82 | 71.84 | 85.24 | 115.32 | 52.26 |
| Ligands | 41.78(Pro) | 46.88(Pro)/86.19(4d) | 45.85(Pro)/66.03(4h) | 57.29(Pro)/68.15(4j) | 78.54(Pro)/99.88(3b) | 36.49(Pro)/52.9(3c) |
| Solvent | 52.88 | 53.62 | 58.58 | 75.61 | 113.26 | 41.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Weeks, S.D.; Juhás, M.; Strelkov, S.V.; Zitko, J.; Van Aerschot, A. Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies. Int. J. Mol. Sci. 2021, 22, 7793. https://doi.org/10.3390/ijms22157793
Pang L, Weeks SD, Juhás M, Strelkov SV, Zitko J, Van Aerschot A. Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies. International Journal of Molecular Sciences. 2021; 22(15):7793. https://doi.org/10.3390/ijms22157793
Chicago/Turabian StylePang, Luping, Stephen D. Weeks, Martin Juhás, Sergei V. Strelkov, Jan Zitko, and Arthur Van Aerschot. 2021. "Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies" International Journal of Molecular Sciences 22, no. 15: 7793. https://doi.org/10.3390/ijms22157793