
Favorable Effects of GLP-1 Receptor Agonist against Pancreatic β-Cell Glucose Toxicity and the Development of Arteriosclerosis: “The Earlier, the Better” in Therapy with Incretin-Based Medicine

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
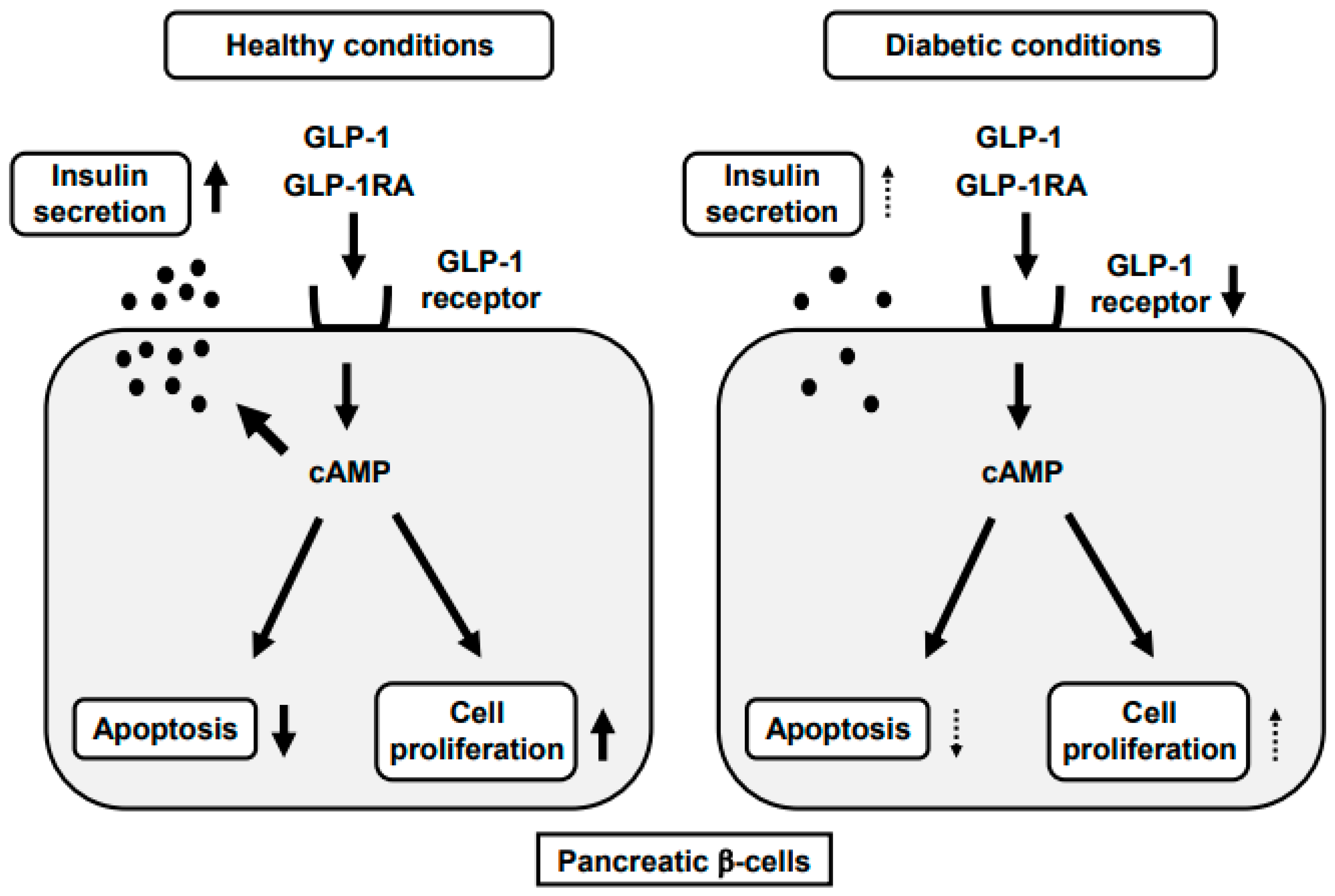
2. Incretin and Pancreatic β-Cells
3. GLP-1-Stimulated Insulin Secretion
4. GLP-1RA and Pancreatic β-Cells
5. GLP-1RA and Arteriosclerosis
5.1. Incretin Signaling and Arterial Cells
5.2. Downregulation of GLP-1R Expression in Arterial Cells under Diabetic Conditions
5.3. Favorable Antiarteriosclerotic Effects of GLP-1RA
5.4. Protective Role of GLP-1RA against Cardiovascular Events in Subjects with T2DM
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Campos, C. Chronic hyperglycemia and glucose toxicity: Pathology and clinical sequelae. Postgrad. Med. 2012, 124, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. beta-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon, C.; Boland, B.B.; Uchizono, Y.; Moore, P.C.; Peterson, B.; Rajan, S.; Rhodes, O.S.; Noske, A.B.; Haataja, L.; Arvan, P.; et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes 2016, 65, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.B.; Brown, C., Jr.; Boland, M.L.; Cann, J.; Sulikowski, M.; Hansen, G.; Grønlund, R.V.; King, W.; Rondinone, C.; Trevaskis, J.; et al. Pancreatic beta-cell rest replenishes insulin secretory capacity and attenuates diabetes in an extreme model of obese type 2 diabetes. Diabetes 2019, 68, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of beta-cell lipo- and glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Hall, E.; Jonsson, J.; Ofori, J.K.; Volkov, P.; Perfilyev, A.; Dekker Nitert, M.; Eliasson, L.; Ling, C.; Bacos, K. Glucolipotoxicity alters insulin secretion via epigenetic changes in human islets. Diabetes 2019, 68, 1965–1974. [Google Scholar] [CrossRef]
- Roma, L.P.; Jonas, J.C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and beta-Cells. J. Mol. Biol. 2020, 432, 1461–1493. [Google Scholar] [CrossRef]
- Benito-Vicente, A.; Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Uribe, K.B.; Martin, S. Molecular mechanisms of lipotoxicity-induced pancreatic beta-cell dysfunction. Int. Rev. Cell Mol. Biol. 2021, 359, 357–402. [Google Scholar] [PubMed]
- Hong, J.H.; Kim, D.H.; Lee, M.K. Glucolipotoxicity and GLP-1 secretion. BMJ Open Diabetes Res. Care 2021, 9, e001905. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, T.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic β-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, Y.; Yamamoto, T.; Benniger, R.K.; Brissova, M.; Guo, M.; Bush, W.; Piston, D.W.; Powers, A.C.; Magnuson, M.; Thurmond, D.C.; et al. The MafA transcription factor becomes essential to islet β-cells soon after birth. Diabetes 2014, 63, 1994–2005. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving MafA expression in diabetic islet β-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Matsuoka, T. Role of pancreatic transcription factors in maintenance of mature β-cell function. Int. J. Mol. Sci. 2015, 16, 6281–6297. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Miyatsuka, T.; Sasaki, S.; Miyashita, K.; Kubo, N.; Shimo, N.; Takebe, S.; Watada, H.; Kaneto, H.; Matsuoka, T.; et al. Recovered expression of Pdx1 improves β-cell failure in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 483, 418–424. [Google Scholar] [CrossRef]
- Zhu, X.; Oguh, A.; Gingerich, M.A.; Soleimanpour, S.A.; Stoffers, D.A.; Gannon, M. Cell cycle regulation of the Pdx1 transcription factor in developing pancreas and insulin-producing beta cells. Diabetes 2021, 70, 903–916. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, C.; Zhu, M.; Qian, L.; Luo, Y.; Cheng, H.; Geng, R.; Xu, X.; Qian, C.; Liu, Y. Saturated fatty acids entrap PDX1 in stress granules and impede islet beta cell function. Diabetologia 2021. [Google Scholar] [CrossRef]
- van Genugten, R.E.; van Raalte, D.H.; Diamant, M. Dipeptidyl peptidase-4 inhibitors and preservation of pancreatic islet-cell function: A critical appraisal of the evidence. Diabetes Obes. Metab. 2012, 14, 101–111. [Google Scholar] [CrossRef]
- Hamamoto, S.; Kanda, Y.; Shimoda, M.; Tatsumi, F.; Kohara, K.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Vildagliptin preserves the mass and function of pancreatic beta cells via the developmental regulation and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetes Obes. Metab. 2013, 15, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Hirukawa, H.; Kaneto, H.; Shimoda, M.; Kimura, T.; Okauchi, S.; Obata, A.; Kohara, K.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; et al. Combination of DPP-4 inhibitor and PPARγ agonist exerts protective effects on pancreatic β-cells in diabetic db/db mice through the augmentation of IRS-2 expression. Mol. Cell. Endocrinol. 2015, 413, 49–60. [Google Scholar] [CrossRef]
- Wei, R.; Cui, X.; Feng, J.; Gu, L.; Lang, S.; Wei, T.; Yang, J.; Liu, J.; Le, Y.; Wang, H.; et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism 2020, 111, 154324. [Google Scholar] [CrossRef]
- Kusakabe, T.; Yokota, S.; Shimizu, M.; Inoue, T.; Tanaka, M.; Ohue-Kitano, R.; Muranaka, K.; Yamakage, H.; Wada, H.; Hasegawa, K.; et al. Differential effects of sodium-glucose cotransporter 2 inhibitor and low-carbohydrate diet on body composition and metabolic profile in obese diabetic db/db mice. BMJ Open Diabetes Res. Care 2020, 8, e001303. [Google Scholar] [CrossRef]
- Hogan, M.F.; Hackney, D.J.; Aplin, A.C.; Mundinger, T.O.; Larmore, M.J.; Castillo, J.J.; Esser, N.; Zraika, S.; Hull, R.L. SGLT2-i improves markers of islet endothelial cell function in db/db diabetic mice. J. Endocrinol. 2021, 248, 95–106. [Google Scholar] [CrossRef]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef]
- Xu, G.; Kaneto, H.; Laybutt, D.R.; Duvivier-Kali, V.; Trivedi, N.; Suzuma, K.; King, G.L.; Weir, G.C.; Bonner-Weir, S. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to the impaired incretin effects in diabetes. Diabetes 2007, 56, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Matsuoka, T.; Kaneto, H.; Tochino, Y.; Kato, K.; Yamamoto, K.; Yamamoto, T.; Matsuhisa, M.; Shimomura, I. Effect of alogliptin, pioglitazone and glargine on pancreatic β-cells in diabetic db/db mice. Biochem. Biophys. Res. Commun. 2011, 404, 534–540. [Google Scholar] [CrossRef]
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735. [Google Scholar] [CrossRef] [Green Version]
- Takamoto, I.; Kubota, N.; Nakaya, K.; Kumagai, K.; Hashimoto, S.; Kubota, T.; Inoue, M.; Kajiwara, E.; Katsuyama, H.; Obata, A.; et al. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia 2014, 57, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, R.K.; Mondragon, A.; Chen, L.; McGinty, J.A.; French, P.M.; Ferrer, J.; Thorens, B.; Hodson, D.J.; Rutter, G.A.; Xavier, G.D. Selective disruption of Tcf7l2 in the pancreatic β cell impairs secretory function and lowers β cell mass. Hum. Mol. Genet. 2015, 24, 1390–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jainandunsing, S.; Koole, H.R.; van Miert, J.N.I.; Rietveld, T.; Wattimena, J.L.D.; Sijbrands, E.J.G.; de Rooij, F.W.M. Transcription factor 7-like 2 gene links increased in vivo insulin synthesis to type 2 diabetes. EBioMedicine 2018, 30, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Tu, M.S.; da Silva Xavier, G.; Leclerc, I.; Rutter, G.A. Transcription factor-7-like 2 (TCF7L2) gene acts downstream of the Lkb1/Stk11 kinase to control mTOR signaling, beta cell growth, and insulin secretion. J. Biol. Chem. 2018, 293, 14178–14189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.H.; Li, Y.L.; Liu, N.J.; Yang, Z.; Tao, X.M.; Du, Y.P.; Wang, X.C.; Lu, B.; Zhang, Z.Y.; Hu, R.M.; et al. TCF7L2 regulates pancreatic beta-cell function through PI3K/AKT signal pathway. Diabetol. Metab. Syndr. 2019, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, L.; Xu, X. The role of transcription factor 7-like 2 in metabolic disorders. Obes. Rev. 2021, 22, e13166. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C.; Jablonski, K.A.; Bayley, N.; Pollin, T.I.; de Bakker, P.I.; Shuldiner, A.R.; Knowler, W.C.; Nathan, D.M.; Altshuler, D. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 2006, 355, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Horikoshi, M.; Hara, K.; Ito, C.; Nagai, R.; Froguel, P.; Kadowaki, T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia 2007, 50, 747–751. [Google Scholar] [CrossRef]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Lyssenko, V.; Lupi, R.; Marchetti, P.; del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjögren, M.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; Jose, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villareal, D.T.; Robertson, H.; Bell, G.I.; Patterson, B.W.; Tran, H.; Wice, B.; Polonsky, K.S. TCF7L2 variant rs7903146 affects the risk of type 2 diabetes by modulating incretin action. Diabetes 2010, 59, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seino, S.; Takahashi, H.; Fujimoto, W.; Shibasaki, T. Roles of cAMP signalling in insulin granule exocytosis. Diabetes Obes. Metab. 2009, 11, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jin, T. New insights into the role of cAMP in the production and function of the incretin hormone glucagon-like peptide-1 (GLP-1). Cell. Signal. 2010, 22, 1–8. [Google Scholar] [CrossRef]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhosaini, K.; Azhar, A.; Alonazi, A.; Al-Zoghaibi, F. GPCRs: The most promiscuous druggable receptor of the mankind. Saudi Pharm. J. 2021, 29, 539–551. [Google Scholar] [CrossRef]
- Shigeto, M.; Ramracheya, R.; Tarasov, A.I.; Cha, C.Y.; Chibalina, M.V.; Hastoy, B.; Philippaert, K.; Reinbothe, T.; Rorsman, N.; Salehi, A.; et al. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J. Clin. Investig. 2015, 125, 4714–4728. [Google Scholar] [CrossRef] [Green Version]
- Kaku, K. New concept of the glucagon-like peptide-1 signaling pathway on pancreatic insulin secretion. J. Diabetes Investig. 2020, 11, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Kanda, Y.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Matsuki, M.; Kaku, K. The human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells via regulation of cell kinetics and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetologia 2011, 54, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Cernea, S.; Raz, I. Therapy in the early stage: Incretins. Diabetes Care 2011, 34, S264–S271. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kaneto, H.; Shimoda, M.; Hirukawa, H.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Protective effects of pioglitazone and/or liraglutide on pancreatic β-cells: Comparison of their effects between in an early and advanced stage of diabetes. Mol. Cell. Endocrinol. 2015, 400, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Minami, K.; Kudo, M.; Iemoto, K.; Takahashi, H.; Seino, S. Liraglutide improves pancreatic beta cell mass and function in Alloxan-induced diabetic mice. PLoS ONE 2015. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Chen, T.; Zhu, Y.; Li, H.-Q.; Deng, X.-L.; Wang, Q.-H.; Zhang, J.-Y.; Chen, L.-L. Liraglutide prevents fast weight gain and β-cell dysfunction in male catch-up growth rats. Exp. Biol. Med. 2015, 240, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapodistria, K.; Tsilibary, E.-P.; Kotsopoulou, E.; Moustardas, P.; Kitsiou, P. Liraglutide, a human glucagon-like peptide-1 analogue, stimulates AKT-dependent survival signalling and inhibits pancreatic β-cell apoptosis. J. Cell. Mol. Med. 2018, 22, 2970–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Deng, X.-L.; Liu, Z.-H.; Song, H.-J.; Zheng, J.; Cui, Z.-H.; Xiao, K.-L.; Chen, L.-L.; Li, H.-Q. Liraglutide protects β-cell function by reversing histone modification of Pdx-1 proximal promoter in catch-up growth male rats. J. Diabetes Complicat. 2018, 32, 985–994. [Google Scholar] [CrossRef]
- Fushimi, Y.; Obata, A.; Sanada, J.; Nogami, Y.; Ikeda, T.; Yamasaki, Y.; Obata, Y.; Shimoda, M.; Nakanishi, S.; Mune, T.; et al. Combination of dipeptidyl peptidase 4 (DPP-4) inhibitor and sodium glucose cotransporter 2 (SGLT2) inhibitor substantially protects pancreatic β-cells especially in early phase of diabetes rather than advanced phase. Sci. Rep. 2021, in press. [Google Scholar]
- Kimura, T.; Obata, A.; Shimoda, M.; Hirukawa, H.; Kanda-Kimura, Y.; Nogami, Y.; Kohara, K.; Nakanishi, S.; Mune, T.; Kaku, K.; et al. Durability of protective effect of dulaglutide on pancreatic β-cells in diabetic mice: GLP-1 receptor expression is not reduced at all even after long-term exposure to dulaglutide. Diabetes Metab. 2018, 44, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Heber, D.; Dodson, R.; Stoskopf, C.; Peterson, M.; Swerdloff, R.S. Pituitary desensitization and the regulation of pituitary gonadotropin-releasing hormone (GnRH) receptors following chronic administration of a superactive GnRH analog and testosterone. Life Sci. 1982, 30, 2301–2308. [Google Scholar] [CrossRef]
- He, L.; Fong, J.; von Zastrow, M.; Whistler, J.L. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell 2002, 108, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Conn, P.M.; Crowley, W.F., Jr. Gonadotropin-releasing hormone and its analogues. N. Engl. J. Med. 1991, 324, 93–103. [Google Scholar]
- Abu Hashim, H. Gonadotrophin-releasing hormone analogues and endometriosis: Current strategies and new insights. Gynecol. Endocrinol. 2012, 28, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Goto, H.; Nomiyama, T.; Mita, T.; Yasunari, E.; Azuma, K.; Komiya, K.; Arakawa, M.; Jin, W.L.; Kanazawa, A.; Kawamori, R.; et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces intimal thickening after vascular injury. Biochem. Biophys. Res. Commun. 2011, 405, 79–84. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, L.; Feng, B.; He, N.; Zhang, Y.; Ye, H. Protective effects of glucagon-likepeptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6kinase pathway in diabetes mellitus. J. Diabetes Investig. 2020, 11, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstadter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schon, S.; Oelze, M.; et al. Endothelial GLP-1 (glucagon-like peptide-1) receptor mediates cardiovascular protection by liraglutide in mice with experimental arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Obata, A.; Shimoda, M.; Okauchi, S.; Hirukawa, H.; Kohara, K.; Kinoshita, T.; Nogami, Y.; Nakanishi, S.; Mune, T.; et al. Decreased GLP-1 receptor expression in endothelial and smooth muscle cells in diabetic db/db mice: TCF7L2 is a possible regulator of vascular GLP-1 receptor. Diabetes Vasc. Dis. Res. 2017, 14, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Obata, A.; Shimoda, M.; Shimizu, I.; da Silva Xavier, G.; Okauchi, S.; Hirukawa, H.; Kohara, K.; Mune, T.; Moriuchi, S.; et al. Down-regulation of vascular GLP-1 receptor expression in human subjects with obesity. Sci. Rep. 2018, 8, 10644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, J.; Obata, A.; Obata, Y.; Fushimi, Y.; Shimoda, M.; Kohara, K.; Nakanishi, S.; Mune, T.; Kaku, K.; Kaneto, H. Dulaglutide exerts more beneficial anti-atherosclerotic effects in an early phase of diabetes rather than in a late phase in ApoE knockout mice with diabetes. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Poulter, N.R.; Bhatt, D.L.; Bain, S.C.; Buse, J.B.; Leiter, L.A.; Nauck, M.A.; Pratley, R.E.; Zinman, B.; Ørsted, D.D.; et al. Effects of liraglutide on cardiovascular outcomes in patients with type 2 diabetes mellitus with or without history of myocardial infarction or stroke. Circulation 2018, 138, 2884–2894. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2017, 394, 121–130. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Kaul, S. Mitigating cardiovascular risk in type 2 diabetes with antidiabetes drugs: A review of principal cardiovascular outcome results of EMPA-REG OUTCOME, LEADER, and SUSTAIN-6 trials. Diabetes Care 2017, 40, 821–831. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneto, H.; Kimura, T.; Shimoda, M.; Obata, A.; Sanada, J.; Fushimi, Y.; Nakanishi, S.; Mune, T.; Kaku, K. Favorable Effects of GLP-1 Receptor Agonist against Pancreatic β-Cell Glucose Toxicity and the Development of Arteriosclerosis: “The Earlier, the Better” in Therapy with Incretin-Based Medicine. Int. J. Mol. Sci. 2021, 22, 7917. https://doi.org/10.3390/ijms22157917
Kaneto H, Kimura T, Shimoda M, Obata A, Sanada J, Fushimi Y, Nakanishi S, Mune T, Kaku K. Favorable Effects of GLP-1 Receptor Agonist against Pancreatic β-Cell Glucose Toxicity and the Development of Arteriosclerosis: “The Earlier, the Better” in Therapy with Incretin-Based Medicine. International Journal of Molecular Sciences. 2021; 22(15):7917. https://doi.org/10.3390/ijms22157917
Chicago/Turabian StyleKaneto, Hideaki, Tomohiko Kimura, Masashi Shimoda, Atsushi Obata, Junpei Sanada, Yoshiro Fushimi, Shuhei Nakanishi, Tomoatsu Mune, and Kohei Kaku. 2021. "Favorable Effects of GLP-1 Receptor Agonist against Pancreatic β-Cell Glucose Toxicity and the Development of Arteriosclerosis: “The Earlier, the Better” in Therapy with Incretin-Based Medicine" International Journal of Molecular Sciences 22, no. 15: 7917. https://doi.org/10.3390/ijms22157917