
Impact of Hypoxia over Human Viral Infections and Key Cellular Processes

 , , and
, , and 
Abstract
:1. Introduction
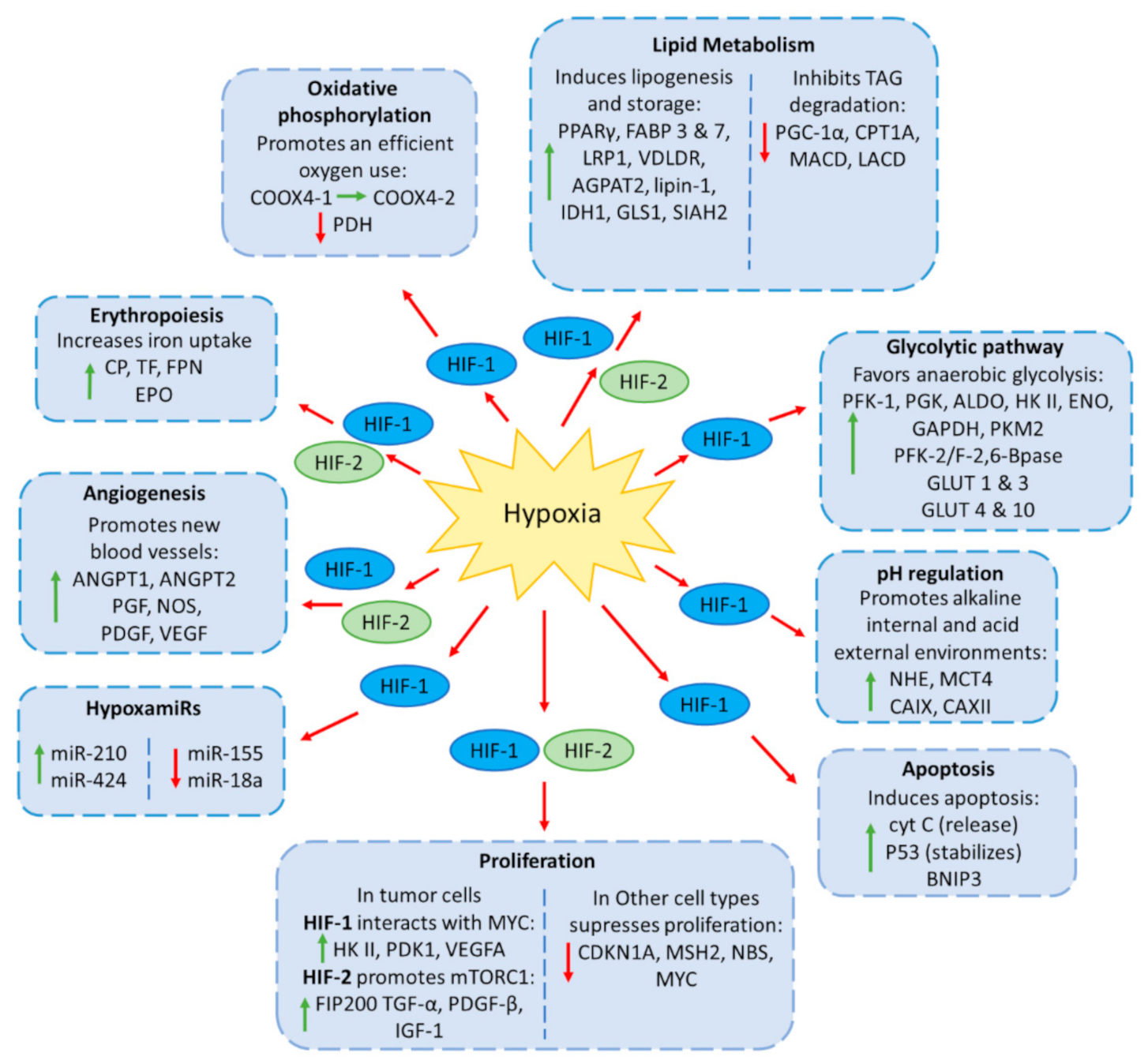
2. Cellular Responses to Hypoxia
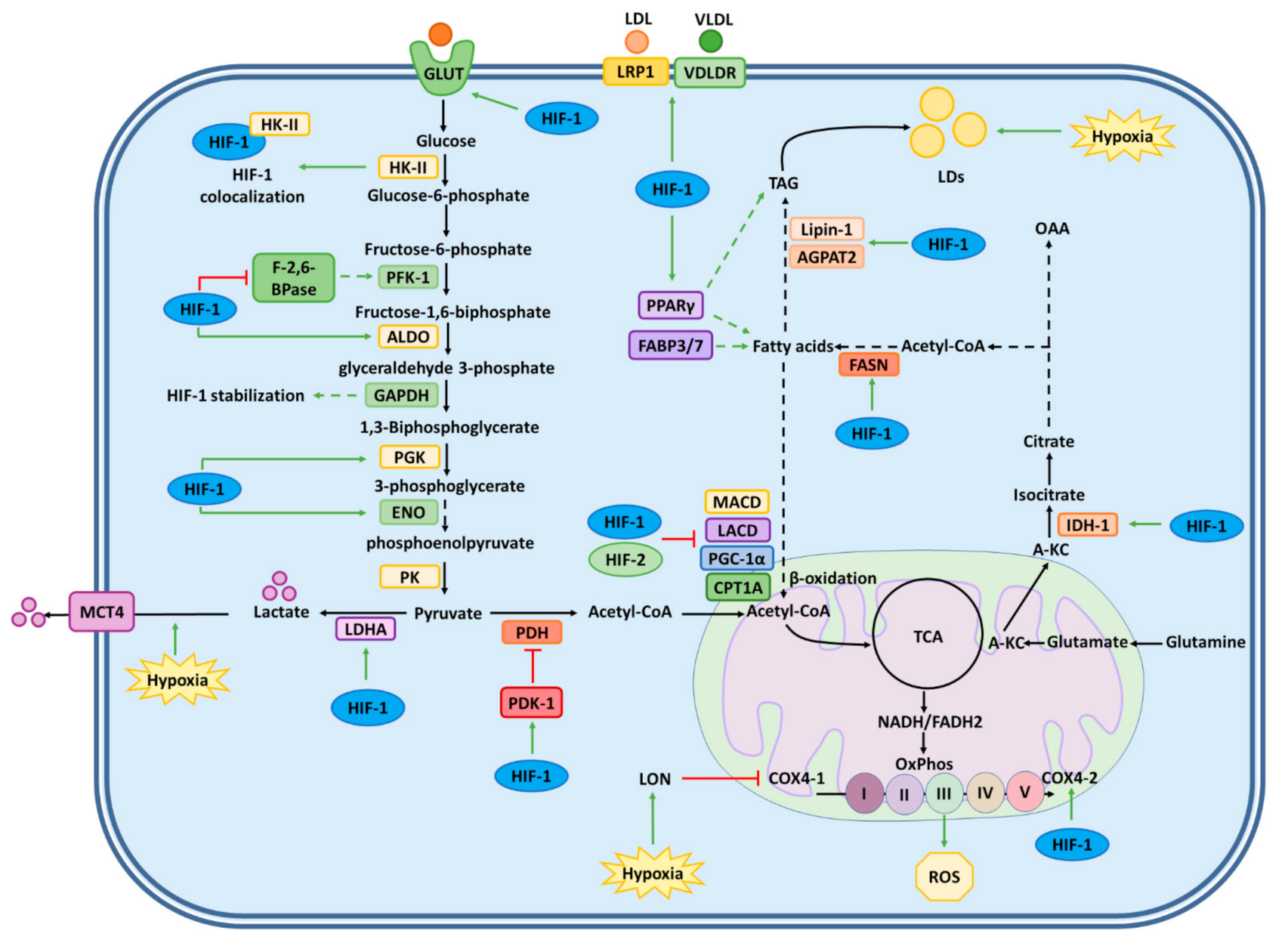
2.1. Regulation of Glucose Metabolism
2.2. Regulation of Lipid Metabolism
2.3. Regulation of Erythropoiesis and Angiogenesis
2.4. Regulation of Apoptosis and Cell Proliferation
2.5. miRNAs Regulated by Hypoxia
3. Effect of Hypoxia over Viral Infections
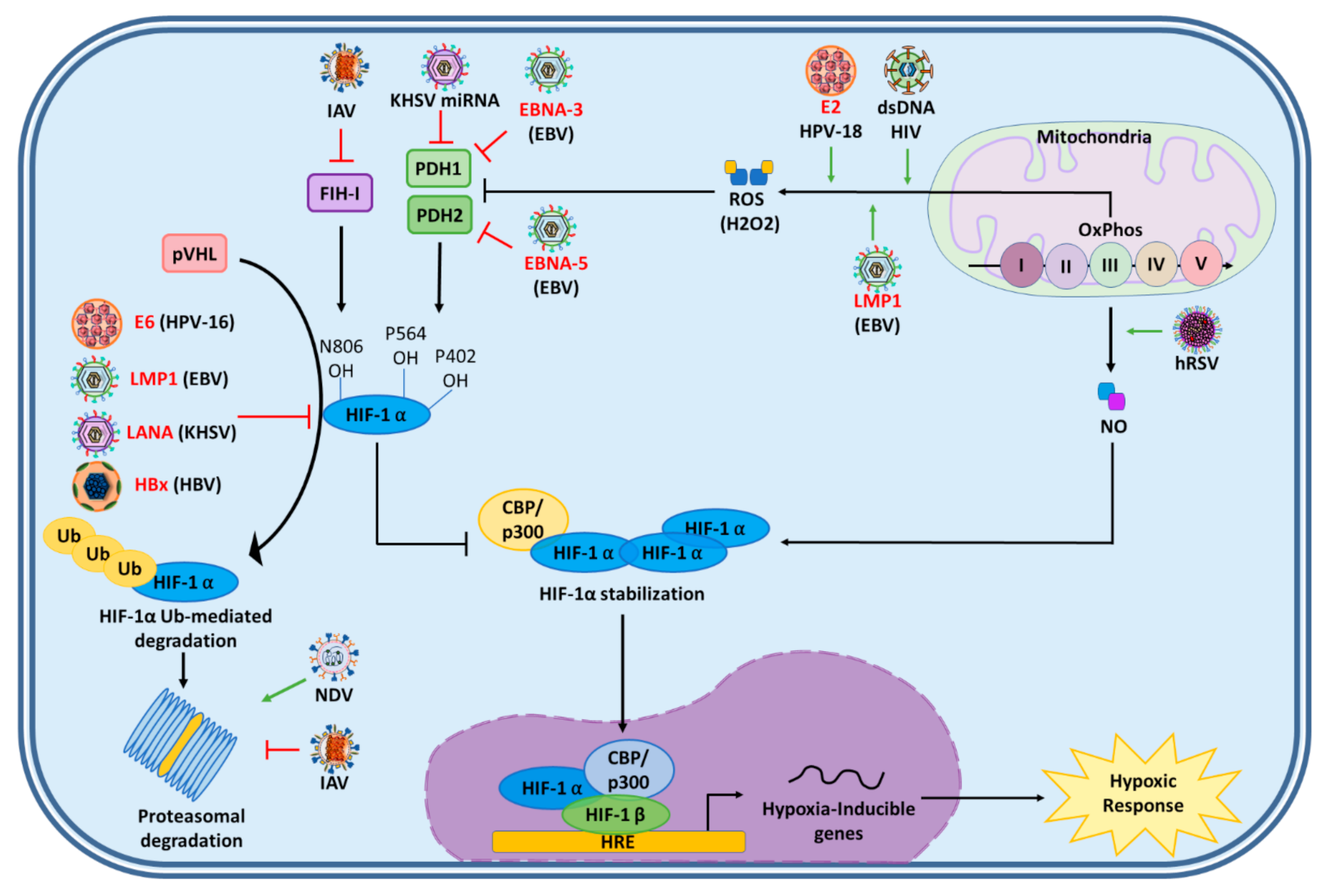
4. Virus Induction of Hypoxia Responses
4.1. Activation of HIF-1α Mediated by Viral Kinases
4.2. Activation of HIF-1α Mediated by Viral Impairment of HIF-1α Inhibitors
4.3. Activation of HIF-1α by Reactive Oxygen Species
5. Virus Inhibition of Hypoxic Responses
Viruses Downregulating HIF-1α
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-mediated biological control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Rhim, T.; Lee, D.Y.; Lee, M. Hypoxia as a target for tissue specific gene therapy. J. Control. Release 2013, 172, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Mahato, R.I.; Lee, M. Hypoxia-specific gene expression for ischemic disease gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Ratcliffe, P. Cellular oxygen sensing in health and disease. Pediatr. Nephrol. 2008, 23, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2007, 409, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer cell metabolism in hypoxia: Role of hif-1 as key regulator and therapeutic target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Yang, S.-L.; Wu, C.; Xiong, Z.-F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [Green Version]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate hif by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Ekietzmann, T.; Emennerich, D.; Dimova, E. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kim, M.S.; Park, J.W. Oxygen-Dependent and -Independent Regulation of HIF-1alpha. J. Korean Med. Sci. 2002, 17, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1α. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [Green Version]
- Casillas, A.L.; Chauhan, S.S.; Toth, R.K.; Sainz, A.G.; Clements, A.N.; Jensen, C.C.; Langlais, P.R.; Miranti, C.K.; Cress, A.E.; Warfel, N.A. Direct phosphorylation and stabilization of HIF-1α by PIM1 kinase drives angiogenesis in solid tumors. Oncogene 2021, 1–11. [Google Scholar] [CrossRef]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [Green Version]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-inducible factor-1a by reactive oxygen species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Sandau, K.B.; Zhou, J.; Kietzmann, T.; Brüne, B. Regulation of the Hypoxia-inducible Factor 1α by the inflammatory mediators nitric oxide and tumor necrosis factor-α in contrast to desferroxamine and phenylarsine oxide. J. Biol. Chem. 2001, 276, 39805–39811. [Google Scholar] [CrossRef] [Green Version]
- Hellwig-Bürgel, T.; Stiehl, D.; Wagner, A.E.; Metzen, E.; Jelkmann, W. Review:Hypoxia-Inducible Factor-1 (HIF-1): A Novel Transcription Factor in Immune Reactions. J. Interf. Cytokine Res. 2005, 25, 297–310. [Google Scholar] [CrossRef]
- Morinet, F.; Parent, M.; Bergeron, C.; Pillet, S.; Capron, C. Oxygen and viruses: A breathing story. J. Gen. Virol. 2015, 96, 1979–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinet, F.; Casetti, L.; François, J.-H.; Capron, C.; Pillet, S. Oxygen tension level and human viral infections. Virology 2013, 444, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werth, N.; Beerlage, C.; Rosenberger, C.; Yazdi, A.S.; Edelmann, M.; Amr, A.; Bernhardt, W.; Von Eiff, C.; Becker, K.; Schäfer, A.; et al. Activation of Hypoxia Inducible Factor 1 Is a General Phenomenon in Infections with Human Pathogens. PLoS ONE 2010, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Frakolaki, E. Virus–host interactions under hypoxia. Microbes Infect. 2017, 19, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Corrales, N.; Gálvez, N.M.S.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Contribution of hypoxia inducible factor-1 during viral infections. Virulence 2020, 11, 1482–1500. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, A.; Joost, H.-G.; Schurmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. J. Parenter. Enter. Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Ancey, P.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Hao, L.; Liu, Q.; Tian, C.; Zhang, D.; Wang, B.; Zhou, D.; Li, Z.; Yuan, Z. Correlation and expression analysis of hypoxia-inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in human gastric cancer. Oncol. Lett. 2019, 18, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Bobarykina, A.Y.; Minchenko, D.; Opentanova, I.L.; Moenner, M.; Caro, J.; Esumi, H.; Minchenko, O. Hypoxic regulation of PFKFB-3 and PFKFB-4 gene expression in gastric and pancreatic cancer cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim. Pol. 2006, 53, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Hu, C.; Wang, L.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1 Alpha (HIF-1 Alpha) and HIF-2 Alpha in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [Green Version]
- Bartrons, R.; Caro, J. Hypoxia, Glucose Metabolism and the Warburg’s Effect. J. Bioenerg. Biomembr. 2007, 39, 223–229. [Google Scholar] [CrossRef]
- Minchenko, O.; Opentanova, I.; Minchenko, D.; Ogura, T.; Esumi, H. Hypoxia induces transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1α activation. FEBS Lett. 2004, 576, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.; Leshchinsky, I.; Opentanova, I.; Sang, N.; Srinivas, V.; Armstead, V.; Caro, J. Hypoxia-inducible Factor-1-mediated Expression of the 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) Gene: Its Possible Role in the Warburg Effect. J. Biol. Chem. 2002, 277, 6183–6187. [Google Scholar] [CrossRef] [Green Version]
- Guzman, G.; Chennuri, R.; Chan, A.; Rea, B.; Quintana, A.; Patel, R.; Xu, P.-Z.; Xie, H.; Hay, N. Evidence for heightened hexokinase ii immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, S.; Arii, S.; Mori, A.; Isobe, N.; Yang, W.; Oe, H.; Fujimoto, A.; Yonenaga, Y.; Sakashita, H.; Imamura, M. Hexokinase II and VEGF expression in liver tumors: Correlation with hypoxia-inducible factor-1α and its significance. J. Hepatol. 2004, 40, 117–123. [Google Scholar] [CrossRef]
- Ramière, C.; Rodriguez, J.; Enache, L.S.; Lotteau, V.; André, P.; Diaz, O. Activity of hexokinase is increased by its interaction with hepatitis c virus protein NS5A. J. Virol. 2014, 88, 3246–3254. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.R.; Qu, Y.; Agrawal, A.; Garofalo, R.P.; Casola, A. HIF-1α Modulates Core Metabolism and Virus Replication in Primary Airway Epithelial Cells Infected with Respiratory Syncytial Virus. Viruses 2020, 12, 1088. [Google Scholar] [CrossRef]
- Bartrons, R.; Simon-Molas, H.; Rodríguez-García, A.; Castaño, E.; Navarro-Sabate, A.; Manzano, A.; Martinez-Outschoorn, U. Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef]
- Yang, H.; Shu, Z.; Jiang, Y.; Mao, W.; Pang, L.; Redwood, A.; Jeter-Jones, S.L.; Jennings, N.B.; Ornelas, A.; Zhou, J.; et al. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase-2 Regulates TP53-Dependent Paclitaxel Sensitivity in Ovarian and Breast Cancers. Clin. Cancer Res. 2019, 25, 5702–5716. [Google Scholar] [CrossRef] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Pommier, S.; Bénéteau, M.; Mondragón, L.; Meynet, O.; Zunino, B.; Mouchotte, A.; Verhoeyen, E.; Guyot, M.; Pagès, G.; et al. GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-κB-dependent induction of HIF-1α. Leukemia 2014, 29, 1163–1176. [Google Scholar] [CrossRef]
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch. Biochem. Biophys. 2011, 509, 1–8. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.-W.; Shimoda, L.; Dang, C.; Semenza, G.L. HIF-1 Regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, P.A.; Yang, J.; Metelo, A.; Pérez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; López-Larrubia, P.; et al. In Vivo HIF-Mediated Reductive Carboxylation Is Regulated by Citrate Levels and Sensitizes VHL-Deficient Cells to Glutamine Deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A. The Plasma Membrane Lactate Transporter MCT4, but Not MCT1, Is Up-regulated by Hypoxia through a HIF-1α-dependent Mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, L.A.; Fallon, M.; Pisarcik, S.; Wang, J.; Semenza, G.L. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, L941–L949. [Google Scholar] [CrossRef] [Green Version]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.; Turley, H.; Talks, K.L.; Maxwell, P.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible factors and the regulation of lipid metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.; Aledo, R.; Sendra, J.; Costales, P.; Juan-Babot, O.; Badimon, L.; Llorente-Cortés, V. Hypoxia stimulates low-density lipoprotein receptor–related protein-1 expression through hypoxia-inducible factor-1α in human vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2011, 31, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Perman, J.C.; Boström, P.; Lindbom, M.; Lidberg, U.; Ståhlman, M.; Hägg, D.; Lindskog, H.; Täng, M.S.; Omerovic, E.; Hultén, L.M.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Investig. 2011, 121, 2625–2640. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis that Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.-Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty Acid Synthase Gene Is Up-regulated by Hypoxia via Activation of Akt and Sterol Regulatory Element Binding Protein. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Triantafyllou, E.-A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1142–1152. [Google Scholar] [CrossRef]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation via HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-Mediated Suppression of Acyl-CoA Dehydrogenases and Fatty Acid Oxidation Is Critical for Cancer Progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Haase, V.H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J. Physiol. Ren. Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef] [Green Version]
- Robach, P.; Fulla, Y.; Westerterp, K.R.; Richalet, J.-P. Comparative Response of EPO and Soluble Transferrin Receptor at High Altitude. Med. Sci. Sports Exerc. 2004, 36, 1493–1498. [Google Scholar] [CrossRef]
- Rolfs, A.; Kvietikova, I.; Gassmann, M.; Wenger, R.H. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J. Biol. Chem. 1997, 272, 20055–20062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, C.K.; Mazumder, B.; Fox, P.L. Role of Hypoxia-inducible Factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000, 275, 21048–21054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G.; Lombardi, C.; Bilo, G.; Revera, M.; Giuliano, A.; Faini, A.; et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [Green Version]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in Wound Healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.A.; Pearson, C.I. Angiogenesis in the pathogenesis of inflammatory joint and lung diseases. Arthritis Res. 2001, 3, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Melillo, G.; Musso, T.; Sica, A.; Taylor, L.S.; Cox, G.W.; Varesio, L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995, 182, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell Type–Specific Regulation of Angiogenic Growth Factor Gene Expression and Induction of Angiogenesis in Nonischemic Tissue by a Constitutively Active Form of Hypoxia-Inducible Factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Gerber, H.-P.; Condorelli, F.; Park, J.; Ferrara, N. Differential Transcriptional Regulation of the Two Vascular Endothelial Growth Factor Receptor Genes. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef] [Green Version]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P.; de Leon, M.; Talasazan, F.; Schoenfeld, A.R.; Davidowitz, E.J.; Burk, R.D. The von Hippel-Lindau Gene Product Inhibits Renal Cell Apoptosis via Bcl-2-dependent Pathways. J. Biol. Chem. 2001, 276, 40599–40605. [Google Scholar] [CrossRef] [Green Version]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct Interactions between HIF-1α and Mdm2 Modulate p53 Function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, J. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [Green Version]
- Kothari, S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.; Bailes, M.; Ens, K.; Kirshenbaum, L.A.; Gibson, S. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene 2003, 22, 4734–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- To, K.; Sedelnikova, O.A.; Samons, M.; Bonner, W.M.; Huang, L.E. The phosphorylation status of PAS-B distinguishes HIF-1α from HIF-2α in NBS1 repression. EMBO J. 2006, 25, 4784–4794. [Google Scholar] [CrossRef] [Green Version]
- Koshiji, M.; To, K.; Hammer, S.; Kumamoto, K.; Harris, A.; Modrich, P.; Huang, L.E. HIF-1α Induces Genetic Instability by Transcriptionally Downregulating MutSα Expression. Mol. Cell 2005, 17, 793–803. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.-J.; Diehl, J.A.; Simon, M.C. HIF-2α Promotes Hypoxic Cell Proliferation by Enhancing c-Myc Transcriptional Activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Corn, P.G.; Ricci, M.S.; Scata, K.A.; Arsham, A.M.; Simon, M.C.; Dicker, D.T.; El-Deiry, W.S. Mxi1 is induced by hypoxia in a HIF-1–dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol. Ther. 2005, 4, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Gan, B.; Melkoumian, Z.K.; Wu, X.; Guan, K.-L.; Guan, J.-L. Identification of FIP200 interaction with the TSC1–TSC2 complex and its role in regulation of cell size control. J. Cell Biol. 2005, 170, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.M.; Watson, I.; Evans, A.J.; Foster, D.A.; Irwin, M.S.; Ohh, M. Suppression of Hypoxia-Inducible Factor 2α Restores p53 Activity via Hdm2 and Reverses Chemoresistance of Renal Carcinoma Cells. Cancer Res. 2009, 69, 9056–9064. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Bertero, T.; Rezzonico, R.; Pottier, N.; Mari, B. Impact of MicroRNAs in the Cellular Response to Hypoxia. Int. Rev. Cell Mol. Biol. 2017, 333, 91–158. [Google Scholar] [CrossRef] [PubMed]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Banerjee, J.; Choi, S.Y.; Sen, C.K. miR-210: The master hypoxamir. Microcirculation 2011, 19, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A Hypoxia-Induced Positive Feedback Loop Promotes Hypoxia-Inducible Factor 1α Stability through miR-210 Suppression of Glycerol-3-Phosphate Dehydrogenase 1-Like. Mol. Cell. Biol. 2011, 31, 2696–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puissegur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2010, 18, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, G.; Subramanian, I.V.; Adhikari, N.; Zhang, X.; Joshi, H.P.; Basi, D.; Chandrashekhar, Y.; Hall, J.L.; Roy, S.; Zeng, Y.; et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Investig. 2010, 120, 4141–4154. [Google Scholar] [CrossRef] [Green Version]
- Bruning, U.; Cerone, L.; Neufeld, Z.; Fitzpatrick, S.F.; Cheong, A.; Scholz, C.C.; Simpson, D.; Leonard, M.O.; Tambuwala, M.M.; Cummins, E.; et al. MicroRNA-155 Promotes resolution of hypoxia-inducible factor 1 activity during prolonged hypoxia. Mol. Cell. Biol. 2011, 31, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Han, F.; Wu, Y. MicroRNA-18a Decreases Choroidal Endothelial Cell Proliferation and Migration by Inhibiting HIF1A Expression. Med. Sci. Monit. 2015, 21, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Yanagisawa, K.; Tanaka, M.; Cao, K.; Matsuyama, Y.; Goto, H.; Takahashi, T. Identification of Hypoxia-Inducible Factor-1α as a Novel Target for miR-17-92 MicroRNA Cluster. Cancer Res. 2008, 68, 5540–5545. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.-H.; Wang, N.; Li, A.; Liao, W.-T.; Pan, Z.-G.; Mai, S.-J.; Li, D.-J.; Zeng, M.; Wen, J.-M.; Zeng, Y.-X. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. J. Clin. Virol. 2006, 37, 98–103. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.-L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus Latent Protein LANA Interacts with HIF-1α to Upregulate RTA Expression during Hypoxia: Latency Control under Low Oxygen Conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.; Davis, D.A.; Wang, V.; Widmer, I.; Yarchoan, R. Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus 8) Contains Hypoxia Response Elements: Relevance to Lytic Induction by Hypoxia. J. Virol. 2003, 77, 6761–6768. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of Kaposi sarcoma–associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghi, M.K.; Liu, T.-C.; Rabkin, S.; Martuza, R.L. Hypoxia Enhances the Replication of Oncolytic Herpes Simplex Virus. Mol. Ther. 2009, 17, 51–56. [Google Scholar] [CrossRef]
- Piña-Oviedo, S.; Khalili, K.; Del Valle, L. Hypoxia inducible factor-1 alpha activation of the JCV promoter: Role in the pathogenesis of Progressive Multifocal Leukoencephalopathy. Acta Neuropathol. 2009, 118, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.H.; Hermiston, T.W. Effect of hypoxia on Ad5 infection, transgene expression and replication. Gene Ther. 2005, 12, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Hallez, C.; Li, X.; Suspène, R.; Thiers, V.; Bouzidi, M.S.; Dorobantu, C.M.; Lucansky, V.; Wain-Hobson, S.; Gaudin, R.; Vartanian, J.-P. Hypoxia-induced human deoxyribonuclease I is a cellular restriction factor of hepatitis B virus. Nat. Microbiol. 2019, 4, 1196–1207. [Google Scholar] [CrossRef]
- Frakolaki, E.; Kaimou, P.; Moraiti, M.; Kalliampakou, K.I.; Karampetsou, K.; Dotsika, E.; Liakos, P.; Vassilacopoulou, D.; Mavromara, P.; Bartenschlager, R.; et al. The Role of Tissue Oxygen Tension in Dengue Virus Replication. Cells 2018, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Amini, S.; Sen, S.; Khalili, K.; Sawaya, B.E. Regulation of the HIV-1 promoter by HIF-1α and Vpr proteins. Virol. J. 2011, 8, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A.; et al. Glut1-mediated glucose transport regulates HIV infection. Proc. Natl. Acad. Sci. USA 2012, 109, 2549–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, S.; Ammosova, T.; Cardenas, J.; Foster, A.; Rotimi, J.; Jerebtsova, M.; Ayodeji, A.A.; Niu, X.; Ray, P.E.; Gordeuk, V.R.; et al. Regulation of HIV-1 transcription at 3% versus 21% oxygen concentration. J. Cell. Physiol. 2009, 221, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, J.H.; Naczki, C.; Koumenis, C.; Lyles, D.S. Replication and Cytopathic Effect of Oncolytic Vesicular Stomatitis Virus in Hypoxic Tumor Cells In Vitro and In Vivo. J. Virol. 2004, 78, 8960–8970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, P.A.; Keeley, T.P.; Zhuang, X.; Lee, J.; Prange-Barczynska, M.; Tsukuda, S.; Morgan, S.; Argles, I.L.; Kurlekar, S.; Noerenberg, M.; et al. Hypoxic and Pharmacological Activation of HIF Inhibits SARS-CoV-2 Infection of Lung Epithelial Cells. SSRN Electron. J. 2020, 35, 109020. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Singh, R.; Lang, F.; Pei, Y.; Jha, H.C.; Robertson, E.S. Metabolic reprogramming of Kaposi’s sarcoma associated herpes virus infected B-cells in hypoxia. PLOS Pathog. 2018, 14, e1007062. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, A.; Montaner, S.; Patel, V.; Zohar, M.; Bais, C.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s Sar-coma-Associated Herpes Virus G Protein-Coupled Receptor up-Regulates Vascular Endothelial Growth Factor Ex-pression and Secretion through Mitogen-Activated Protein Kinase and P38 Pathways Acting on Hypoxia-Inducible Factor 1α. Cancer Res. 2000, 17, 4873–4880. [Google Scholar]
- Pipiya, T.; Sauthoff, H.; Huang, Y.Q.; Chang, B.; Cheng, J.; Heitner, S.; Chen, S.; Rom, W.; Hay, J. Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther. 2005, 12, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V. Tripartite Combination of Candidate Pandemic Mitigation Agents: Vitamin D, Quercetin, and Estradiol Manifest Properties of Medicinal Agents for Targeted Mitigation of the COVID-19 Pandemic Defined by Genomics-Guided Tracing of SARS-CoV-2 Targets in Human Cells. Biomedicine 2020, 8, 129. [Google Scholar] [CrossRef]
- Arias-Reyes, C.; Zubieta-DeUrioste, N.; Poma-Machicao, L.; Aliaga-Raduan, F.; Carvajal-Rodriguez, F.; Dutschmann, M.; Schneider-Gasser, E.M.; Zubieta-Calleja, G.; Soliz, J. Does the pathogenesis of SARS-CoV-2 virus decrease at high-altitude? Respir. Physiol. Neurobiol. 2020, 277, 103443. [Google Scholar] [CrossRef]
- Clarke, N.E.; Belyaev, N.D.; Lambert, D.W.; Turner, A.J. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin. Sci. 2014, 126, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Wollenzien, H.; Leclerc, E.; Jarajapu, Y.P. Hypoxic regulation of angiotensin-converting enzyme 2 and Mas receptor in human CD34 + cells. J. Cell. Physiol. 2019, 234, 20420–20431. [Google Scholar] [CrossRef] [PubMed]
- Ana Campos Codo, A.; Gastã Davanzo, G.; Campos Codo, A.; de Brito Monteiro, L.; Fabiano de Souza, G.; fan-ie Primon Muraro, S.; Victor Virgilio-da-Silva, J.; Silveira Prodonoff, J.; Corasolla Carregari, V.; Alberto Oliveira de Biagi Junior, C.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1a/ Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Panchenko, M.V.; Farber, H.; Korn, J.H. Induction of heme oxygenase-1 by hypoxia and free radicals in human dermal fibroblasts. Am. J. Physiol. Physiol. 2000, 278, C92–C101. [Google Scholar] [CrossRef] [PubMed]
- Eyssen-Hernandez, R.; Ladoux, A.; Frelin, C. Differential regulation of cardiac heme oxygenase-1 and vascular endothelial growth factor mRNA expressions by hemin, heavy metals, heat shock and anoxia. FEBS Lett. 1996, 382, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Perrella, M.A.; Lee, M.E.; Kourembanas, S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.; Laderoute, K.; Short, S.; Sutherland, R. The identification of heme oxygenase as a major hypoxic stress protein in Chinese hamster ovary cells. Br. J. Cancer 1991, 64, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, M.; Ogawa, S. Physiology and Pathology of Astrocytes. Stress response of cultured astrocytes to hypoxia/reoxygenation. Folia Pharmacol. Jpn. 1997, 109, 145–151. [Google Scholar] [CrossRef]
- Ibáñez, F.J.; Farías, M.A.; Retamal-Díaz, A.; Espinoza, J.A.; Kalergis, A.; González, P.A. Pharmacological Induction of Heme Oxygenase-1 Impairs Nuclear Accumulation of Herpes Simplex Virus Capsids upon Infection. Front. Microbiol. 2017, 8, 2108. [Google Scholar] [CrossRef]
- Espinoza, J.A.; León, M.A.; Céspedes-Donoso, P.F.; Gómez, R.S.; Canedo-Marroquín, G.; Riquelme, S.A.; Salazar-Echegarai, F.J.; Blancou, P.; Simon, T.; Anegon, I.; et al. Heme Oxygenase-1 Modulates Human Respiratory Syncytial Virus Replication and Lung Pathogenesis during Infection. J. Immunol. 2017, 199, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, J.A.; Gonzalez, P.A.; Kalergis, A.M. Modulation of Antiviral Immunity by Heme Oxygenase-1. Am. J. Pathol. 2017, 187, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of Human Papillomavirus Type 16 Oncoproteins Enhances Hypoxia-Inducible Factor 1α Protein Accumulation and Vascular Endothelial Growth Factor Expression in Human Cervical Carcinoma Cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef] [Green Version]
- Wakisaka, N.; Kondo, S.; Yoshizaki, T.; Murono, S.; Furukawa, M.; Pagano, J.S. Epstein-Barr Virus Latent Membrane Protein 1 Induces Synthesis of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2004, 24, 5223–5234. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.-W.; Chu, Y.-C.; Chen, P.-R.; Liao, M.-H.; Lee, J.-W. Positive regulation of HIF-1A expression by EBV oncoprotein LMP1 in nasopharyngeal carcinoma cells. Cancer Lett. 2016, 382, 21–31. [Google Scholar] [CrossRef]
- Jham, B.C.; Ma, T.; Hu, J.; Chaisuparat, R.; Friedman, E.R.; Pandolfi, P.P.; Schneider, A.; Sodhi, A.; Montaner, S. Amplification of the Angiogenic Signal through the Activation of the TSC/mTOR/HIF Axis by the KSHV vGPCR in Kaposi’s Sarcoma. PLoS ONE 2011, 6, e19103. [Google Scholar] [CrossRef] [Green Version]
- De Wit, R.H.; Mujić-Delić, A.; van Senten, J.R.; Fraile-Ramos, A.; Siderius, M.; Smit, M.J. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1α/PKM2 axis in glioblastoma cells. Oncotarget 2016, 7, 67966–67985. [Google Scholar] [CrossRef] [Green Version]
- McFarlane, S.; Nicholl, M.J.; Sutherland, J.S.; Preston, C.M. Interaction of the human cytomegalovirus particle with the host cell induces hypoxia-inducible factor 1 alpha. Virology 2011, 414, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.-G.; Oh, S.H.; Park, E.S.; Cho, H.; Lee, N.; Park, H.; Kim, D.K.; Yu, D.-Y.; Seong, J.K.; Lee, M.-O. Hepatitis B Virus X Protein Enhances Transcriptional Activity of Hypoxia-inducible Factor-1α through Activation of Mitogen-activated Protein Kinase Pathway. J. Biol. Chem. 2003, 278, 39076–39084. [Google Scholar] [CrossRef] [Green Version]
- Nasimuzzaman, N.; Waris, G.; Mikolon, D.; Stupack, D.G.; Siddiqui, A. Hepatitis C Virus Stabilizes Hypoxia-Inducible Factor 1α and Stimulates the Synthesis of Vascular Endothelial Growth Factor. J. Virol. 2007, 81, 10249–10257. [Google Scholar] [CrossRef] [Green Version]
- Moin, S.M.; Chandra, V.; Arya, R.; Jameel, S. The hepatitis E virus ORF3 protein stabilizes HIF-1α and enhances HIF-1-mediated transcriptional activity through p300/CBP. Cell. Microbiol. 2009, 11, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human Papillomavirus 16 E6 Contributes HIF-1α Induced Warburg Effect by Attenuating the VHL-HIF-1α Interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [Green Version]
- Kondo, S.; Seo, S.Y.; Yoshizaki, T.; Wakisaka, N.; Furukawa, M.; Joab, I.; Jang, K.L.; Pagano, J.S. EBV Latent Membrane Protein 1 Up-regulates Hypoxia-Inducible Factor 1α through Siah1-Mediated Down-regulation of Prolyl Hydroxylases 1 and 3 in Nasopharyngeal Epithelial Cells. Cancer Res. 2006, 66, 9870–9877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darekar, S.; Georgiou, K.; Yurchenko, M.; Yenamandra, S.P.; Chachami, G.; Simos, G.; Klein, G.; Kashuba, E. Epstein-Barr Virus Immortalization of Human B-Cells Leads to Stabilization of Hypoxia-Induced Factor 1 Alpha, Congruent with the Warburg Effect. PLoS ONE 2012, 7, e42072. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.-L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S Ubiquitin Complex Is Recruited by KSHV Latent Antigen LANA for Degradation of the VHL and p53 Tumor Suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi’s Sarcoma Herpesvirus MicroRNAs Induce Metabolic Transformation of Infected Cells. PLoS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Zhang, W.; Han, P.; Zhang, J.; Zhu, Y.; Meng, X.; Zhang, J.; Hu, Y.; Yi, Z.; Wang, R. Influenza A virus (H1N1) triggers a hypoxic response by stabilizing hypoxia-inducible factor-1α via inhibition of proteasome. Virology 2019, 530, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.; Jeong, C.; Jeong, J.; Kim, K.R.; Yu, D.; Murakami, S.; Kim, C.W.; Kim, K. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1α. FASEB J. 2003, 18, 1–16. [Google Scholar] [CrossRef]
- Yoo, Y.-G.; Na, T.-Y.; Seo, H.-W.; Seong, J.K.; Park, C.K.; Shin, Y.K.; Lee, M.-O. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 2008, 27, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Tan, C.L.; Gunaratne, J.; Quek, L.S.; Nei, W.L.; Thierry, F.; Bellanger, S. Localization of HPV-18 E2 at Mitochondrial Membranes Induces ROS Release and Modulates Host Cell Metabolism. PLoS ONE 2013, 8, e75625. [Google Scholar] [CrossRef] [Green Version]
- Kilani, M.M.; Mohammed, K.A.; Nasreen, N.; Tepper, R.S.; Antony, V.B. RSV Causes HIF-1α Stabilization via NO Release in Primary Bronchial Epithelial Cells. Inflammation 2004, 28, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Duette, G.; Gerber, P.P.; Rubione, J.; Perez, P.S.; Landay, A.L.; Crowe, S.M.; Liao, Z.; Witwer, K.W.; Holgado, M.P.; Salido, J.; et al. Induction of HIF-1α by HIV-1 infection in CD4 + T Cells promotes viral replication and drives extracellular vesicle-mediated inflammation. mBio 2018, 9, e00757-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Mukerjee, R.; Fan, S.; Del Valle, L.; Michiels, C.; Sweet, T.; Rom, I.; Khalili, K.; Rappaport, J.; Amini, S.; et al. Activation of the Oxidative Stress Pathway by HIV-1 Vpr Leads to induction of hypoxia-inducible factor 1α expression. J. Biol. Chem. 2009, 284, 11364–11373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd-Aziz, N.; Stanbridge, E.J.; Shafee, N. Newcastle disease virus degrades HIF-1α through proteasomal pathways independent of VHL and p53. J. Gen. Virol. 2016, 97, 3174–3182. [Google Scholar] [CrossRef]
- Hotani, T.; Tachibana, M.; Mizuguchi, H.; Sakurai, F. Reovirus double-stranded RNA genomes and polyI:C induce down-regulation of hypoxia-inducible factor 1α. Biochem. Biophys. Res. Commun. 2015, 460, 1041–1046. [Google Scholar] [CrossRef]
- Bussiere, L.D.; Miller, C.L. Inhibition of HIF-1α accumulation in prostate cancer cells is initiated during early stages of mammalian orthoreovirus infection. Virology 2021, 558, 38–48. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Mita, M.M.; Mita, A.C.; Nawrocki, S.T. Oncolytic reovirus inhibits angiogenesis through induction of CXCL10/IP-10 and abrogation of HIF activity in soft tissue sarcomas. Oncotarget 2017, 8, 86769–86783. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.-R.; Koh, S.S.; Min, H.-J.; Park, E.-H.; Ratakorn, S.; Jhun, B.H.; Jeong, S.H.; Yoo, Y.H.; Youn, H.; Johnston, R.N.; et al. Down-regulation of HIF-1α by oncolytic reovirus infection independently of VHL and p53. Cancer Gene Ther. 2010, 17, 365–372. [Google Scholar] [CrossRef]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Cellular Process Modulated | Outcome | Model | References |
|---|---|---|---|---|
| DNA Viruses | ||||
| Epstein–Barr virus (EBV) | Hypoxia increases the expression of the EBV transcription factor Zta in a B-lymphoblast cell line. | Increases EBV replication. | In vitro | [111] |
| HIF-1α interacts with the latent-lytic switch BZLF1 gene Zp in HEK 293T cells. | Increase EBV lytic gene expression. | In vitro | [112] | |
| Kaposi’s Sarcoma-associated herpesvirus (KSHV) | HIF-1α binds to the latency-associated nuclear antigen (LANA) of KSHV. The HIF-1α-LANA complex induces Rta gene transcription in primary effusion lymphoma (PEL) cells. | Increases KSHV replication. | In vitro | [113] |
| HIF-1α interacts with the promoter of the viral ORF34 gene in Hep3B cells and induces vGPCR expression in B cells. | Increase KSHV replication. | In vitro | [114] | |
| Hypoxia induces the expression of the viral inducer TPA, producing an increase in the levels of IL-6 expression in KSHV-infected primary effusion lymphoma B-cell lines. | Increase KSHV replication. | In vitro | [115] | |
| Herpes simplex virus type 1 (HSV-1) mutant G207 (γ34.5 gene deletion) | Hypoxia increases GADD34 expression in U87 human glioma cells. | Increase of HSV-1 mutant G207 replication. | In vitro | [116] |
| John Cunningham virus (JCV) | HIF-1α interacts with the JCV control region in glial cells. | Increases JCV replication. | In vitro | [117] |
| Adenovirus | Hypoxia decreases the expression of adenovirus protein E1A. | Decreases adenovirus replication in HEK293 and numerous cancer cell lines. | In vitro | [118] |
| HBV | Hypoxia induces DNase 1 expression, which is encapsidated in HBV particles. | HBV genome copy number reduction in HepG2 cells. | In vitro | [119] |
| RNA Viruses | ||||
| DENV | HIF-1α and HIF-2α induce serine/threonine AKT signaling and ROS production in liver cells. | Increase DENV replication. | In vitro | [120] |
| HIV | HIF-1α interacts with VPR inducing HIV gene expression in human kidney cells. | Increases HIV production. | In vitro | [121] |
| Hypoxia induces IL-17 expression that upregulates GLUT1 expression, favoring glucose uptake in T cells. | Increases HIV production. | In vitro | [122] | |
| Hypoxia decreases CDK9/cyclin T1 and Sp1 expression, producing a reduction in HIV Tat expression. | Reduction in HIV production. | In vitro | [123] | |
| VSV | Hypoxia reduces VSV mRNA at early time points after infection of HeLa cells, but at later times after infection, VSV reduces eIF2α phosphorylation, promoting viral protein synthesis. | Increase in VSV replication. | In vitro | [124] |
| SARS-CoV-2 | HIF-1α downregulates the expression of ACE2 and TMPRSS2 SARS-CoV-2 receptors. | Decrease of SARS-CoV-2 infection by a reduction of the release of viral particles in lung epithelial cells. | In vitro and in vivo in C57BL/6 mice | [125] |
| HIF-1α induces Angiotensin II, which inhibits ACE2 synthesis. | Decrease of SARS-CoV-2 replication in human pulmonary artery smooth muscle cells. | In vitro | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, A.; Duarte, L.F.; Farías, M.A.; Tognarelli, E.; Kalergis, A.M.; Bueno, S.M.; González, P.A. Impact of Hypoxia over Human Viral Infections and Key Cellular Processes. Int. J. Mol. Sci. 2021, 22, 7954. https://doi.org/10.3390/ijms22157954
Reyes A, Duarte LF, Farías MA, Tognarelli E, Kalergis AM, Bueno SM, González PA. Impact of Hypoxia over Human Viral Infections and Key Cellular Processes. International Journal of Molecular Sciences. 2021; 22(15):7954. https://doi.org/10.3390/ijms22157954
Chicago/Turabian StyleReyes, Antonia, Luisa F. Duarte, Mónica A. Farías, Eduardo Tognarelli, Alexis M. Kalergis, Susan M. Bueno, and Pablo A. González. 2021. "Impact of Hypoxia over Human Viral Infections and Key Cellular Processes" International Journal of Molecular Sciences 22, no. 15: 7954. https://doi.org/10.3390/ijms22157954
APA StyleReyes, A., Duarte, L. F., Farías, M. A., Tognarelli, E., Kalergis, A. M., Bueno, S. M., & González, P. A. (2021). Impact of Hypoxia over Human Viral Infections and Key Cellular Processes. International Journal of Molecular Sciences, 22(15), 7954. https://doi.org/10.3390/ijms22157954