
Systematic Modification and Evaluation of Enzyme-Loaded Chitosan Nanoparticles

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
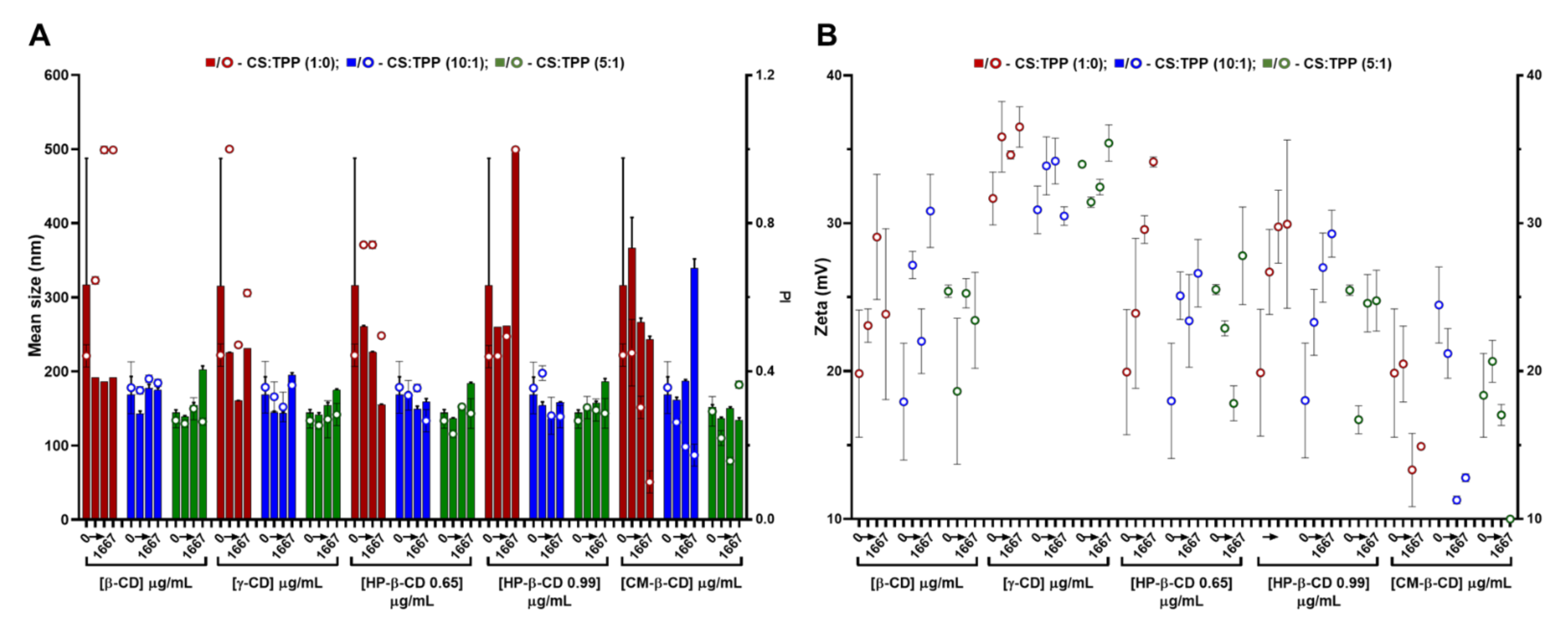
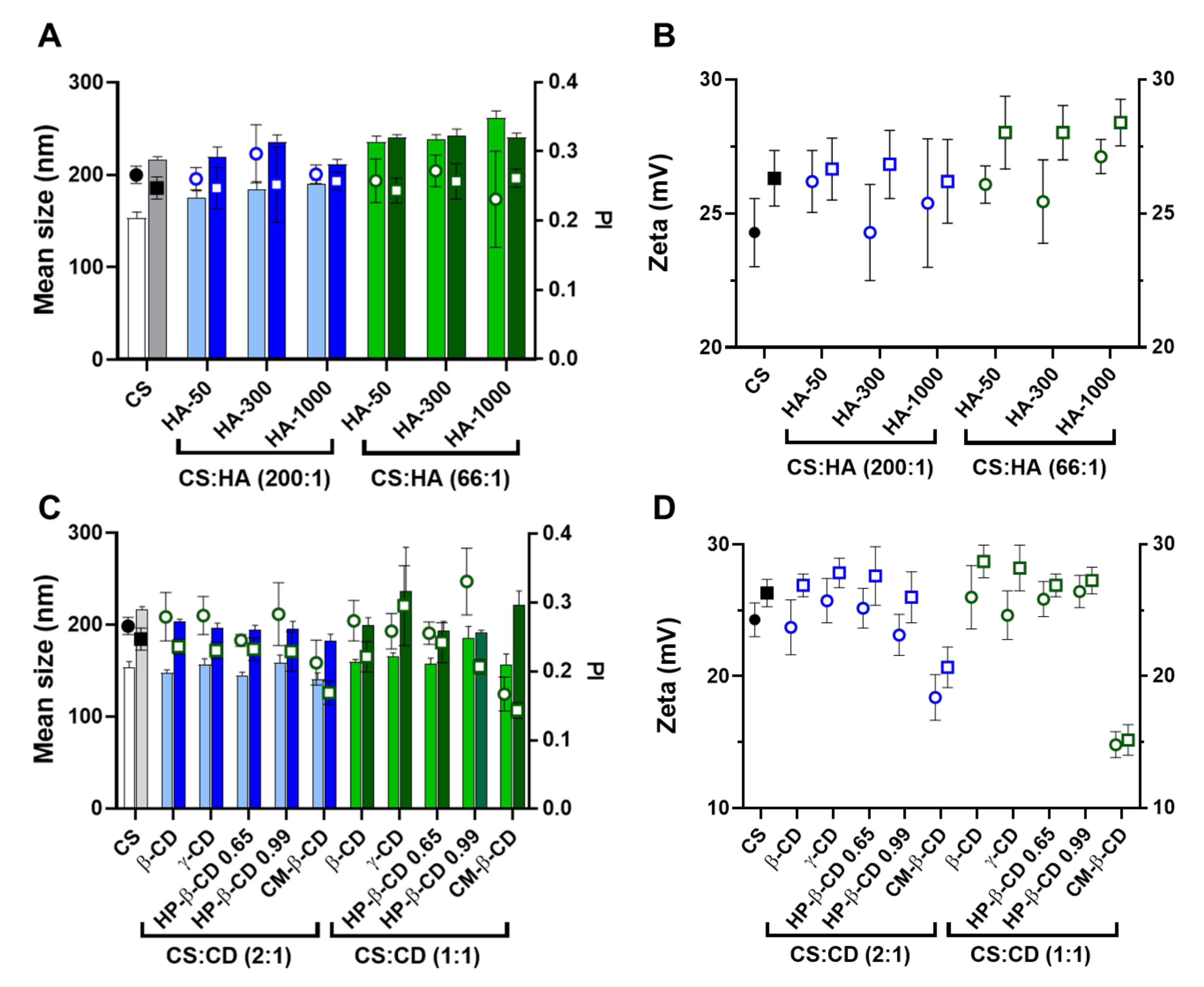
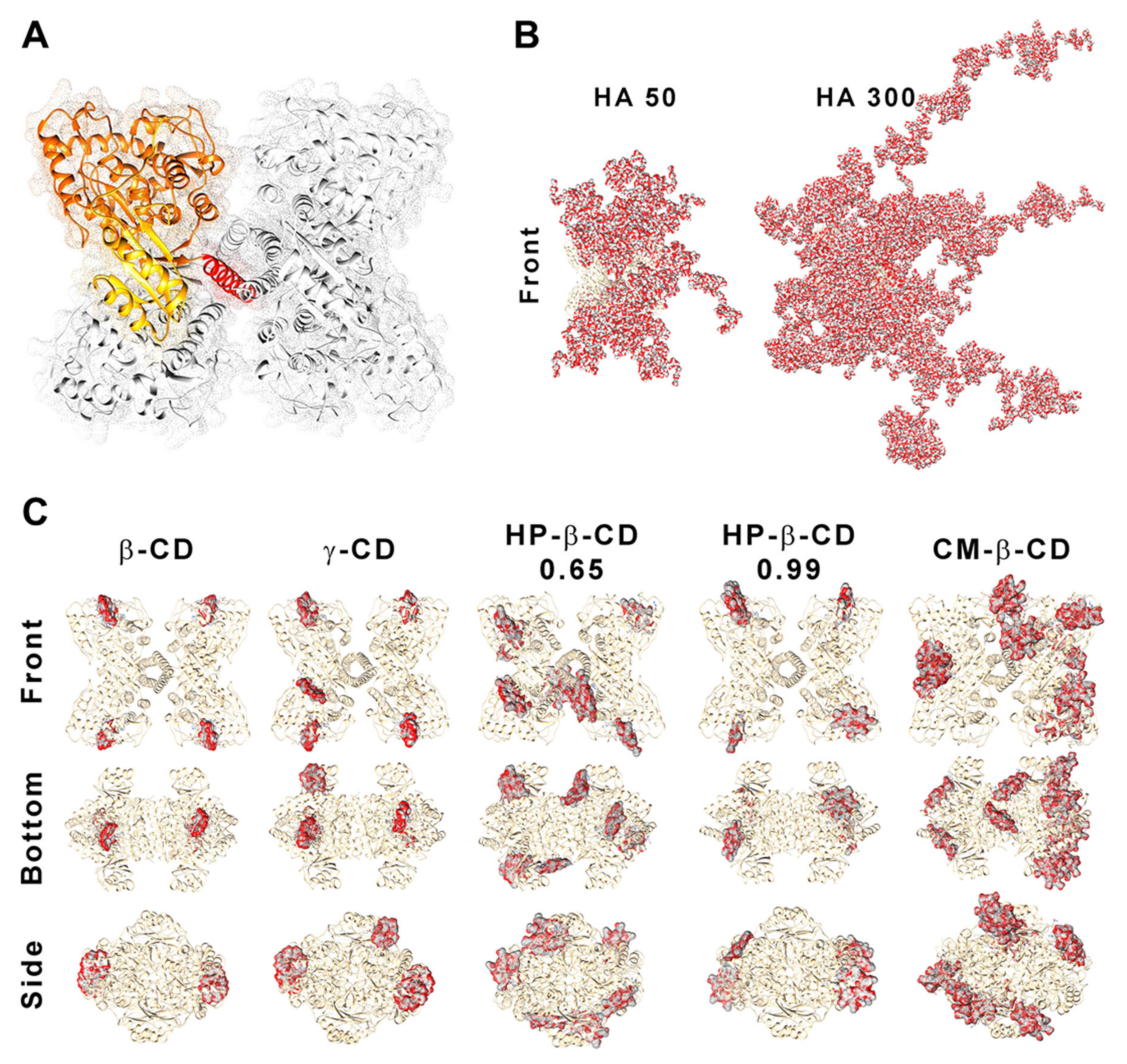
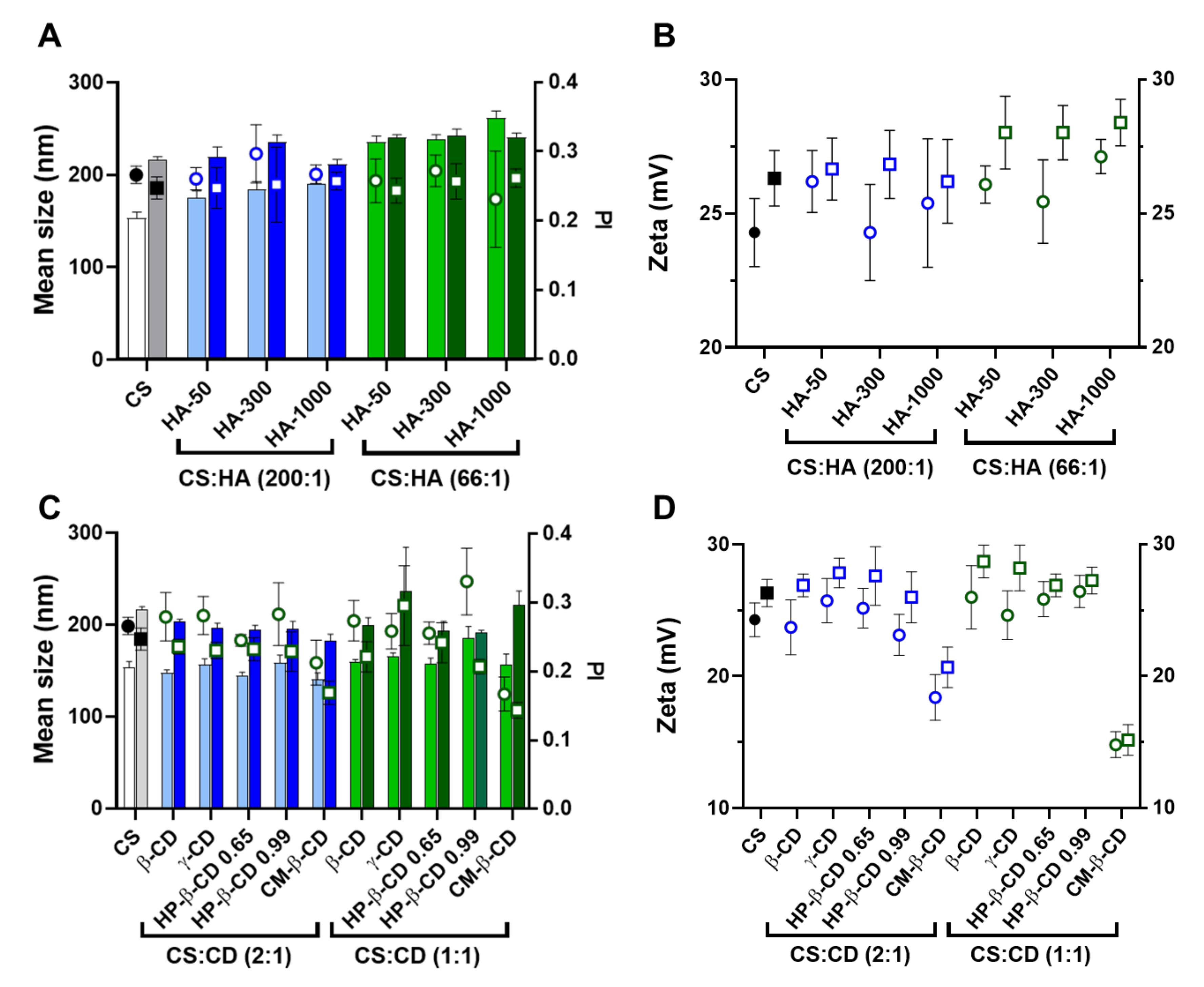
2.1. CS-TPP Matrix Modification with Hyaluronic Acid
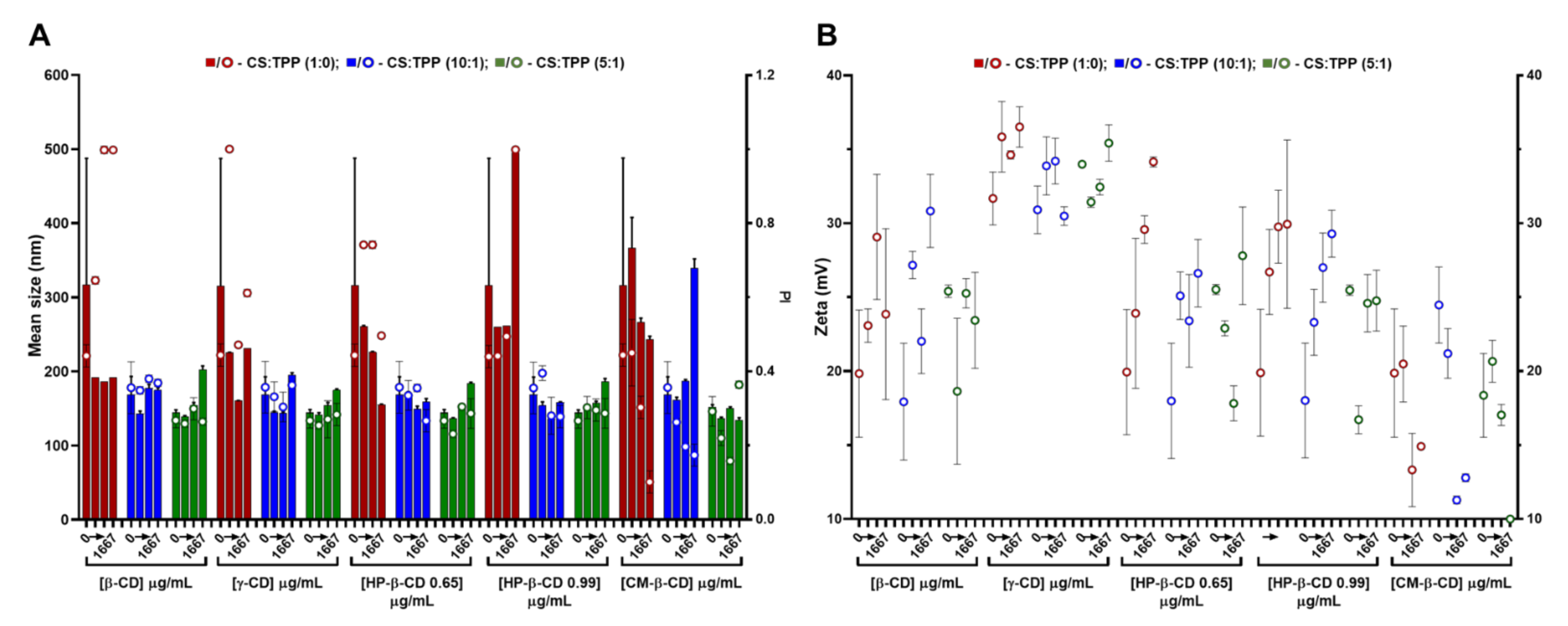
2.2. CS-TPP Matrix Modification with Cyclodextrins
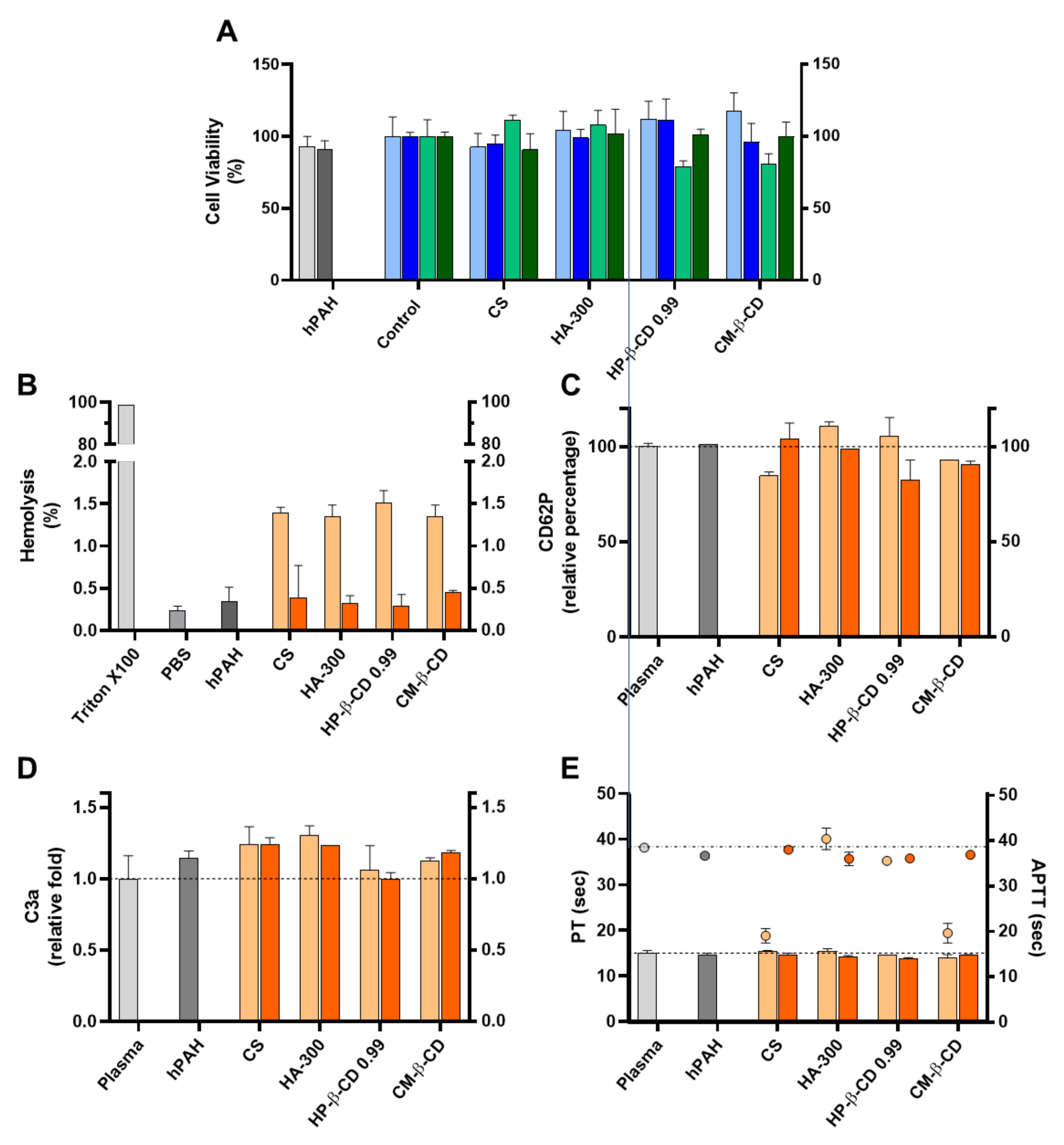
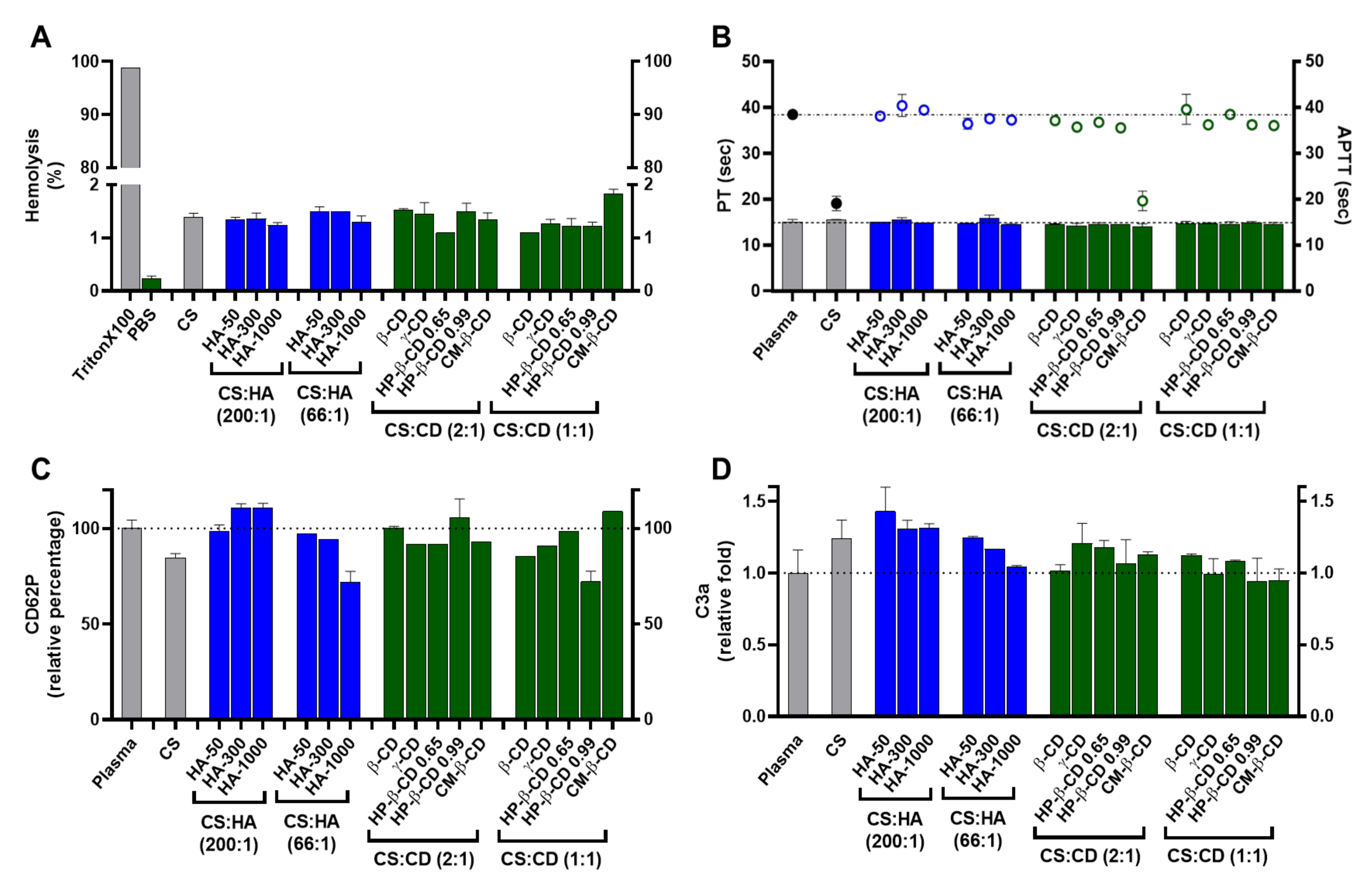
2.3. Cyto and Haemocompatibility of the Modified CS-TPP Matrix
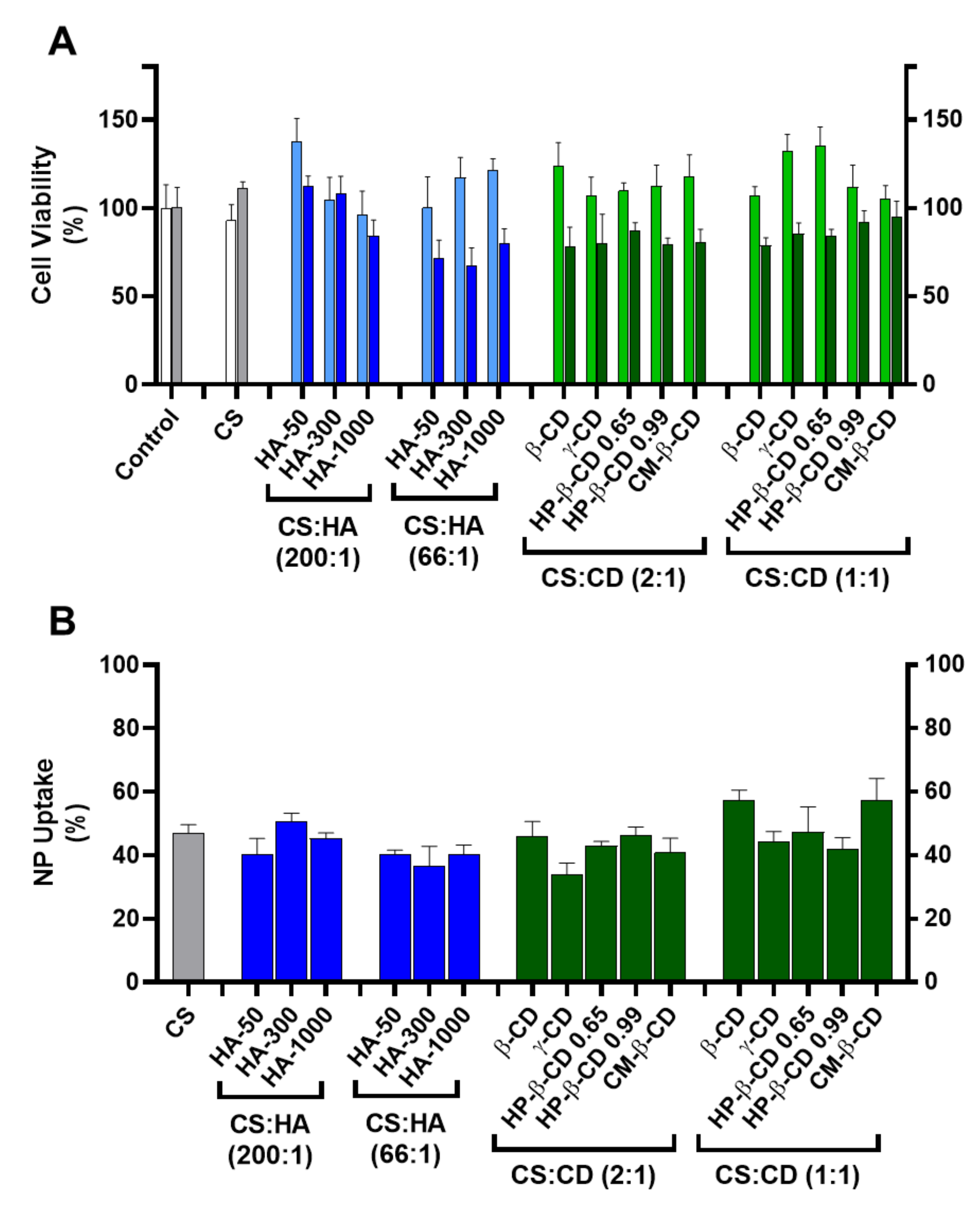
2.3.1. Cytotoxicity and Nanoparticle Uptake
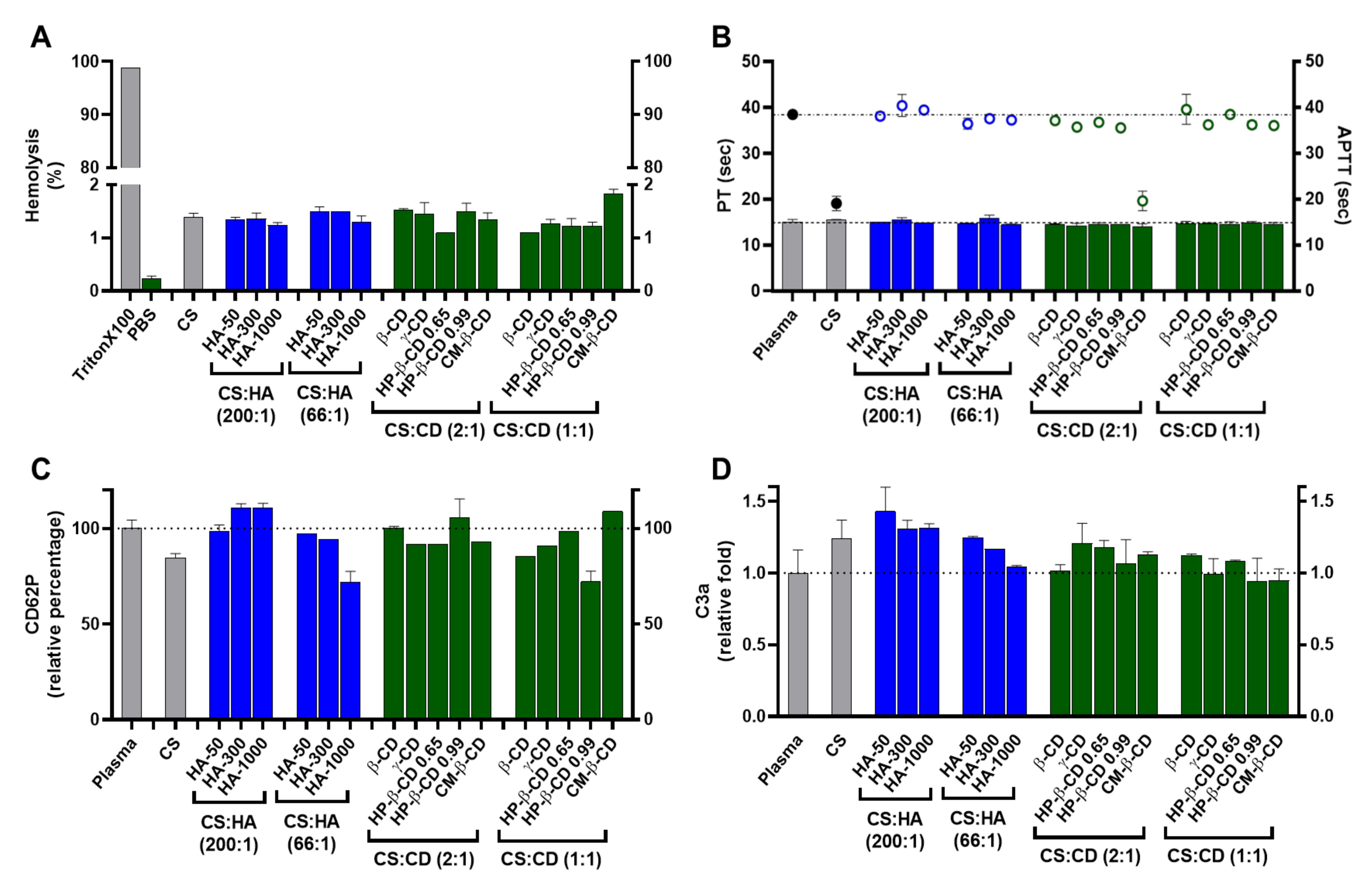
2.3.2. Haemocompatibility
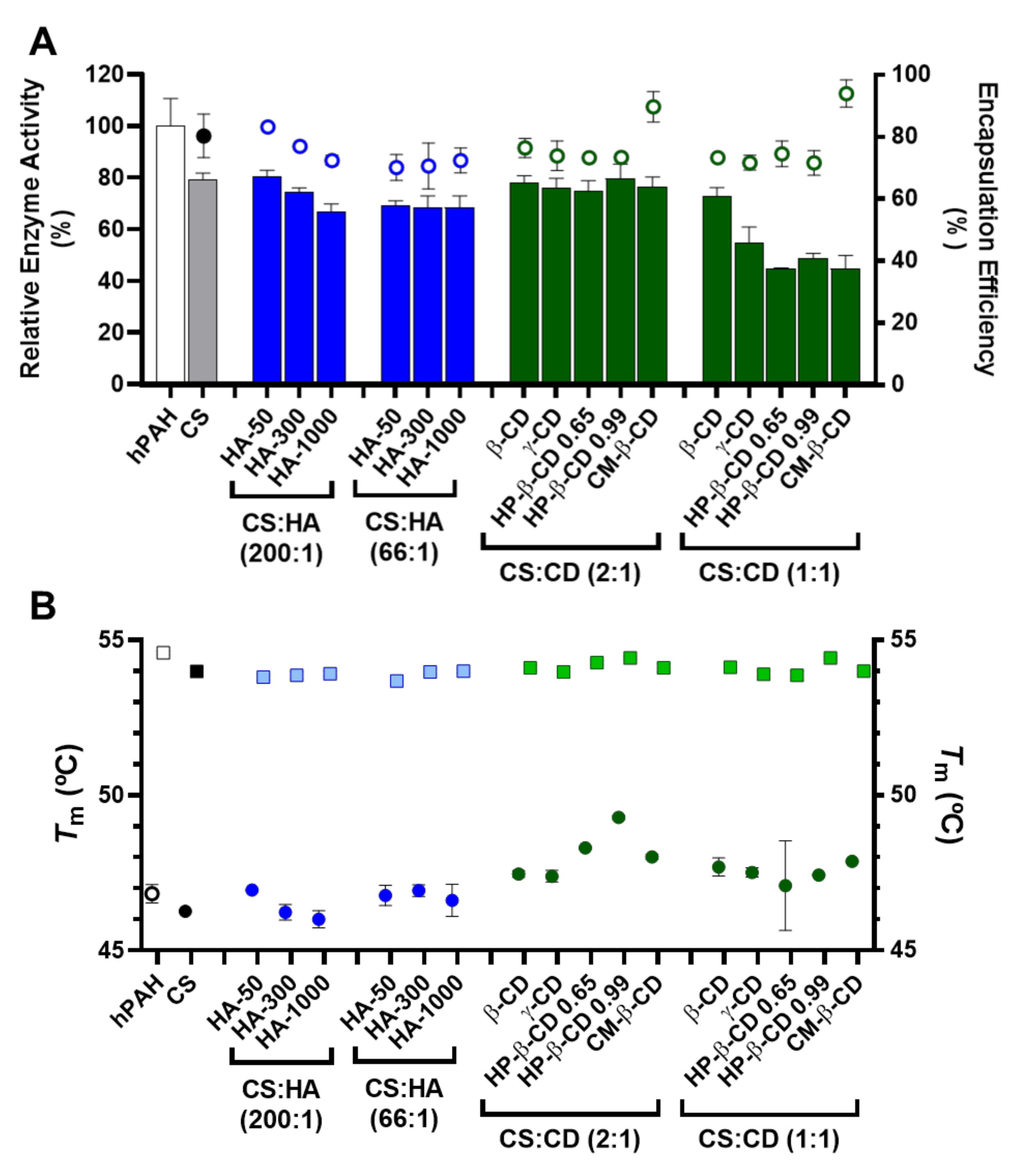
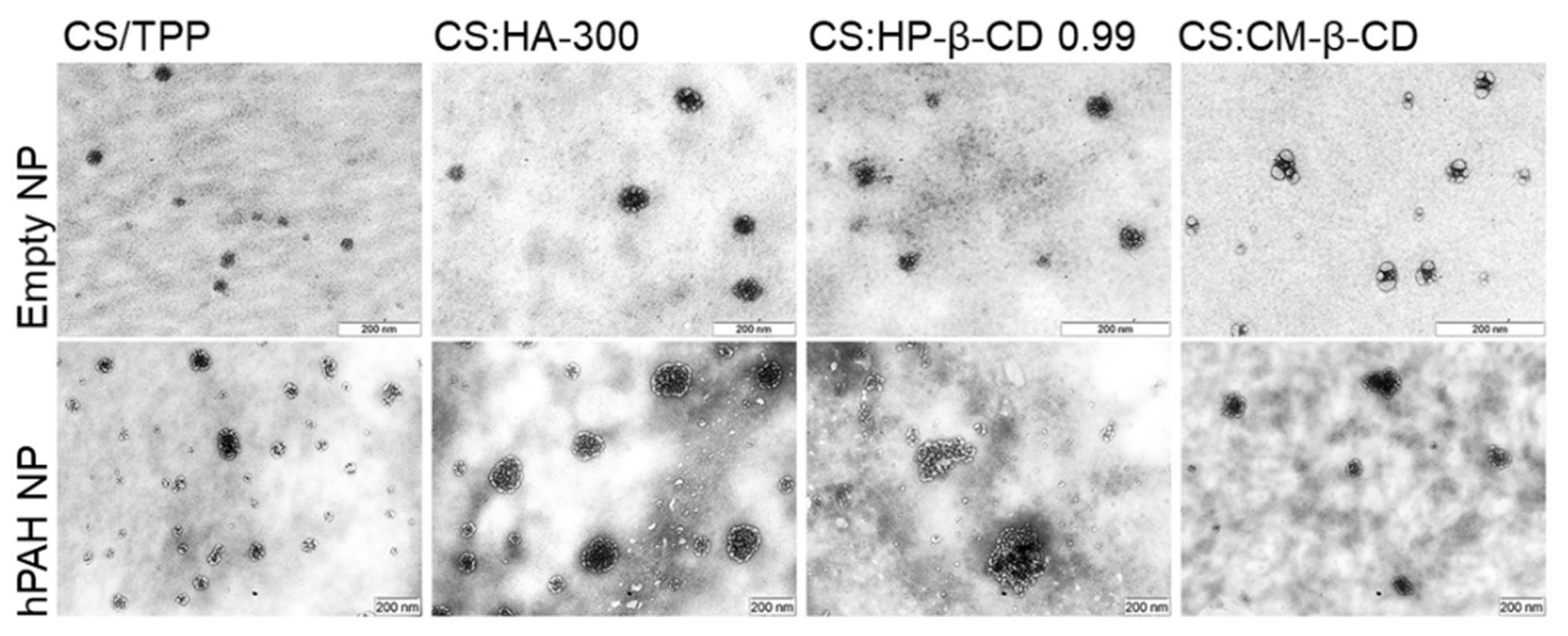
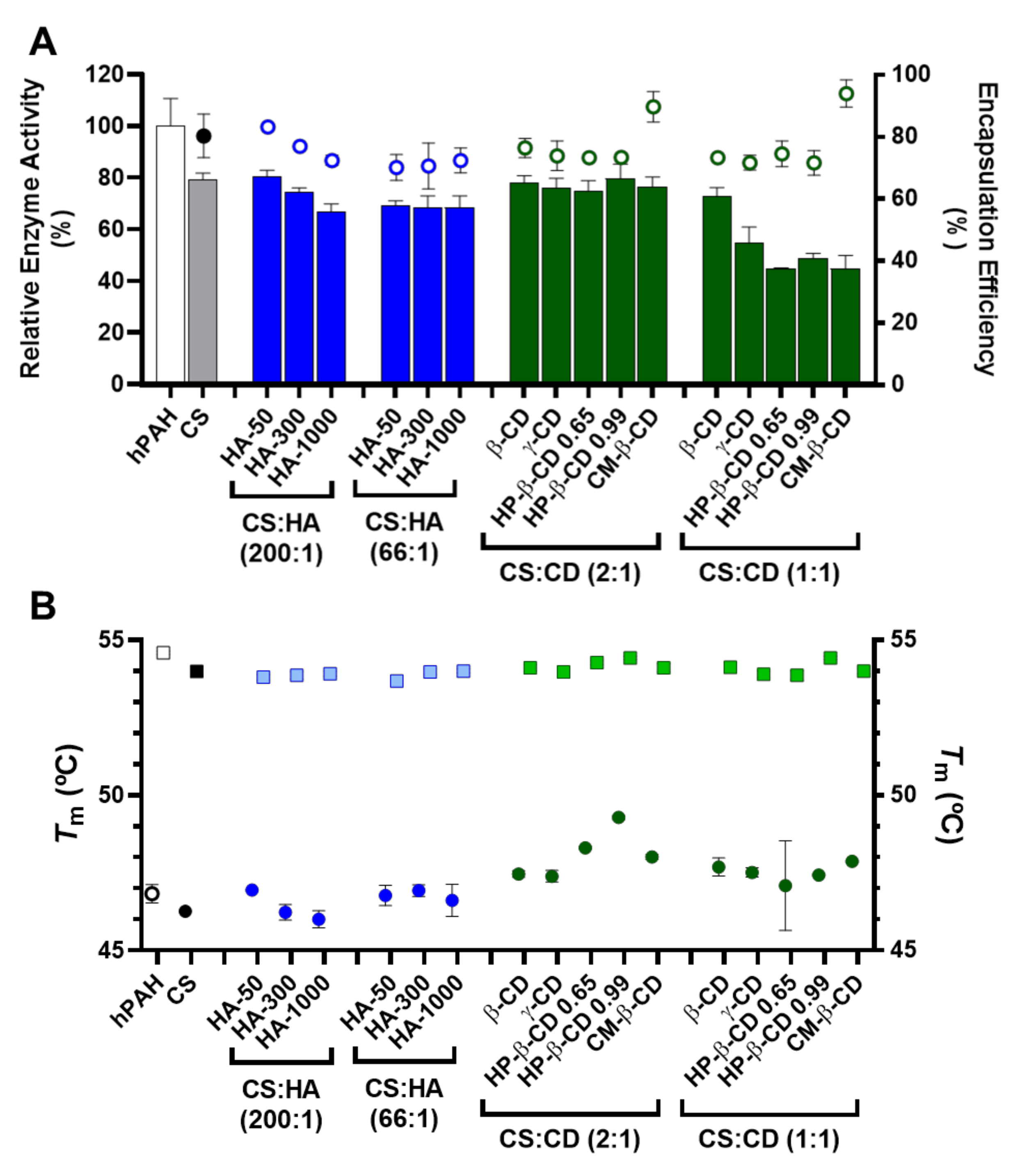
2.4. hPAH Nanoencapsulation in Selected CS Modified Systems
2.5. Encapsulation Efficiency, Enzyme Activity and Thermal Stability of hPAH Loaded Nanoparticles
2.6. Nano-Bio Interface with Selected hPAH-Loaded Nanoparticles
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Expression and Purification of Recombinant hPAH Tetramers
3.3. Preparation of CS Nanoparticles
3.4. Particle Size, Distribution, Zeta Potential and Encapsulation Efficiency
3.5. Transmission Electron Microscopy (TEM)
3.6. Enzymatic Activity of hPAH Loaded Nanoparticles
3.7. Differential Scanning Fluorimetry
3.8. In Silico Modelling
3.9. Cell Viability
3.10. Assessment of the Nanoparticles Uptake Profile
3.11. Haemocompatibility Assays
3.12. Controls and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, W.; Ohtake, S. Science and art of protein formulation development. Int. J. Pharm. 2019, 568, 118505. [Google Scholar] [CrossRef] [PubMed]
- Dingman, R.; Balu-Iyer, S.V. Immunogenicity of protein pharmaceuticals. J. Pharm. Sci. 2019, 108, 1637–1654. [Google Scholar] [CrossRef]
- Miao, T.; Wang, J.; Zeng, Y.; Liu, G.; Chen, X. Polysaccharide-based controlled release systems for therapeutics delivery and tissue engineering: From bench to bedside. Adv. Sci. 2018, 5, 1700513. [Google Scholar] [CrossRef]
- Dovedytis, M.; Liu, Z.J.; Bartlett, S. Hyaluronic acid and its biomedical applications: A review. Eng. Regen. 2020, 1, 102–113. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Nicolas, J.; Mura, S.; Brambilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors controlling nanoparticle pharmacokinetics: An integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into cellular uptake and intracellular trafficking of nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Kim, S. Competitive biological activities of chitosan and its derivatives: Antimicrobial, antioxidant, anticancer, and anti-inflammatory activities. Int. J. Polym. Sci. 2018, 2018, 1708172. [Google Scholar] [CrossRef]
- Lino, P.R.; Leandro, J.; Amaro, M.; Goncalves, L.M.D.; Leandro, P.; Almeida, A.J. In silico and In Vitro tailoring of a chitosan nanoformulation of a human metabolic enzyme. Pharmaceutics 2021, 13, 329. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Jesus, S.; Marques, A.P.; Duarte, A.; Soares, E.; Costa, J.P.; Colaco, M.; Schmutz, M.; Som, C.; Borchard, G.; Wick, P.; et al. Chitosan nanoparticles: Shedding light on immunotoxicity and hemocompatibility. Front. Bioeng. Biotechnol. 2020, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Brasselet, C.; Pierre, G.; Dubessay, P.; Dols-Lafargue, M.; Coulon, J.; Maupeu, J.; Vallet-Courbin, A.; de Baynast, H.; Doco, T.; Michaud, P.; et al. Modification of chitosan for the generation of functional derivatives. Appl. Sci. 2019, 9, 1321. [Google Scholar] [CrossRef] [Green Version]
- Casalini, T. Not only in silico drug discovery: Molecular modeling towards in silico drug delivery formulations. J. Control. Release 2021, 332, 390–417. [Google Scholar] [CrossRef]
- Pagolu, R.; Singh, R.; Shanmugam, R.; Kondaveeti, S.; Patel, S.K.S.; Kalia, V.C.; Lee, J.K. Site-directed lysine modification of xylanase for oriented immobilization onto silicon dioxide nanoparticles. Bioresour. Technol. 2021, 331, 125063. [Google Scholar] [CrossRef]
- Pedroso-Santana, S.; Fleitas-Salazar, N. Ionotropic gelation method in the synthesis of nanoparticles/microparticles for biomedical purposes. Polym. Int. 2020, 69, 443–447. [Google Scholar] [CrossRef]
- Lallana, E.; de la Rosa, J.M.R.; Tirella, A.; Pelliccia, M.; Gennari, A.; Stratford, I.J.; Puri, S.; Ashford, M.; Tirelli, N. Chitosan/Hyaluronic acid nanoparticles: Rational design revisited for RNA delivery. Mol. Pharm. 2017, 14, 2422–2436. [Google Scholar] [CrossRef]
- Turcsanyi, A.; Varga, N.; Csapo, E. Chitosan-modified hyaluronic acid-based nanosized drug carriers. Int. J. Biol. Macromol. 2020, 148, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Krauland, A.H.; Alonso, M.J. Chitosan/cyclodextrin nanoparticles as macromolecular drug delivery system. Int. J. Pharm. 2007, 340, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Saha, K.; Kim, C.; Rotello, V.M. The role of surface functionality in determining nanoparticle cytotoxicity. Acc. Chem. Res. 2013, 46, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, S.; Yeh, Y.C.; Rotello, V.M. Engineering the nanoparticle-protein interface: Applications and possibilities. Curr. Opin. Chem. Biol. 2010, 14, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ruan, Y.; Xing, X.; Chen, Q.; Peng, Y.; Cai, J. Curcumin induced nanoscale CD44 molecular redistribution and antigen-antibody interaction on HepG2 cell surface. Anal. Chim. Acta 2011, 697, 83–89. [Google Scholar] [CrossRef]
- Kiss, T.; Fenyvesi, F.; Bacskay, I.; Varadi, J.; Fenyvesi, E.; Ivanyi, R.; Szente, L.; Tosaki, A.; Vecsernyes, M. Evaluation of the cytotoxicity of beta-cyclodextrin derivatives: Evidence for the role of cholesterol extraction. Eur. J. Pharm. Sci. 2010, 40, 376–380. [Google Scholar] [CrossRef]
- Barbero, F.; Russo, L.; Vitali, M.; Piella, J.; Salvo, I.; Borrajo, M.L.; Busquets-Fite, M.; Grandori, R.; Bastus, N.G.; Casals, E.; et al. Formation of the protein corona: The interface between nanoparticles and the immune system. Semin. Immunol. 2017, 34, 52–60. [Google Scholar] [CrossRef] [PubMed]
- De la Harpe, K.M.; Kondiah, P.P.D.; Choonara, Y.E.; Marimuthu, T.; du Toit, L.C.; Pillay, V. The hemocompatibility of nanoparticles: A review of cell-nanoparticle interactions and hemostasis. Cells 2019, 8, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brash, J.L. Chapter 2—Blood compatibility of nanomaterials. In Drug Delivery Nanosystems for Biomedical Applications; Sharma, C.P., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 13–31. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Standard Test Method for Analysis of Hemolytic Properties of Nanoparticles. Method for Analysis of Hemolytic Properties of Nanoparticles; ASTM International: West Conshohocken, PA, USA, 2013. [Google Scholar] [CrossRef]
- Nadesh, R.; Narayanan, D.; Vadakumpully, S.; Mony, U.; Koyakkutty, M.; Nair, S.V.; Menon, D. Hematotoxicological analysis of surface-modified and -unmodified chitosan nanoparticles. J. Biomed. Mater. Res. A 2013, 101, 2957–2966. [Google Scholar] [CrossRef]
- Patel, S.K.S.; Kim, J.H.; Kalia, V.C.; Lee, J.K. Antimicrobial activity of amino-derivatized cationic polysaccharides. Indian J. Microbiol. 2019, 59, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; López-Viota, M.; Sáez-Fernández, E.; Ruiz, M.A.; Delgado, Á.V. Engineering of an antitumor (core/shell) magnetic nanoformulation based on the chemotherapy agent ftorafur. Colloids Surf. A Physicochem. Eng. Asp. 2011, 384, 157–163. [Google Scholar] [CrossRef]
- Marchand, C.; Bachand, J.; Perinet, J.; Baraghis, E.; Lamarre, M.; Rivard, G.E.; De Crescenzo, G.; Hoemann, C.D. C3, C5, and factor B bind to chitosan without complement activation. J. Biomed. Mater. Res. A 2010, 93, 1429–1441. [Google Scholar] [CrossRef]
- Neun, B.; Rodriguez, J.; Ilinskaya, A.; Dobrovolskaia, M. NCL Method ITA-12; NCI Hub: Bethesda, MD, USA, 2020. [Google Scholar] [CrossRef]
- Chen, F.; Wang, G.; Griffin, J.I.; Brenneman, B.; Banda, N.K.; Holers, V.M.; Backos, D.S.; Wu, L.; Moghimi, S.M.; Simberg, D. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat. Nanotechnol. 2017, 12, 387–393. [Google Scholar] [CrossRef]
- Owens, D.E., 3rd.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Neun, B.; Cedrone, E.; Dobrovolskaia, M. NCL Method ITA-5.2; NCI Hub: Bethesda, MD, USA, 2020. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Reca, R.; Wysoczynski, M.; Kucia, M.; Baran, J.T.; Allendorf, D.J.; Ratajczak, J.; Ross, G.D. Transplantation studies in C3-deficient animals reveal a novel role of the third complement component (C3) in engraftment of bone marrow cells. Leukemia 2004, 18, 1482–1490. [Google Scholar] [CrossRef]
- Tome, C.S.; Lopes, R.R.; Sousa, P.M.F.; Amaro, M.P.; Leandro, J.; Mertens, H.D.T.; Leandro, P.; Vicente, J.B. Structure of full-length wild-type human phenylalanine hydroxylase by small angle X-ray scattering reveals substrate-induced conformational stability. Sci. Rep. 2019, 9, 13615. [Google Scholar] [CrossRef] [Green Version]
- Leandro, P.; Rivera, I.; Lechner, M.C.; de Almeida, I.T.; Konecki, D. The V388M mutation results in a kinetic variant form of phenylalanine hydroxylase. Mol. Genet. Metab. 2000, 69, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, C.; Leandro, J.; Lino, P.R.; Ramos, L.; Almeida, A.J.; de Almeida, I.T.; Leandro, P. Polyol additives modulate the in vitro stability and activity of recombinant human phenylalanine hydroxylase. Appl. Biochem. Biotechnol. 2010, 162, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, F.; Leandro, J.; Farias, G.D.V.F.; Lino, P.R.; Guedes, R.C.; Vicente, J.B.; Leandro, P.; Gois, P.M.P. Phenylalanine iminoboronates as new phenylalanine hydroxylase modulators. RSC Adv. 2014, 4, 61022–61027. [Google Scholar] [CrossRef]
- Lopes, R.R.; Tome, C.S.; Russo, R.; Paterna, R.; Leandro, J.; Candeias, N.R.; Goncalves, L.M.D.; Teixeira, M.; Sousa, P.M.F.; Guedes, R.C.; et al. Modulation of human phenylalanine hydroxylase by 3-hydroxyquinolin-2(1H)-one derivatives. Biomolecules 2021, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Cadete, A.; Figueiredo, L.; Lopes, R.; Calado, C.C.; Almeida, A.J.; Goncalves, L.M. Development and characterization of a new plasmid delivery system based on chitosan-sodium deoxycholate nanoparticles. Eur. J. Pharm. Sci. 2012, 45, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Neun, B.; Ilinskaya, A.; Dobrovolskaia, M. NCL Method ITA-1; NCI Hub: Bethesda, MD, USA, 2020. [Google Scholar] [CrossRef]
- Dash, B.C.; Rethore, G.; Monaghan, M.; Fitzgerald, K.; Gallagher, W.; Pandit, A. The influence of size and charge of chitosan/polyglutamic acid hollow spheres on cellular internalization, viability and blood compatibility. Biomaterials 2010, 31, 8188–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lino, P.R.; Leandro, J.; Figueiredo, L.; Amaro, M.P.; Gonçalves, L.M.D.; Leandro, P.; Almeida, A.J. Systematic Modification and Evaluation of Enzyme-Loaded Chitosan Nanoparticles. Int. J. Mol. Sci. 2021, 22, 7987. https://doi.org/10.3390/ijms22157987
Lino PR, Leandro J, Figueiredo L, Amaro MP, Gonçalves LMD, Leandro P, Almeida AJ. Systematic Modification and Evaluation of Enzyme-Loaded Chitosan Nanoparticles. International Journal of Molecular Sciences. 2021; 22(15):7987. https://doi.org/10.3390/ijms22157987
Chicago/Turabian StyleLino, Paulo R., João Leandro, Lara Figueiredo, Mariana P. Amaro, Lídia M. D. Gonçalves, Paula Leandro, and António J. Almeida. 2021. "Systematic Modification and Evaluation of Enzyme-Loaded Chitosan Nanoparticles" International Journal of Molecular Sciences 22, no. 15: 7987. https://doi.org/10.3390/ijms22157987
APA StyleLino, P. R., Leandro, J., Figueiredo, L., Amaro, M. P., Gonçalves, L. M. D., Leandro, P., & Almeida, A. J. (2021). Systematic Modification and Evaluation of Enzyme-Loaded Chitosan Nanoparticles. International Journal of Molecular Sciences, 22(15), 7987. https://doi.org/10.3390/ijms22157987