
The Essential Role of PRAK in Preserving Cardiac Function and Insulin Resistance in High-Fat Diet-Induced Diabetes

 ,
,  ,
,  ,
,
Abstract
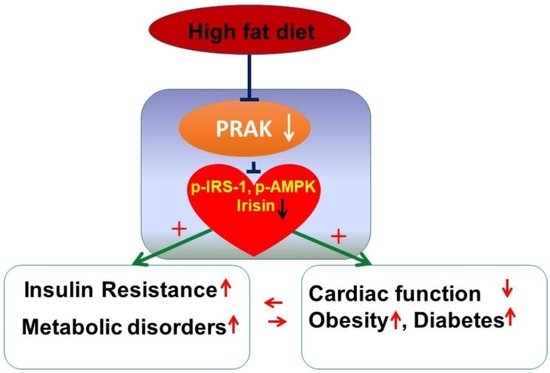
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PRAK Knockout-Enhanced HFD Induces Glucose Intolerance
2.2. PRAK Knockout-Enhanced HFD Induces Left Ventricular Dysfunction
2.3. PRAK Deletion Increased Interstitial Fibrosis and Enhanced Myocyte Hypertrophy in the Myocardium
2.4. PRAK Deletion Resulted in a Decrease in Phosphorylation of IRS-1, AMPKα, and ERK1/2 and in a Reduction of Irisin
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies and Animals
4.2. Animal and Experimental Protocol
4.3. Metabolic Measurements
4.4. Echocardiographic Measurements
4.5. Histological Analysis
4.6. Real-Time Polymerase Chain Reaction (PCR)
4.7. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Assay
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.L.; Zhu, T.B.; Yin, H.; Huang, J.; Wang, L.S.; Cao, K.J.; Yang, Z.J. Inhibition of MAPK signaling by eNOS gene transfer improves ventricular remodeling after myocardial infarction through reduction of inflammation. Mol. Biol. Rep. 2010, 37, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.C.; Taher, M.M.; Valerie, K.C.; Kukreja, R.C. p38 Triggers late preconditioning elicited by anisomycin in heart: Involvement of NF-kappaB and iNOS. Circ. Res. 2001, 89, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.C.; Hines, D.S.; Kukreja, R.C. Adenosine-induced late preconditioning in mouse hearts: Role of p38 MAP kinase and mitochondrial K (ATP) channels. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1278–H1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.C.; Du, J.; Zhuang, S.; Liu, P.; Zhang, L.X. HDAC inhibition elicits myocardial protective effect through modulation of MKK3/Akt-1. PLoS ONE 2013, 8, e65474. [Google Scholar] [CrossRef] [Green Version]
- Jialal, I.; Adams-Huet, B.; Pahwa, R. Selective increase in monocyte p38 mitogen-activated protein kinase activity in metabolic syndrome. Diab. Vasc. Dis. Res. 2016, 13, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, L.; Cristina de Souza, J.; Harari, A.A.; Backs, J.; Olson, E.N.; Tabas, I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013, 18, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Hemi, R.; Yochananov, Y.; Barhod, E.; Kasher-Meron, M.; Karasik, A.; Tirosh Kanety, H. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: A novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes 2011, 60, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Moens, U.; Kostenko, S. Structure and function of MK5/PRAK: The loner among the mitogen-activated protein kinase-activated protein kinases. Biol. Chem. 2013, 394, 1115–1132. [Google Scholar] [CrossRef]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.; Wang, X.S.; Diener, K.; Yao, Z. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 1998, 243, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Perander, M.; Keyse, S.M.; Seternes, O.M. New insights into the activation, interaction partners and possible functions of MK5/PRAK. Front. Biosci. 2016, 21, 374–384. [Google Scholar]
- Zheng, M.; Wang, Y.H.; Wu, X.N.; Wu, S.Q.; Lu, B.J.; Dong, M.Q.; Zhang, H.; Sun, P.; Lin, S.C.; Guan, K.L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, E.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell. 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Hardie, D.G. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008, 582, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, Y.; Tsurumaki, H.; Aoki-Saito, H.; Sato, M.; Yatomi, M.; Takehara, K.; Hisada, T. Roles of Cyclic AMP Response Element Binding Activation inthe ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int. J. Mol. Sci. 2019, 20, 1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.H.; Gavin, T.P.; Shulman, G.I.; Fernandez-Hernando, C.; et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Terán, B.; Matesanz, N.; Nikolic, I.; Verdugo, M.A.; Sreeramkumar, V.; Hernández-Cosido, L.; Mora, A.; Crainiciuc, G.; Sáiz, M.L.; Bernardo, E.; et al. p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016, 35, 536–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.T.; Du, J.; Yano, N.; Wang, H.; Wang, J.; Dubielecka, P.M.; Zhang, L.X.; Qin, G.; Zhuang, S.; Liu, P.Y.; et al. p38-Regulated/activated protein kinase plays a pivotal role in protecting heart against ischemia-reperfusion injury and preserving cardiac performance. Am. J. Physiol. Cell Physiol. 2019, 317, C525–C533. [Google Scholar] [CrossRef]
- Rajesh, M.; Bátkai, S.; Kechrid, M.; Mukhopadhyay, P.; Lee, W.S.; Horváth, B.; Holovac, E.; Cinar, R.; Liaudet, L.; Mackie, K.; et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes 2012, 61, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Huang, Z.; Gu, J.; Yan, X.; Lu, X.; Zhou, S.; Wang, S.; Shao, M.; Zhang, F.; Cheng, P.; et al. Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signalling pathway. Diabetologia 2015, 58, 1937–1948. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, H.; Zhang, J.; Zhang, X.; Xin, C.; Zhang, F.; Lee, Y.; Zhang, L.; Lian, K.; Yan, W.; et al. Irisin improves endothelial function in type 2 diabetes through reducing oxidative/nitrative stresses. J. Mol. Cell Cardiol. 2015, 87, 138–147. [Google Scholar] [CrossRef]
- Park, M.J.; Kim, D.I.; Choi, J.H.; Heo, Y.R.; Park, S.H. New role of irisin in hepatocytes: The protective effect of hepatic steatosis in vitro. Cell Signal. 2015, 27, 1831–1839. [Google Scholar] [CrossRef]
- Wright, D.C.; Han, D.H.; Garcia-Roves, P.M.; Geiger, P.C.; Jones, T.E.; Holloszy, J.O. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J. Biol. Chem. 2007, 282, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teyssier, C.; Ma, H.; Emter, R.; Kralli Aand Stallcup, M.R. Activation of nuclear receptor coactivator PGC-1α by arginine methylation. Genes Dev. 2005, 19, 1466–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Rhee, J.; Lin, J.D.; Wu, Z.D.; Yoon, C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; BLowell, B.; Spiegelman, B.M. Cytokine Stimulation of Energy Expenditure through p38 MAP Kinase Activation of PPARγ Coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Cao, W.H.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Spiegelman, B.M. Irisin ERKs the fat. Diabetes 2014, 63, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Sanchis-Gomar, F.; Perez-Quilis, C. p38–PGC-1α–irisin–betatrophin axisExploring new pathways in insulin resistance. Adipocyte 2014, 3, 67–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.W.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.X.; Wang, X.L.; Cui, T.X.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.L.; Zhu, H.H.; Xu, J.Q.; Yang, L.H.; Liu, L.J.; Li, J. β-arrestin-2 is involved in irisin induced glucose metabolism in type 2 diabetes via p38 MAPK signaling. Exp. Cell Res. 2017, 360, 199–204. [Google Scholar] [CrossRef]
- Yano, N.; Zhang, L.; Wei, D.; Dubielecka, P.M.; Wei, L.; Zhuang, S.; Zhu, P.; Qin, G.; Liu, P.Y.; Chin, Y.E.; et al. Irisin counteracts high glucose and fatty acid-induced cytotoxicity by preserving the AMPK-insulin receptor signaling axis in C2C12 myoblasts. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E791–E805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Yano, N.; Wang, H.; Zhao, Y.T.; Dubielecka, P.M.; Zhuang, S.; Chin, Y.E.; Qin, G.; Zhao, T.C. Sodium Butyrate Protects -Against High Fat Diet-Induced Cardiac Dysfunction and Metabolic Disorders in Type II Diabetic Mice. J. Cell Biochem. 2017, 118, 2395–2408. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Zhao, Y.T.; Wang, H.; Zhang, L.X.; Qin, G.; Zhuang, S.; Kadin, M.; Chin, Y.E.; Liu, P.Y.; Zhao, T.C. The Essential Role of PRAK in Preserving Cardiac Function and Insulin Resistance in High-Fat Diet-Induced Diabetes. Int. J. Mol. Sci. 2021, 22, 7995. https://doi.org/10.3390/ijms22157995
Du J, Zhao YT, Wang H, Zhang LX, Qin G, Zhuang S, Kadin M, Chin YE, Liu PY, Zhao TC. The Essential Role of PRAK in Preserving Cardiac Function and Insulin Resistance in High-Fat Diet-Induced Diabetes. International Journal of Molecular Sciences. 2021; 22(15):7995. https://doi.org/10.3390/ijms22157995
Chicago/Turabian StyleDu, Jianfeng, Yu Tina Zhao, Hao Wang, Ling X. Zhang, Gangjian Qin, Shougang Zhuang, Marshall Kadin, Y. Eugene Chin, Paul Y. Liu, and Ting C. Zhao. 2021. "The Essential Role of PRAK in Preserving Cardiac Function and Insulin Resistance in High-Fat Diet-Induced Diabetes" International Journal of Molecular Sciences 22, no. 15: 7995. https://doi.org/10.3390/ijms22157995
APA StyleDu, J., Zhao, Y. T., Wang, H., Zhang, L. X., Qin, G., Zhuang, S., Kadin, M., Chin, Y. E., Liu, P. Y., & Zhao, T. C. (2021). The Essential Role of PRAK in Preserving Cardiac Function and Insulin Resistance in High-Fat Diet-Induced Diabetes. International Journal of Molecular Sciences, 22(15), 7995. https://doi.org/10.3390/ijms22157995