
Computational Insights into the Deleterious Impacts of Missense Variants on N-Acetyl-d-glucosamine Kinase Structure and Function

 ,
, 
 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
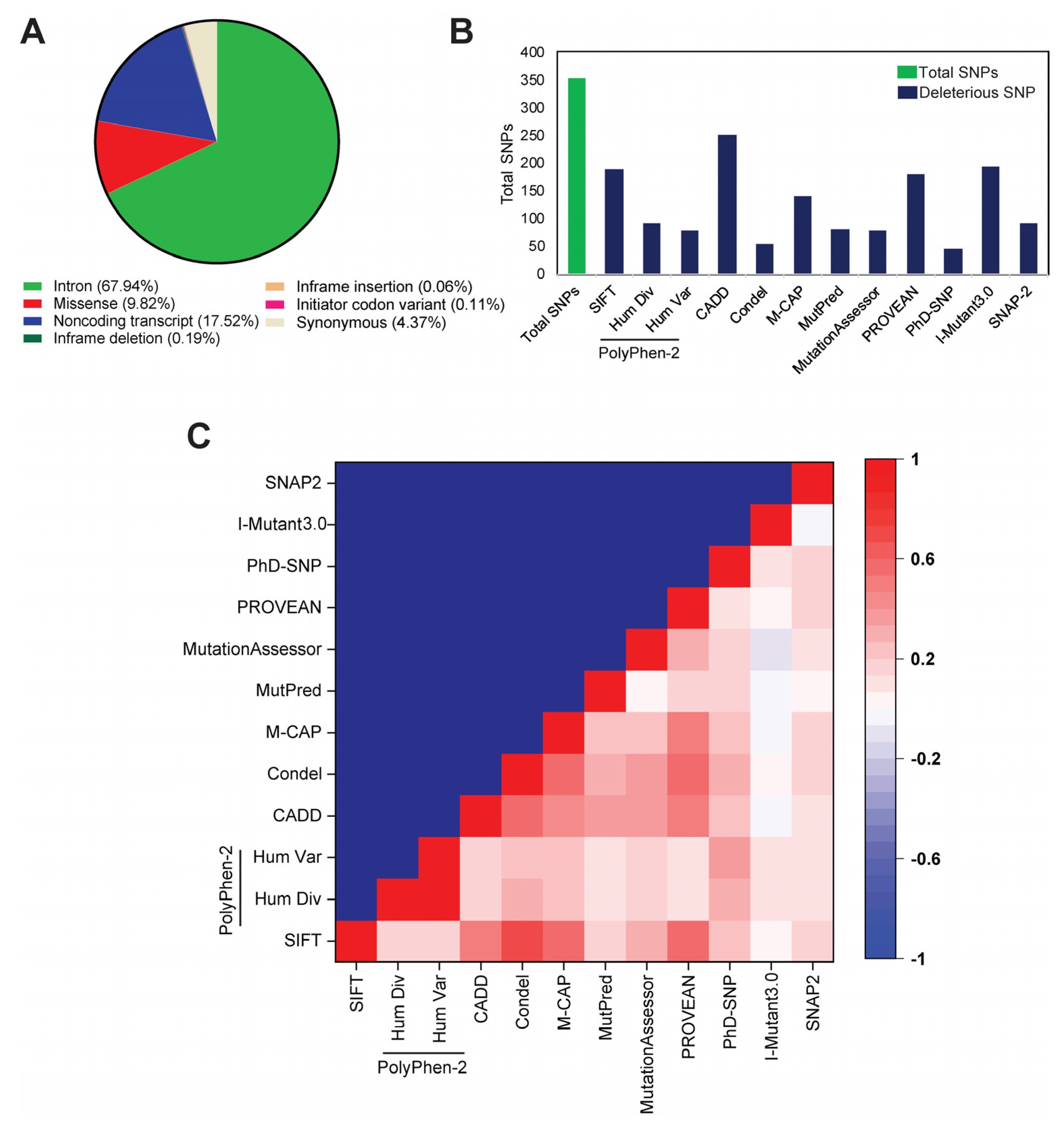
2.1. Identification of Deleterious nsSNP
2.2. Conservation Analysis
2.3. Molecular Dynamics (MD) Simulation
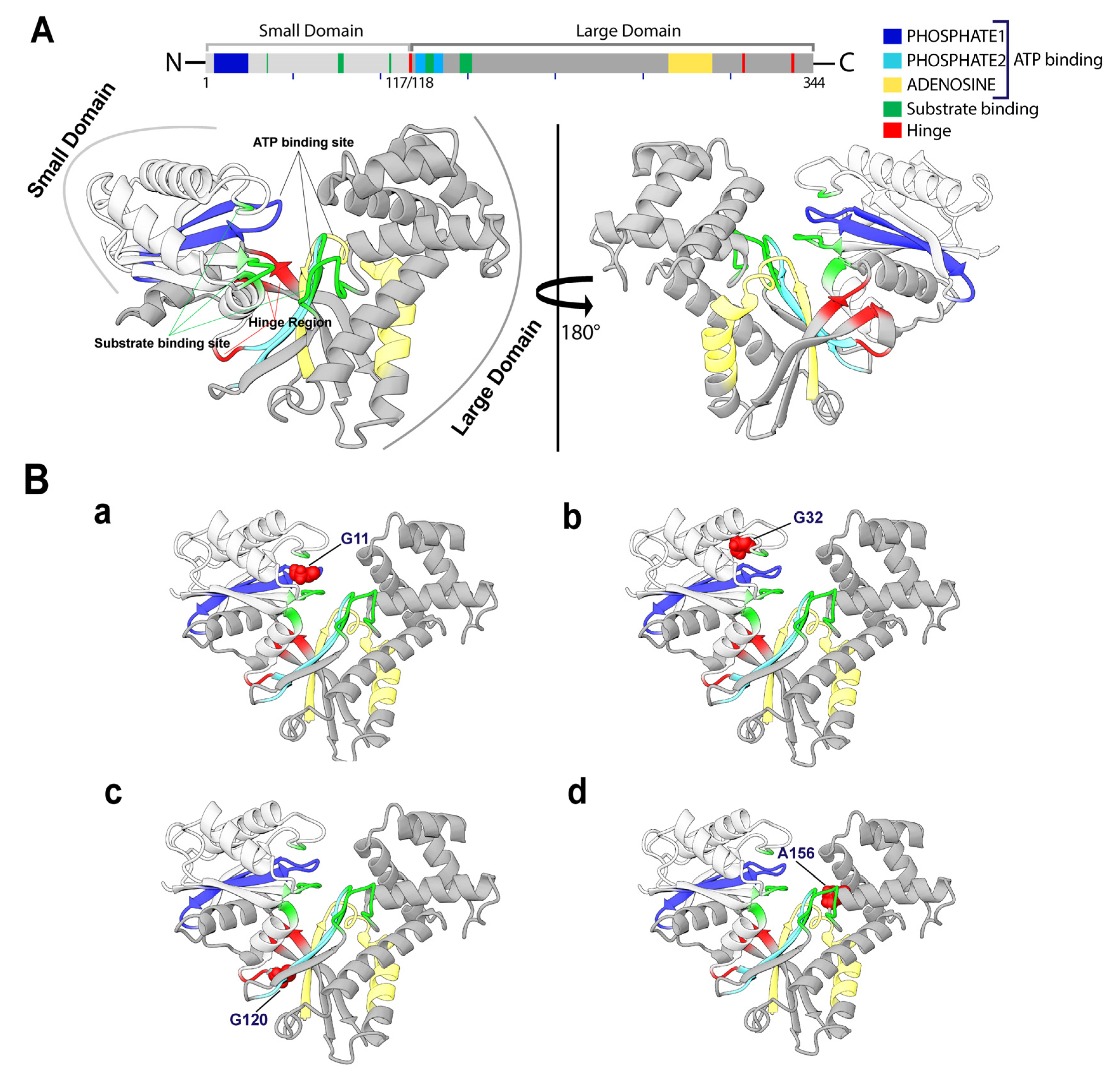
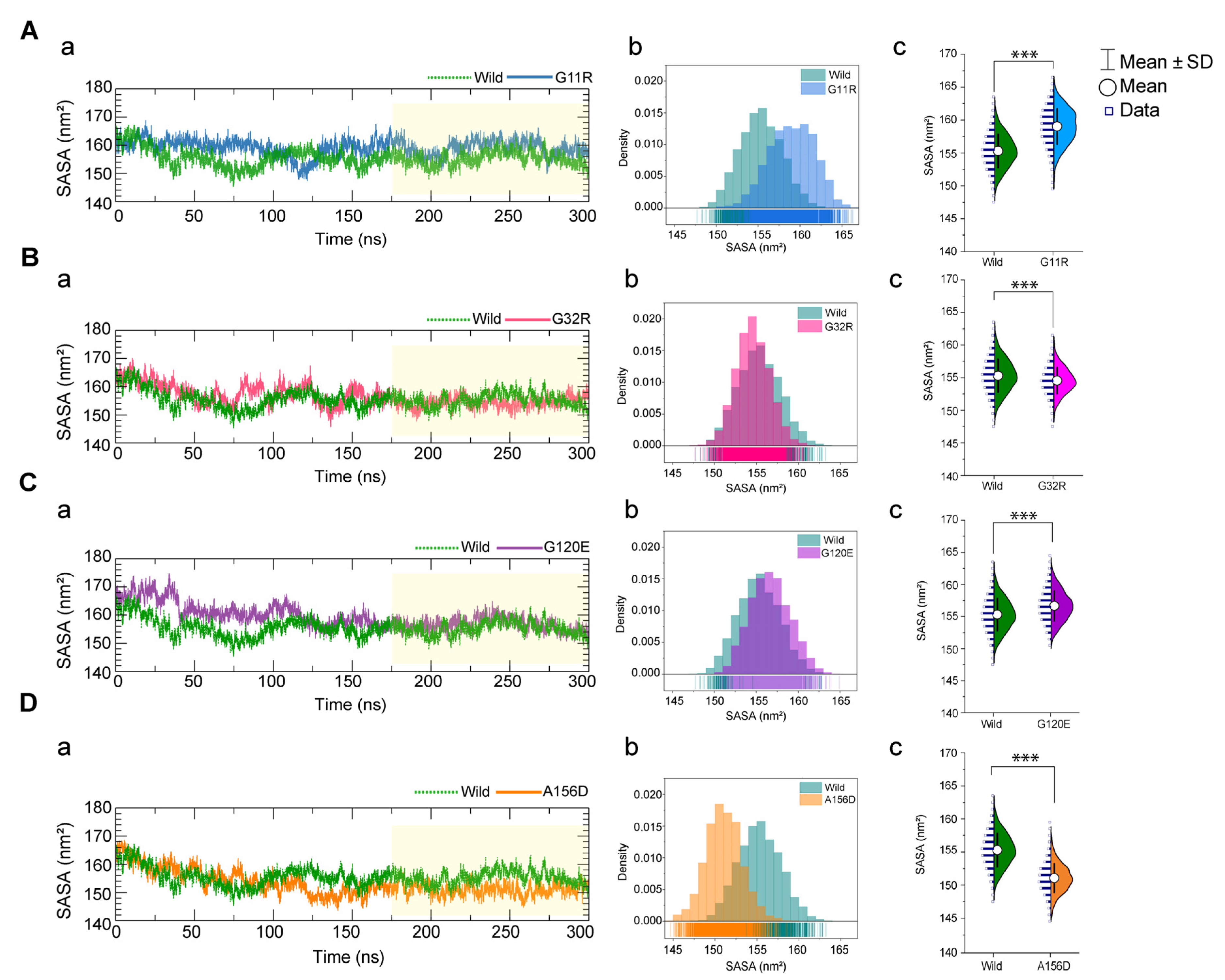
2.3.1. Effects of Variants on Conformational Dynamics
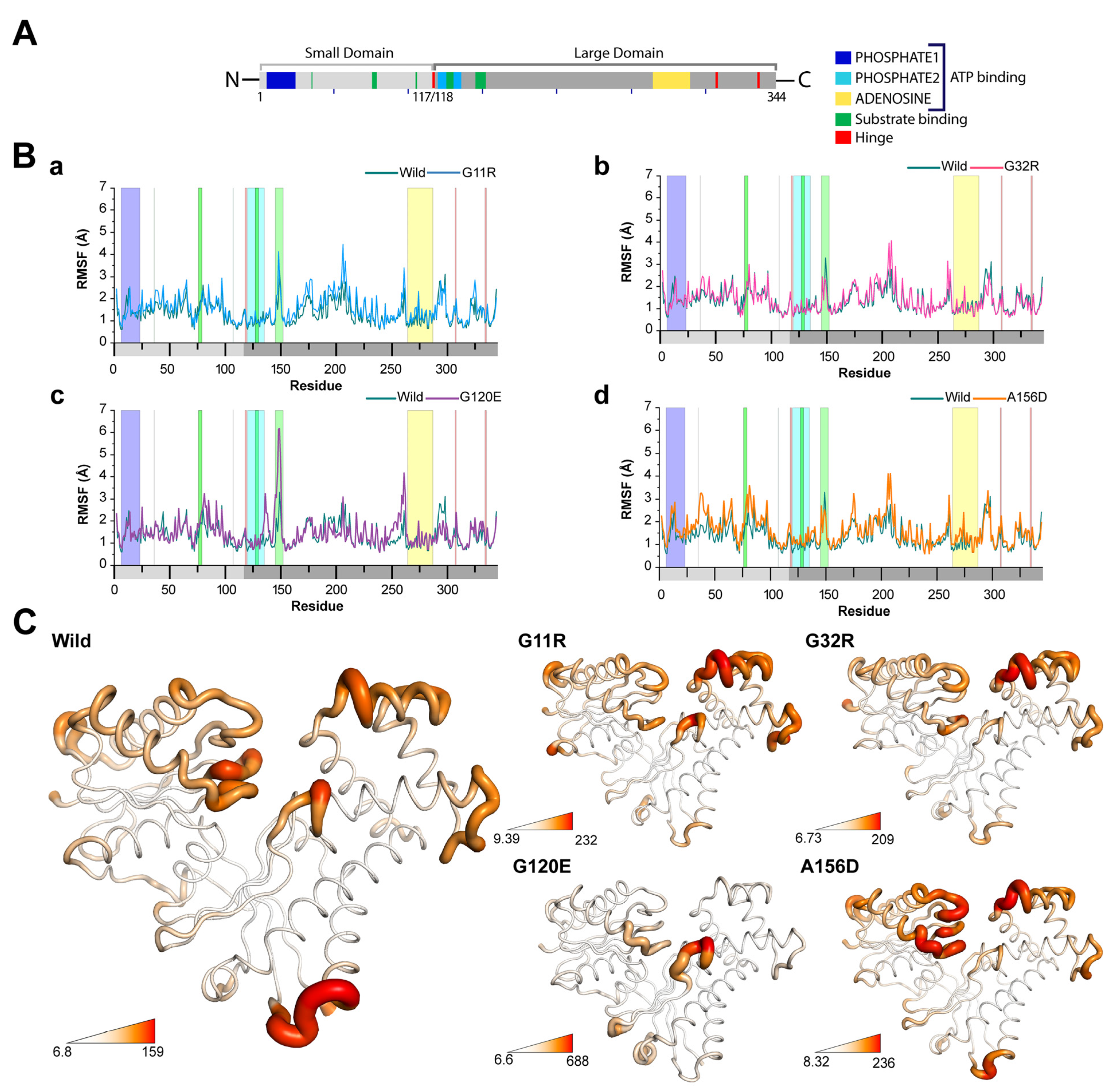
2.3.2. Effects of Variants on Protein Dynamics
2.3.3. Variants Alters NAGK Secondary Structural Organization
2.4. Impacts on Non-Canonical Functions
3. Discussion
4. Materials and Methods
4.1. Data Collection and Identification of Deleterious SNPs
4.2. Conservation Analysis
4.3. Molecular Dynamics (MD) Simulation
4.3.1. Preparation of Simulation System
4.3.2. Protein–Protein Docking and MM-GBSA Calculation
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharif, S.R.; Islam, A.; Moon, I.S. N-Acetyl-D-Glucosamine Kinase Interacts with Dynein-Lis1-NudE1 Complex and Regulates Cell Division. Mol. Cells 2016, 39, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderlich, S.; Oetke, C.; Pawlita, M. Biochemical engineering of sialic acids. In Handbook of Carbohydrate Engineering; CRC Press: Boca Raton, FL, USA, 2005; pp. 425–444. [Google Scholar]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [Green Version]
- Wells, L.; Vosseller, K.; Hart, G.W. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell. Mol. Life Sci. CMLS 2003, 60, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, S.R.; Lee, H.; Islam, M.A.; Seog, D.H.; Moon, I.S. N-acetyl-D-glucosamine kinase is a component of nuclear speckles and paraspeckles. Mol. Cells 2015, 38, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Cho, S.J.; Moon, I.S. The non-canonical effect of N-acetyl-D-glucosamine kinase on the formation of neuronal dendrites. Mol. Cells 2014, 37, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.; Chen, H.; Reutter, W.; Hinderlich, S. Structure and function of N-acetylglucosamine kinase. Identification of two active site cysteines. Eur. J. Biochem. 2002, 269, 4212–4218. [Google Scholar] [CrossRef]
- Blume, A.; Berger, M.; Benie, A.J.; Peters, T.; Hinderlich, S. Characterization of ligand binding to N-acetylglucosamine kinase studied by STD NMR. Biochemistry 2008, 47, 13138–13146. [Google Scholar] [CrossRef]
- Hurley, J.H. The sugar kinase/heat shock protein 70/actin superfamily: Implications of conserved structure for mechanism. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 137–162. [Google Scholar] [CrossRef]
- Lee, H.; Dutta, S.; Moon, I.S. Upregulation of dendritic arborization by N-acetyl-D-glucosamine kinase is not dependent on its kinase activity. Mol. Cells 2014, 37, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Sharif, S.R.; Lee, H.; Moon, I.S. N-acetyl-D-glucosamine kinase promotes the axonal growth of developing neurons. Mol. Cells 2015, 38, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.A.; Choi, H.J.; Dash, R.; Sharif, S.R.; Oktaviani, D.F.; Seog, D.H.; Moon, I.S. N-acetyl-D-Glucosamine kinase interacts with NudC and Lis1 in dynein motor complex and promotes cell migration. Int. J. Mol. Sci. 2020, 22, 129. [Google Scholar] [CrossRef]
- Ripon, M.K.H.; Lee, H.; Dash, R.; Choi, H.J.; Oktaviani, D.F.; Moon, I.S.; Haque, M.N. N-acetyl-D-glucosamine kinase binds dynein light chain roadblock 1 and promotes protein aggregate clearance. Cell Death Dis. 2020, 11, 619. [Google Scholar] [CrossRef] [PubMed]
- Arifuzzaman, M.; Mitra, S.; Das, R.; Hamza, A.; Absar, N.; Dash, R. In silico analysis of nonsynonymous single-nucleotide polymorphisms (nsSNPs) of the SMPX gene. Ann. Hum. Genet. 2020, 84, 54–71. [Google Scholar] [CrossRef]
- Dash, R.; Choi, H.J.; Moon, I.S. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Sci. Rep. 2020, 10, 3663. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Ali, M.C.; Rana, M.L.; Munni, Y.A.; Barua, L.; Jahan, I.; Haque, M.F.; Hannan, M.A.; Moon, I.S. Computational SNP Analysis and Molecular Simulation Revealed the Most Deleterious Missense Variants in the NBD1 Domain of Human ABCA1 Transporter. Int. J. Mol. Sci. 2020, 21, 7606. [Google Scholar] [CrossRef]
- Mah, J.T.; Low, E.S.; Lee, E. In silico SNP analysis and bioinformatics tools: A review of the state of the art to aid drug discovery. Drug Discov. Today 2011, 16, 800–809. [Google Scholar] [CrossRef]
- Sneha, P.; George Priya Doss, C. Chapter Seven—Molecular Dynamics: New Frontier in Personalized Medicine. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 102, pp. 181–224. [Google Scholar]
- Pal, L.R.; Moult, J. Genetic Basis of Common Human Disease: Insight into the Role of Missense SNPs from Genome-Wide Association Studies. J. Mol. Biol. 2015, 427, 2271–2289. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Dorajoo, R.; Chang, X.; Wang, L.; Khor, C.-C.; Sim, X.; Cheng, C.-Y.; Shi, Y.; Tham, Y.C.; Zhao, W.; et al. Genome-wide association study identifies a missense variant at APOA5 for coronary artery disease in Multi-Ethnic Cohorts from Southeast Asia. Sci. Rep. 2017, 7, 17921. [Google Scholar] [CrossRef] [Green Version]
- Kakar, M.U.; Matloob, M.; Dai, R.; Deng, Y.; Ullah, K.; Kakar, I.U.; Khaliq, G.; Umer, M.; Bhutto, Z.A.; Fazlani, S.A.; et al. In silico screening and identification of deleterious missense SNPs along with their effects on CD-209 gene: An insight to CD-209 related-diseases. PLoS ONE 2021, 16, e0247249. [Google Scholar] [CrossRef]
- Lee, P.H.; Shatkay, H. Ranking single nucleotide polymorphisms by potential deleterious effects. AMIA Annu. Symp. Proc. 2008, 2008, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.H.; Chrysina, E.D.; Irons, L.I.; Papageorgiou, A.C.; Acharya, K.R.; Brew, K. Role of conserved residues in structure and stability: Tryptophans of human serum retinol-binding protein, a model for the lipocalin superfamily. Protein Sci. A Publ. Protein Soc. 2001, 10, 2301–2316. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, M.A.; Blundell, T.L. Sequence and structure conservation in a protein core. Proteins 1998, 33, 358–366. [Google Scholar] [CrossRef]
- Hossain, M.S.; Roy, A.S.; Islam, M.S. In silico analysis predicting effects of deleterious SNPs of human RASSF5 gene on its structure and functions. Sci. Rep. 2020, 10, 14542. [Google Scholar] [CrossRef]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002, 65, 031910. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lazim, R. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci. Rep. 2017, 7, 44651. [Google Scholar] [CrossRef] [Green Version]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [Green Version]
- Ahrari, S.; Khosravi, F.; Osouli, A.; Sakhteman, A.; Nematollahi, A.; Ghasemi, Y.; Savardashtaki, A. MARK4 protein can explore the active-like conformations in its non-phosphorylated state. Sci. Rep. 2019, 9, 12967. [Google Scholar] [CrossRef]
- Amadei, A.; Ceruso, M.A.; Di Nola, A. On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins’ molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 1999, 36, 419–424. [Google Scholar] [CrossRef]
- Anwar, M.A.; Choi, S. Structure-Activity Relationship in TLR4 Mutations: Atomistic Molecular Dynamics Simulations and Residue Interaction Network Analysis. Sci. Rep. 2017, 7, 43807. [Google Scholar] [CrossRef] [Green Version]
- Yesudhas, D.; Anwar, M.A.; Panneerselvam, S.; Durai, P.; Shah, M.; Choi, S. Structural Mechanism behind Distinct Efficiency of Oct4/Sox2 Proteins in Differentially Spaced DNA Complexes. PLoS ONE 2016, 11, e0147240. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Sharif, S.R.; Lee, H.; Seog, D.H.; Moon, I.S. N-acetyl-D-glucosamine kinase interacts with dynein light-chain roadblock type 1 at Golgi outposts in neuronal dendritic branch points. Exp. Mol. Med. 2015, 47, e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanwar, H.; Kumar, D.T.; Doss, C.G.P.; Zayed, H. Bioinformatics classification of mutations in patients with Mucopolysaccharidosis IIIA. Metab. Brain Dis. 2019, 34, 1577–1594. [Google Scholar] [CrossRef] [Green Version]
- Feder, M.E.; Mitchell-Olds, T. Evolutionary and ecological functional genomics. Nat. Rev. Genet. 2003, 4, 649–655. [Google Scholar] [CrossRef]
- Adebali, O.; Reznik, A.O.; Ory, D.S.; Zhulin, I.B. Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations. Genet. Med. 2016, 18, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihofen, W.A.; Berger, M.; Chen, H.; Saenger, W.; Hinderlich, S. Structures of Human N-Acetylglucosamine Kinase in Two Complexes with N-Acetylglucosamine and with ADP/Glucose: Insights into Substrate Specificity and Regulation. J. Mol. Biol. 2006, 364, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Curtis, R.A.; Warwicker, J. Soluble expression of proteins correlates with a lack of positively-charged surface. Sci. Rep. 2013, 3, 3333. [Google Scholar] [CrossRef]
- Dean, H.B.; Roberson, E.D.; Song, Y. Neurodegenerative Disease-Associated Variants in TREM2 Destabilize the Apical Ligand-Binding Region of the Immunoglobulin Domain. Front. Neurol. 2019, 10, 1252. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Dakal, T.C.; Kala, D.; Dhiman, G.; Yadav, V.; Krokhotin, A.; Dokholyan, N.V. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci. Rep. 2017, 7, 6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Ali Mohamoud, H.S.; Manwar Hussain, M.R.; El-Harouni, A.A.; Shaik, N.A.; Qasmi, Z.U.; Merican, A.F.; Baig, M.; Anwar, Y.; Asfour, H.; Bondagji, N.; et al. First Comprehensive n Silico Analysis of the Functional and Structural Consequences of SNPs in Human GalNAc-T1 Gene. Comput. Math. Methods Med. 2014, 2014, 904052. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc. Hum. Genet. 2013, 76. [Google Scholar] [CrossRef] [Green Version]
- Nailwal, M.; Chauhan, J.B. In silico analysis of non-synonymous single nucleotide polymorphisms in human DAZL gene associated with male infertility. Syst. Biol. Reprod. Med. 2017, 63, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Sunyaev, S.R.; Eisenhaber, F.; Rodchenkov, I.V.; Eisenhaber, B.; Tumanyan, V.G.; Kuznetsov, E.N. PSIC: Profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. 1999, 12, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.M. Identification and analysis of pathogenic nsSNPs in human LSP1 gene. Bioinformation 2019, 15, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, B.; Krishnan, V.G.; Mort, M.E.; Xin, F.; Kamati, K.K.; Cooper, D.N.; Mooney, S.D.; Radivojac, P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 2009, 25, 2744–2750. [Google Scholar] [CrossRef] [Green Version]
- Hepp, D.; Gonçalves, G.L.; de Freitas, T.R.O. Prediction of the damage-associated non-synonymous single nucleotide polymorphisms in the human MC1R gene. PLoS ONE 2015, 10, e0121812. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Frousios, K.; Iliopoulos, C.S.; Schlitt, T.; Simpson, M.A. Predicting the functional consequences of non-synonymous DNA sequence variants—Evaluation of bioinformatics tools and development of a consensus strategy. Genomics 2013, 102, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, H.A.; Albkrye, A.M.S.; AbdAlla, B.M.; Khair, M.A.M.; Abdelwahid, N.; Elnasri, H.A. Computational determination of human PPARG gene: SNPs and prediction of their effect on protein functions of diabetic patients. Clin. Transl. Med. 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Huang, C.; Wang, Z.; Lv, H.; Li, X. In silico analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in the human GJA3 gene associated with congenital cataract. BMC Mol. Cell Biol. 2020, 21, 12. [Google Scholar] [CrossRef]
- Bromberg, Y.; Yachdav, G.; Rost, B. SNAP predicts effect of mutations on protein function. Bioinformatics 2008, 24, 2397–2398. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.A.; Solayman, M.; Paul, S.; Saha, M.; Khalil, M.I.; Gan, S.H. Impacts of Nonsynonymous Single Nucleotide Polymorphisms of Adiponectin Receptor 1 Gene on Corresponding Protein Stability: A Computational Approach. Biomed. Res. Int. 2016, 2016, 9142190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.; Ali, M.C.; Dash, N.; Azad, M.A.K.; Hosen, S.M.Z.; Hannan, M.A.; Moon, I.S. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int. J. Mol. Sci. 2019, 20, 6256. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.; Arifuzzaman, M.; Mitra, S.; Abdul Hannan, M.; Absar, N.; Hosen, S.M.Z. Unveiling the Structural Insights into the Selective Inhibition of Protein Kinase D1. Curr. Pharm. Des. 2019, 25, 1059–1074. [Google Scholar] [CrossRef]
- Ali, M.C.; Khatun, M.S.; Jahan, S.I.; Das, R.; Munni, Y.A.; Rahman, M.M.; Dash, R. In silico design of epitope-based peptide vaccine against non-typhoidal Salmonella through immunoinformatic approaches. J. Biomol. Struct. Dyn. 2021, 1–19. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Haque, M.N.; Hannan, M.A.; Dash, R.; Choi, S.M.; Moon, I.S. The potential LXRβ agonist stigmasterol protects against hypoxia/reoxygenation injury by modulating mitophagy in primary hippocampal neurons. Phytomed. Int. J. Phytother. Phytopharm. 2021, 81, 153415. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices - from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Hosen, S.Z.; Dash, R.; Junaid, M.; Mitra, S.; Absar, N. Identification and structural characterization of deleterious non-synonymous single nucleotide polymorphisms in the human SKP2 gene. Comput. Biol. Chem. 2019, 79, 127–136. [Google Scholar] [CrossRef]
- Dash, R.; Junaid, M.; Mitra, S.; Arifuzzaman, M.; Hosen, S.Z. Structure-based identification of potent VEGFR-2 inhibitors from in vivo metabolites of a herbal ingredient. J. Mol. Model. 2019, 25, 98. [Google Scholar] [CrossRef]
- Mitra, S.; Dash, R.J. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J. Mol. Graph. Model. 2018, 83, 42–52. [Google Scholar]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Dash, R.; Haque, M.N.; Choi, S.M.; Moon, I.S. Integrated System Pharmacology and In Silico Analysis Elucidating Neuropharmacological Actions of Withania somnifera in the Treatment of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 541–556. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G.J. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G.J. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K.J. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dash, R.; Das, R.; Junaid, M.; Akash, M.F.C.; Islam, A.; Hosen, S.Z. In silico-based vaccine design against Ebola virus glycoprotein. Adv. Appl. Bioinform. Chem. AABC 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Fukuda, I.; Nakamura, H. A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer–DNA Complex. PLoS ONE 2014, 9, e112419. [Google Scholar] [CrossRef] [Green Version]
- Merlino, A.; Vitagliano, L.; Ceruso, M.A.; Mazzarella, L. Subtle functional collective motions in pancreatic-like ribonucleases: From ribonuclease A to angiogenin. Proteins Struct. Funct. Bioinform. 2003, 53, 101–110. [Google Scholar] [CrossRef]
- Skjærven, L.; Yao, X.-Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2000, 62, 8438–8448. [Google Scholar] [CrossRef] [Green Version]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.G.; Fernandez-Recio, J.; Bates, P.A. SwarmDock: A server for flexible protein-protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [Green Version]
- Moal, I.H.; Bates, P.A. SwarmDock and the use of normal modes in protein-protein docking. Int. J. Mol. Sci. 2010, 11, 3623–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, R.; Mitra, S.; Munni, Y.A.; Choi, H.J.; Ali, M.C.; Barua, L.; Jang, T.J.; Moon, I.S. Computational Insights into the Deleterious Impacts of Missense Variants on N-Acetyl-d-glucosamine Kinase Structure and Function. Int. J. Mol. Sci. 2021, 22, 8048. https://doi.org/10.3390/ijms22158048
Dash R, Mitra S, Munni YA, Choi HJ, Ali MC, Barua L, Jang TJ, Moon IS. Computational Insights into the Deleterious Impacts of Missense Variants on N-Acetyl-d-glucosamine Kinase Structure and Function. International Journal of Molecular Sciences. 2021; 22(15):8048. https://doi.org/10.3390/ijms22158048
Chicago/Turabian StyleDash, Raju, Sarmistha Mitra, Yeasmin Akter Munni, Ho Jin Choi, Md. Chayan Ali, Largess Barua, Tae Jung Jang, and Il Soo Moon. 2021. "Computational Insights into the Deleterious Impacts of Missense Variants on N-Acetyl-d-glucosamine Kinase Structure and Function" International Journal of Molecular Sciences 22, no. 15: 8048. https://doi.org/10.3390/ijms22158048