
Characterization of the Heat-Stable Proteome during Seed Germination in Arabidopsis with Special Focus on LEA Proteins

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
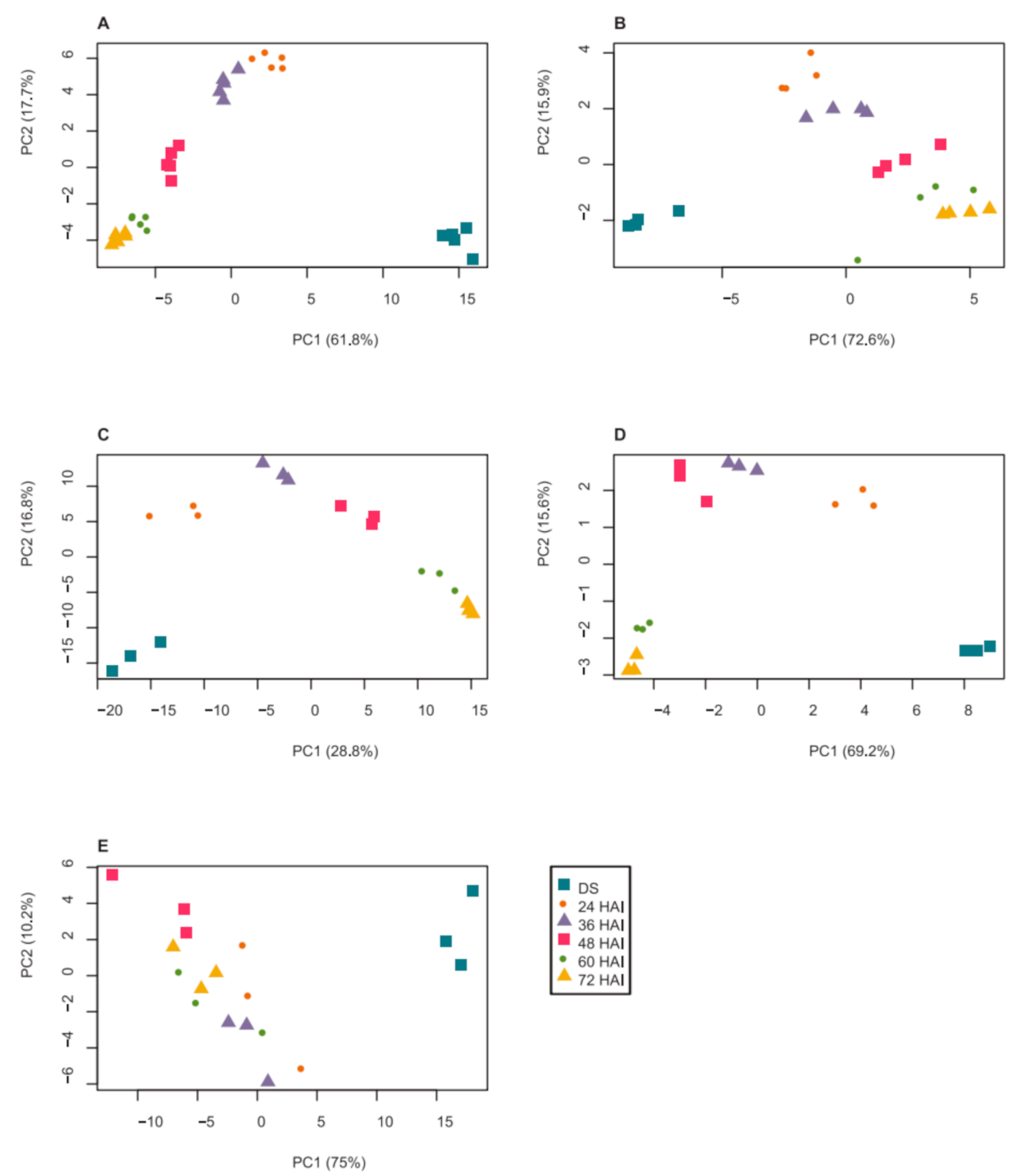
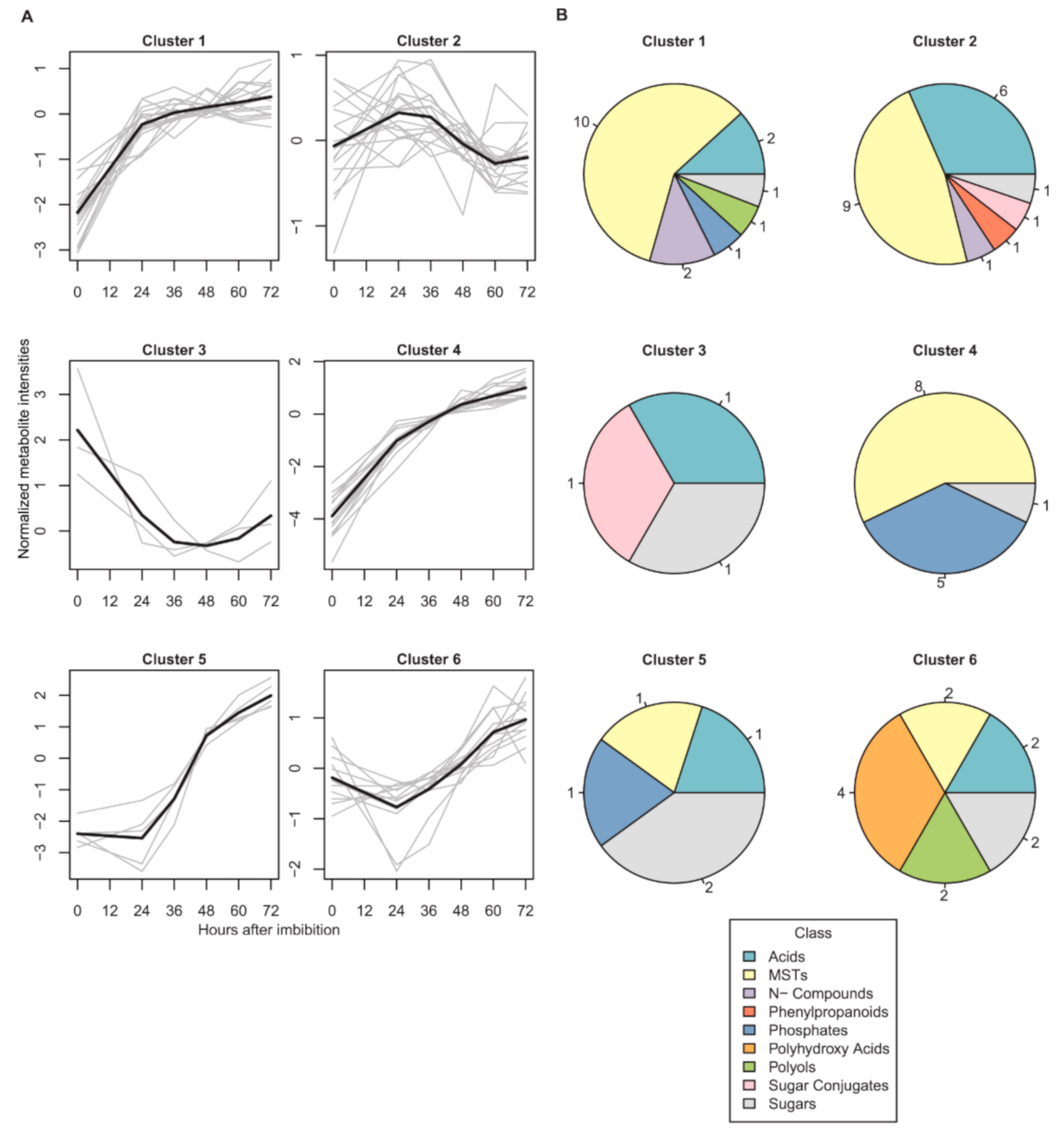
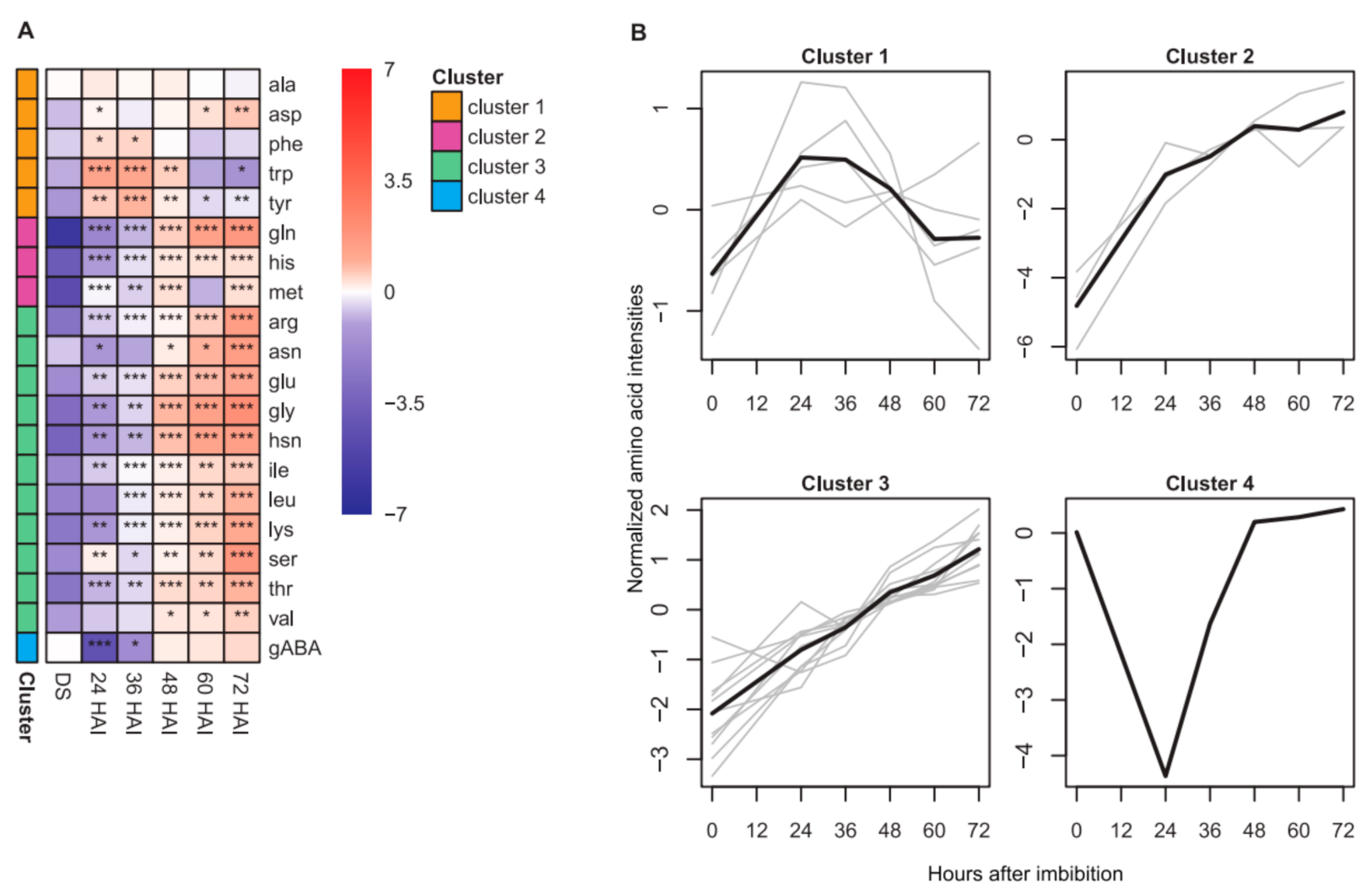
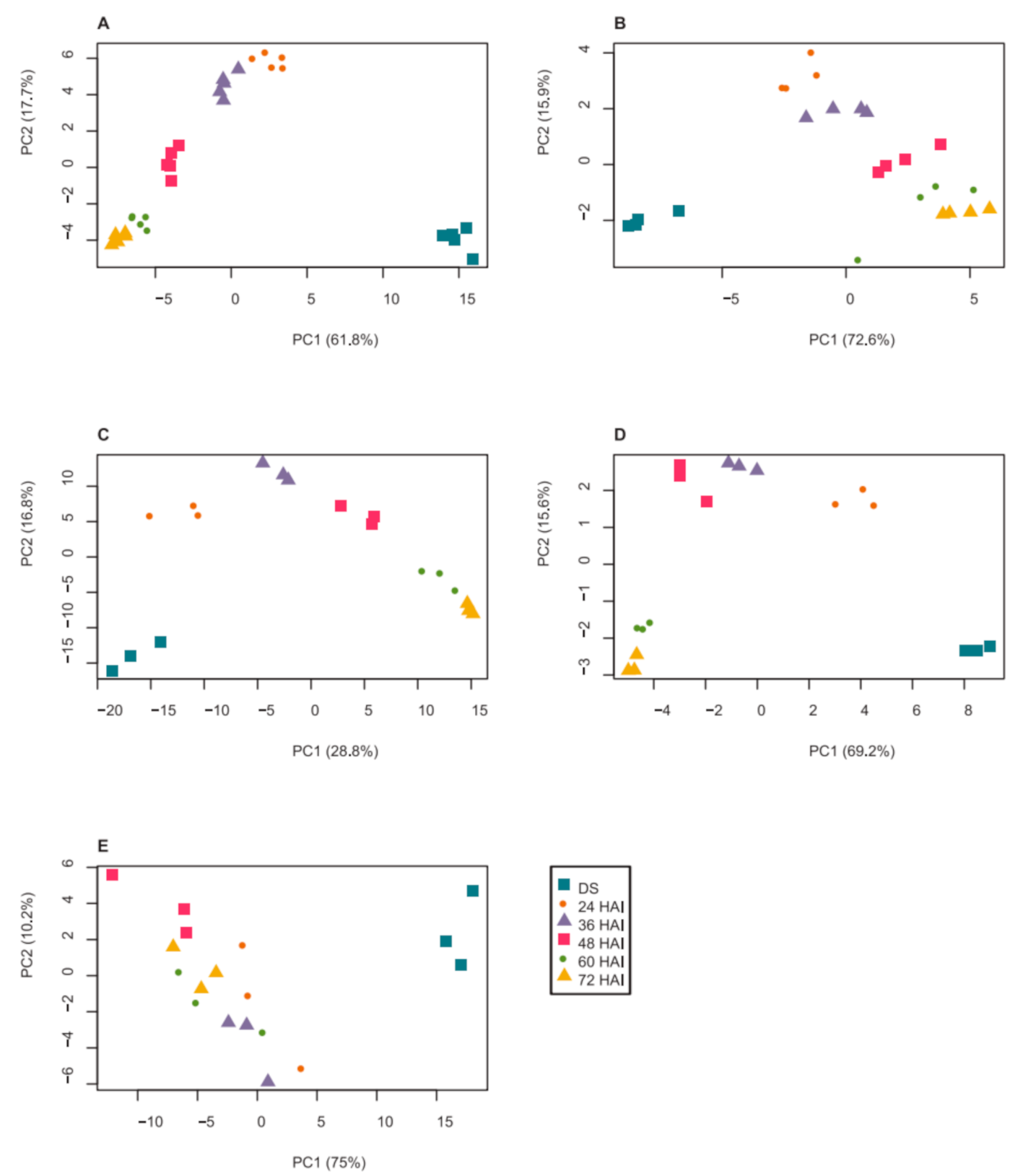
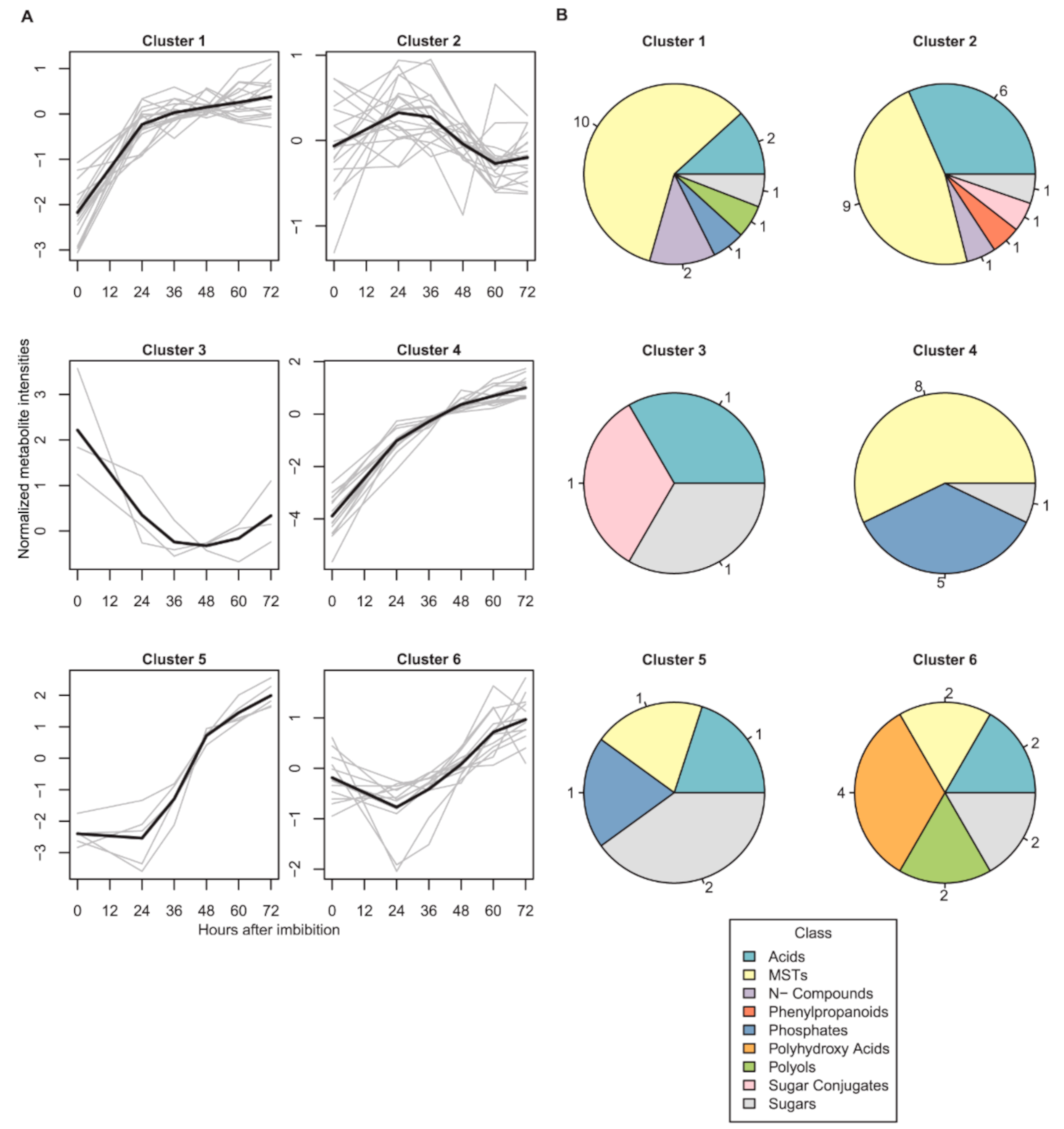
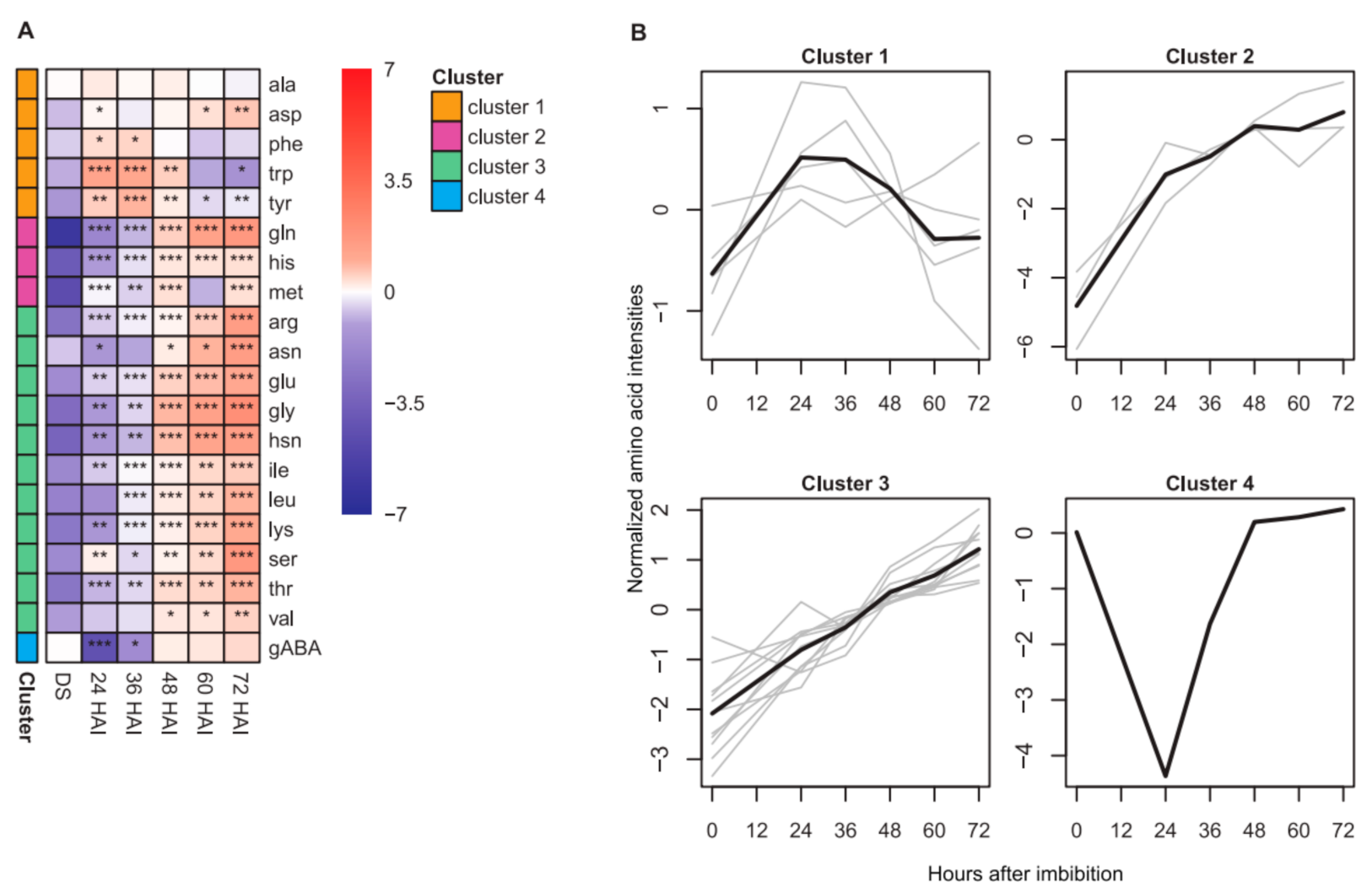
2.1. Dynamics of Seed Metabolites and Amino Acids after Imbibition
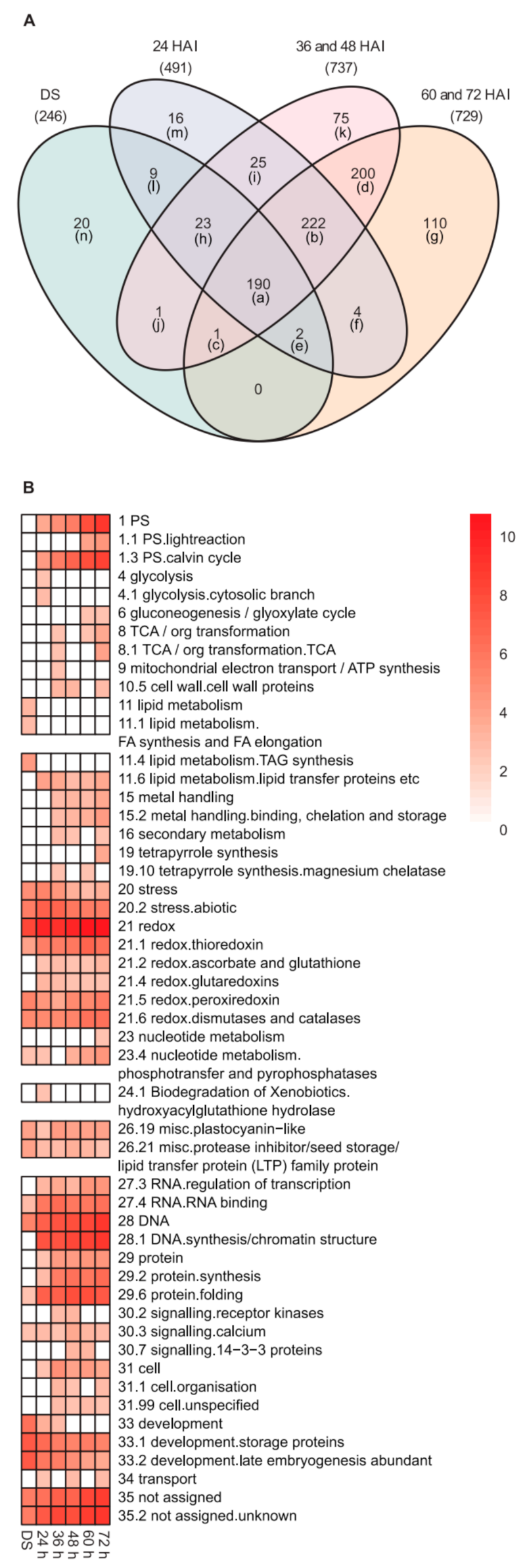
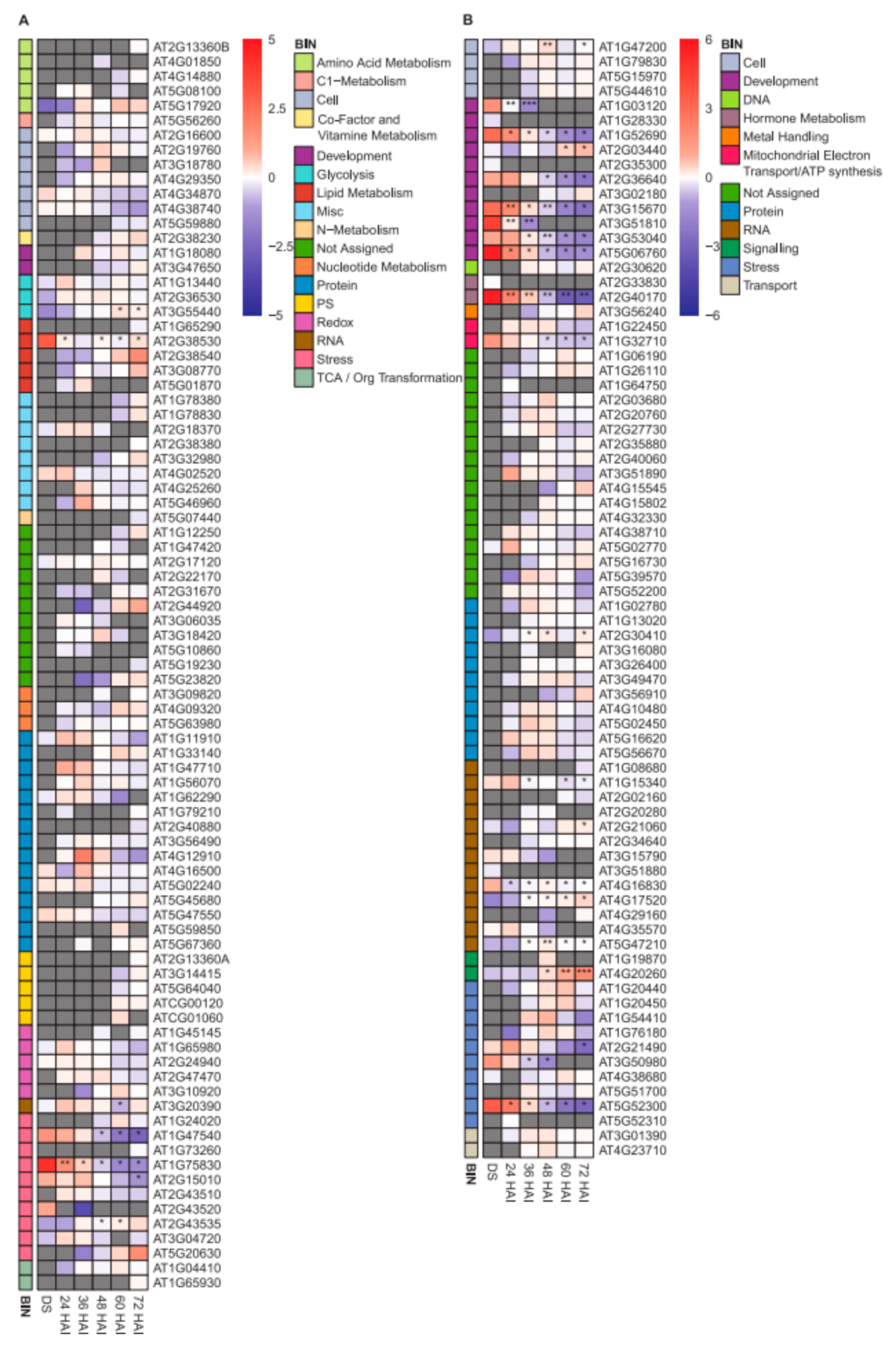
2.2. Changes in the Heat-Stable Proteome during Germination and Seedling Development
2.3. The Heat-Stable Proteome Exhibited an Equal Distribution of Intrinsically Ordered and Disordered Proteins
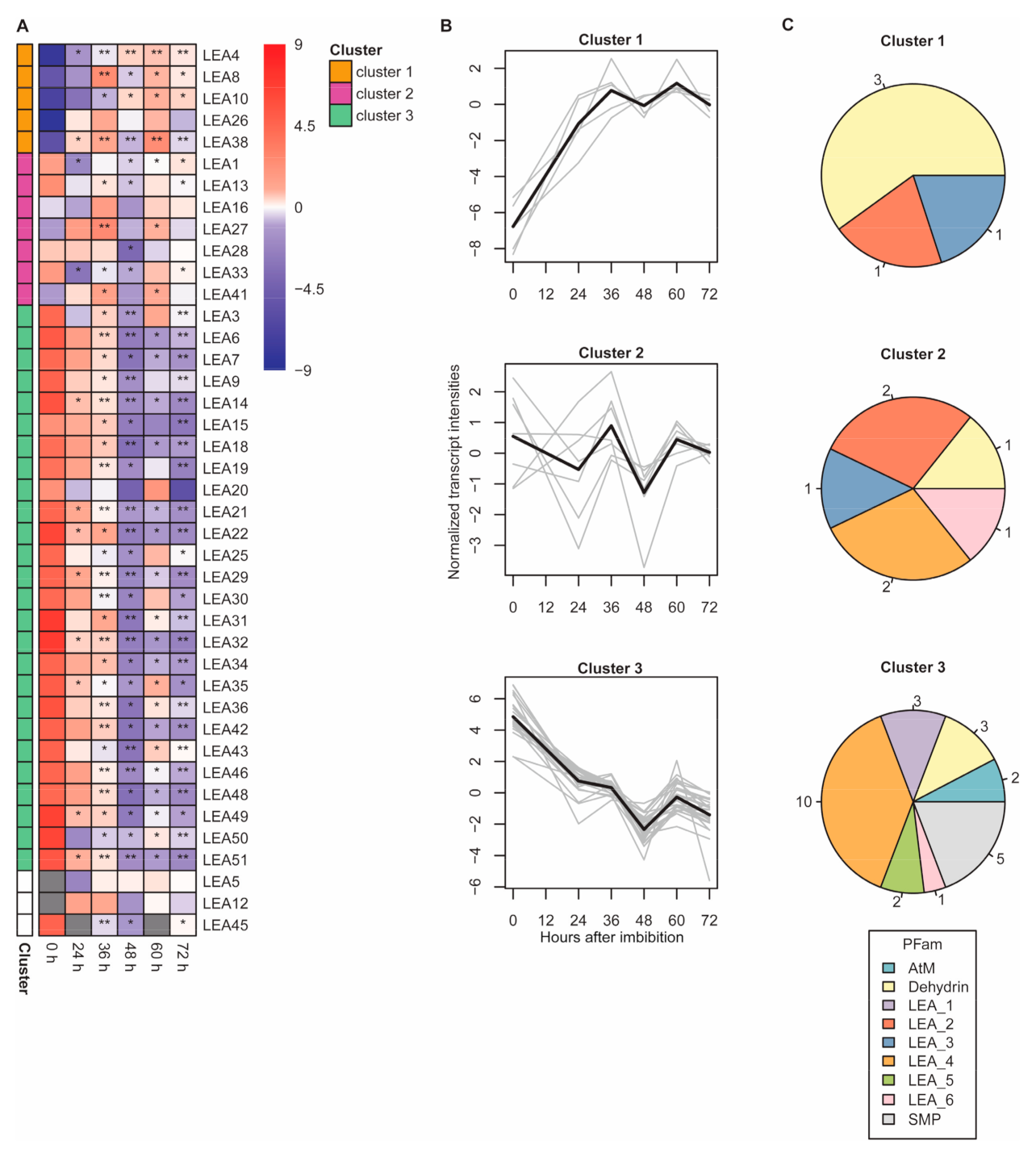
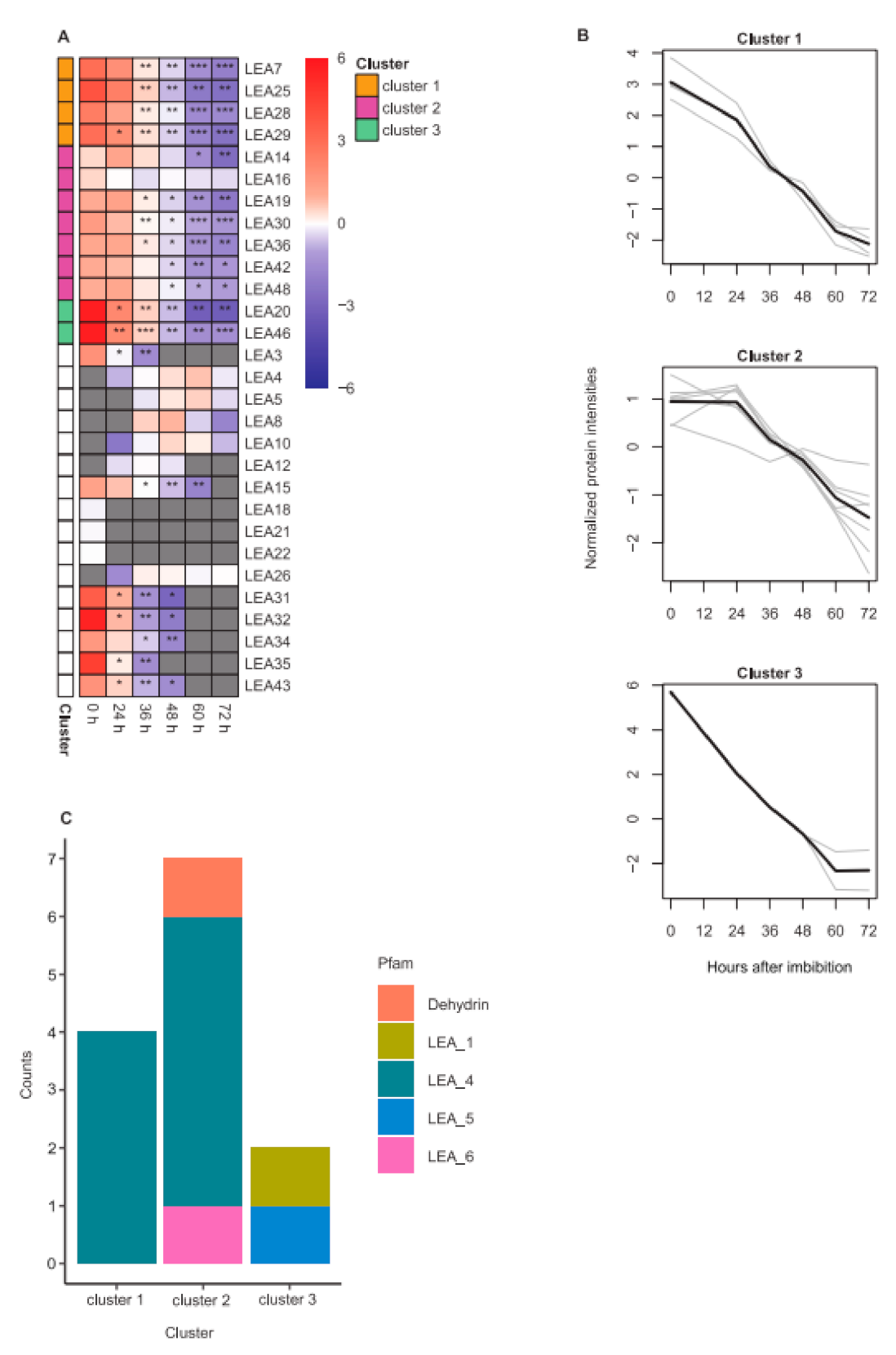
2.4. LEA Protein Dynamics during Seed Germination
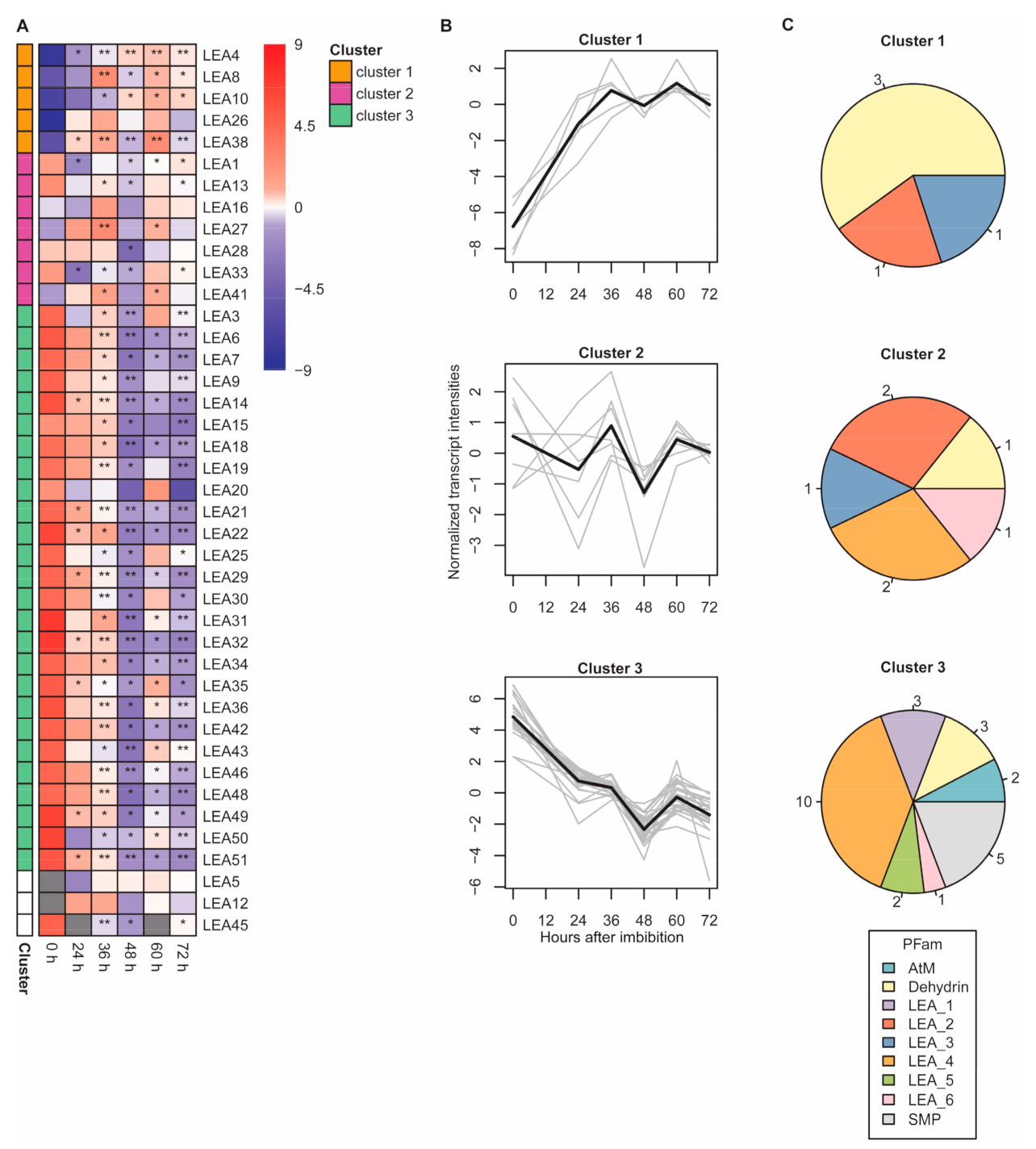
2.5. Transcript Abundance of Genes Encoding LEA Proteins
2.6. Protein–Metabolite Network Analyses Reveal a LEA Cluster with High Connectivity
3. Discussion
3.1. Degradation of Storage Compounds to Central Metabolites Contributes to the Transition from Heterotrophic to Autotrophic Growth
3.2. Thermal Stability of Proteins Is Related to Biological Processes
3.3. An Equal Distribution of Fully Ordered and Fully Intrinsically Disordered Proteins Characterized a Part of the Heat-Stable Proteome of Germinating Seeds
3.4. Dehydrins Were Identified as Seed Germination-Specific LEA Proteins
3.5. LEA Proteins Build a Highly Correlated Cluster in the Seed Germination Specific Common Protein–Metabolite Network
4. Materials and Methods
4.1. Plant Materials and Experimental Conditions
4.2. Metabolite Profiling
4.3. Amino Acid Analysis
4.4. Proteomic Analysis of Heat-Stable Proteins
4.5. qRT-PCR Analysis
4.6. Statistical and Other Analyses
4.7. Network Analysis of Common Metabolites, Amino Acids, Heat-Stable Proteins
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maia, J.; Dekkers, B.J.W.; Provart, N.J.; Ligterink, W.; Hilhorst, H.W.M. The Re-Establishment of Desiccation Tolerance in Germinated Arabidopsis thaliana Seeds and Its Associated Transcriptome. PLoS ONE 2011, 6, e29123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daws, M.; Bolton, S.; Burslem, D.F.R.P.; Garwood, N.C.; Mullins, C.E. Loss of desiccation tolerance during germinationin neo-tropical pioneer seeds: Implications forseed mortality and germination characteristics. Seed Sci. Res. 2007, 17, 273–281. [Google Scholar] [CrossRef]
- Buitink, J.; Vu, B.L.; Satour, P.; Leprince, O. The re-establishment of desiccation tolerance in germinated radicles ofMedicago truncatulaGaertn. seeds. Seed Sci. Res. 2003, 13, 273–286. [Google Scholar] [CrossRef]
- Oliver, M.J.; Farrant, J.M.; Hilhorst, H.W.; Mundree, S.; Williams, B.; Bewley, J.D. Desiccation Tolerance: Avoiding Cellular Damage During Drying and Rehydration. Annu. Rev. Plant Biol. 2020, 71, 435–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Nonogaki, H.; Bassel, G.; Bewley, D. Germination—Still a mystery. Plant Sci. 2010, 179, 574–581. [Google Scholar] [CrossRef]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Gorlach, J. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Fait, A.; Angelovici, R.; Less, H.; Ohad, I.; Urbanczyk-Wochniak, E.; Fernie, A.R.; Galili, G. Arabidopsis Seed Development and Germination Is Associated with Temporally Distinct Metabolic Switches. Plant Physiol. 2006, 142, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasulu, N.; Usadel, B.; Winter, A.; Radchuk, V.; Scholz, U.; Stein, N.; Weschke, W.; Strickert, M.; Close, T.J.; Stitt, M.; et al. Barley Grain Maturation and Germination: Metabolic Pathway and Regulatory Network Commonalities and Differences Highlighted by New MapMan/PageMan Profiling Tools. Plant Physiol. 2008, 146, 1738–1758. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Moing, A.; Ebbels, T.M.; Maucourt, M.; Tomos, A.D.; Rolin, D.; Hooks, M.A. Correlation Network Analysis reveals a sequential reorganization of metabolic and transcriptional states during germination and gene-metabolite relationships in developing seedlings of Arabidopsis. BMC Syst. Biol. 2010, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.T.; Ligterink, W.; Hilhorst, H.W.M. Metabolite profiling and associated gene expression reveal two metabolic shifts during the seed-to-seedling transition in Arabidopsis thaliana. Plant Mol. Biol. 2017, 95, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, K.; Okamoto, M.; Koshiba, T.; Kamiya, Y.; Nambara, E. Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: Epigenetic and genetic regulation of transcription in seed. Plant J. 2005, 41, 697–709. [Google Scholar] [CrossRef]
- Silva, A.T.; Ribone, P.A.; Chan, R.L.; Ligterink, W.; Hilhorst, H.W. A Predictive Coexpression Network Identifies Novel Genes Controlling the Seed-to-Seedling Phase Transition in Arabidopsis thaliana. Plant Physiol. 2016, 170, 2218–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, B.; Peviani, A.; Van Der Horst, S.; Gamm, M.; Snel, B.; Bentsink, L.; Hanson, J. Extensive translational regulation during seed germination revealed by polysomal profiling. New Phytol. 2016, 214, 233–244. [Google Scholar] [CrossRef]
- Bai, B.; Van Der Horst, S.; Cordewener, J.H.; America, T.A.H.P.; Hanson, J.; Bentsink, L. Seed-Stored mRNAs that Are Specifically Associated to Monosomes Are Translationally Regulated during Germination. Plant Physiol. 2020, 182, 378–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Zhen, S.; Zhu, G.; Bian, Y.; Yan, Y. Comparative metabolome analysis of wheat embryo and endosperm reveals the dynamic changes of metabolites during seed germination. Plant Physiol. Biochem. 2017, 115, 320–327. [Google Scholar] [CrossRef]
- Kazmi, R.H.; Willems, L.A.J.; Joosen, R.V.L.; Khan, N.; Ligterink, W.; Hilhorst, H.W.M. Metabolomic analysis of tomato seed germination. Metabolomics 2017, 13, 145. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, K.; Job, C.; Groot, S.P.; Puype, M.; Demol, H.; Vandekerckhove, J.; Job, D. Proteomic Analysis of Arabidopsis Seed Germination and Priming. Plant Physiol. 2001, 126, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Rajjou, L.; Gallardo, K.; Debeaujon, I.; Vandekerckhove, J.; Job, C.; Job, D. The Effect of α-Amanitin on the Arabidopsis Seed Proteome Highlights the Distinct Roles of Stored and Neosynthesized mRNAs during Germination. Plant Physiol. 2004, 134, 1598–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Wang, B.-C.; Jin, X.; Li, H.-B.; Han, P.; Wei, K.-H.; Zhang, X.-M.; Zhu, Y.-X. Proteomic Analysis and Extensive Protein Identification from Dry, Germinating Arabidopsis Seeds and Young Seedlings. BMB Rep. 2005, 38, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Li, X.; Wang, X.; Chen, H.; Chen, F.; Shen, S. Proteomic analysis of rice (Oryza sativa) seeds during germination. Proteomics 2007, 7, 3358–3368. [Google Scholar] [CrossRef]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic Proteomics Emphasizes the Importance of Selective mRNA Translation and Protein Turnover during Arabidopsis Seed Germination. Mol. Cell. Proteom. 2014, 13, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Chao, H.; Gan, L.; Guo, L.; Zhang, K.; Li, Y.; Wang, H.; Raboanatahiry, N.; Li, M. Proteomic Dissection of Seed Germination and Seedling Establishment in Brassica napus. Front. Plant Sci. 2016, 7, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, F.K.; Doner, N.M.; Krawczyk, H.E.; Scholz, P.; Schmitt, K.; Valerius, O.; Braus, G.H.; Mullen, R.T.; Ischebeck, T. Identification of Low-Abundance Lipid Droplet Proteins in Seeds and Seedlings. Plant Physiol. 2020, 182, 1326–1345. [Google Scholar] [CrossRef] [Green Version]
- Sano, N.; Permana, H.; Kumada, R.; Shinozaki, Y.; Tanabata, T.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Proteomic Analysis of Embryonic Proteins Synthesized from Long-Lived mRNAs During Germination of Rice Seeds. Plant Cell Physiol. 2012, 53, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Takebayashi, Y.; To, A.; Mhiri, C.; Rajjou, L.; Nakagami, H.; Kanekatsu, M. Shotgun Proteomic Analysis Highlights the Roles of Long-Lived mRNAs and De Novo Transcribed mRNAs in Rice Seeds upon Imbibition. Plant Cell Physiol. 2019, 60, 2584–2596. [Google Scholar] [CrossRef] [PubMed]
- Obendorf, R.L. Oligosaccharides and galactosyl cyclitols in seed desiccation tolerance. Seed Sci. Res. 1997, 7, 63–74. [Google Scholar] [CrossRef]
- Ballesteros, D.; Walters, C. Detailed characterization of mechanical properties and molecular mobility within dry seed glasses: Relevance to the physiology of dry biological systems. Plant J. 2011, 68, 607–619. [Google Scholar] [CrossRef]
- Ballesteros, D.; Walters, C. Solid-State Biology and Seed Longevity: A Mechanical Analysis of Glasses in Pea and Soybean Embryonic Axes. Front. Plant Sci. 2019, 10, 920. [Google Scholar] [CrossRef] [Green Version]
- Buitink, J.; Leprince, O. Glass formation in plant anhydrobiotes: Survival in the dry state. Cryobiology 2004, 48, 215–228. [Google Scholar] [CrossRef]
- Roberts, J.K.; DeSimone, N.A.; Lingle, W.L.; Dure, L., 3rd. Cellular concentrations and uniformity of cell-type accumulation of two LEA proteins in cotton embryos. Plant Cell 1993, 5, 769–780. [Google Scholar] [CrossRef]
- Dure, L.; Chlan, C. Developmental biochemistry of cottonseed embryogenesis and germination: Xii. Purification and properties of principal storage proteins. Plant Physiol. 1981, 68, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Dure, L.; Galau, G.A. Developmental biochemistry of cottonseed embryogenesis and germination: Xiii. Regulation of biosynthesis of principal storage proteins. Plant Physiol. 1981, 68, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Dure, L., 3rd; Greenway, S.C.; Galau, G.A. Developmental biochemistry of cottonseed embryogenesis and germination: Changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry 1981, 20, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Tunnacliffe, A.; Wise, M.J. The continuing conundrum of the LEA proteins. Naturwissenschaften 2007, 94, 791–812. [Google Scholar] [CrossRef] [PubMed]
- Leprince, O.; Pellizzaro, A.; Berriri, S.; Buitink, J. Late seed maturation: Drying without dying. J. Exp. Bot. 2016, 68, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Hand, S.C.; Menze, M.A.; Toner, M.; Boswell, L.; Moore, D. LEA proteins during water stress: Not just for plants anymore. Annu. Rev. Physiol. 2011, 73, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Hincha, D.K.; Thalhammer, A. LEA proteins: IDPs with versatile functions in cellular dehydration tolerance. Biochem. Soc. Trans. 2012, 40, 1000–1003. [Google Scholar] [CrossRef] [Green Version]
- Tunnacliffe, A.; Hincha, D.; Leprince, O.; Macherel, D. LEA proteins: Versatility of forms and function. In Dormancy and Resistance in Harsh Environments; Lubzens, E., Cerda, J., Clark, M., Eds.; Springer: Berlin, Germany, 2010; pp. 91–108. [Google Scholar]
- Blackman, S.A.; Wettlaufer, S.H.; Obendorf, R.L.; Leopold, A.C. Maturation Proteins Associated with Desiccation Tolerance in Soybean. Plant Physiol. 1991, 96, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudet, J.; Buitink, J.; Hoekstra, F.A.; Rogniaux, H.; Larre, C.; Satour, P.; Leprince, O. Comparative Analysis of the Heat Stable Proteome of Radicles of Medicago truncatula Seeds during Germination Identifies Late Embryogenesis Abundant Proteins Associated with Desiccation Tolerance. Plant Physiol. 2006, 140, 1418–1436. [Google Scholar] [CrossRef] [Green Version]
- Hundertmark, M.; Hincha, D.K. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Knox-Brown, P.; Rindfleisch, T.; Günther, A.; Balow, K.; Bremer, A.; Walther, D.; Miettinen, M.S.; Hincha, D.K.; Thalhammer, A. Similar Yet Different–Structural and Functional Diversity among Arabidopsis thaliana LEA_4 Proteins. Int. J. Mol. Sci. 2020, 21, 2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Rikkerink, E.; Jones, W.T.; Uversky, V.N. Multifarious Roles of Intrinsic Disorder in Proteins Illustrate Its Broad Impact on Plant Biology. Plant Cell 2013, 25, 38–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.A.; Romero-Pérez, P.S.; Cuevas-Velazquez, C.L.; Rendón-Luna, D.F. The functional diversity of structural disorder in plant proteins. Arch. Biochem. Biophys. 2020, 680, 108229. [Google Scholar] [CrossRef] [PubMed]
- Graether, S.P.; Boddington, K.F. Disorder and function: A review of the dehydrin protein family. Front. Plant Sci. 2014, 5, 576. [Google Scholar] [CrossRef] [Green Version]
- Chatelain, E.; Hundertmark, M.; Leprince, O.; Le Gall, S.; Satour, P.; Deligny-Penninck, S.; Rogniaux, H.; Buitink, J. Temporal profiling of the heat-stable proteome during late maturation of Medicago truncatula seeds identifies a restricted subset of late embryogenesis abundant proteins associated with longevity. Plant Cell Environ. 2012, 35, 1440–1455. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Hirai, M.; Fujiwara, T.; Naito, S.; Noji, M.; Saito, K. Proteomic and transcriptomic analysis of Arabidopsis seeds: Molecular evidence for successive processing of seed proteins and its implication in the stress response to sulfur nutrition. Plant J. 2006, 48, 557–571. [Google Scholar] [CrossRef]
- Receveur-Brechot, V.; Bourhis, J.-M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins: Struct. Funct. Bioinform. 2005, 62, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Peng, Z.; Kurgan, L. Mfdp2: Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord Proteins 2013, 1, e24428. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Vidigal, D.D.S.; Willems, L.; Van Arkel, J.; Dekkers, B.J.; Hilhorst, H.W.; Bentsink, L. Galactinol as marker for seed longevity. Plant Sci. 2016, 246, 112–118. [Google Scholar] [CrossRef]
- Salvi, P.; Saxena, S.C.; Petla, B.P.; Kamble, N.U.; Kaur, H.; Verma, P.; Rao, V.; Ghosh, S.; Majee, M. Differentially expressed galactinol synthase(s) in chickpea are implicated in seed vigor and longevity by limiting the age induced ROS accumulation. Sci. Rep. 2016, 6, 35088. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-H.; Shang, Y.; Kang, H.K.; Kim, S.Y.; Kim, B.H.; Nam, K.H. Arabidopsis galactinol synthases 1 (AtGOLS1) negatively regulates seed germination. Plant Sci. 2018, 267, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gennari, L.; Felletti, M.; Blasa, M.; Angelino, D.; Celeghini, C.; Corallini, A.; Ninfali, P. Total extract of Beta Vulgaris var. Cicla seeds versus its purified phenolic components: Antioxidant activities and antiproliferative effects against colon cancer cells. Phytochem. Anal. 2011, 22, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-Y.; Zheng, H.-C.; Zhang, H.-W.; Zhang, J.-Y.; Ma, C.-M. Comparison of Antioxidant Constituents of Agriophyllum squarrosum Seed with Conventional Crop Seeds. J. Food Sci. 2018, 83, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Mucciarelli, M.; Gallino, M.; Maffei, M.; Scannerini, S. Effects of 3,4-dihydroxybenzoic acid on tobacco (Nicotiana tabacum L.) culturedin vitro. Growth regulation in callus and organ cultures. Plant Biosyst. Int. J. Deal. All Asp. Plant Biol. 2000, 134, 185–192. [Google Scholar] [CrossRef]
- Ghimire, B.K.; Hwang, M.H.; Sacks, E.J.; Yu, C.Y.; Kim, S.H.; Chung, I.M. Screening of Allelochemicals in Miscanthus sacchariflorus Extracts and Assessment of Their Effects on Germination and Seedling Growth of Common Weeds. Plants 2020, 9, 1313. [Google Scholar] [CrossRef]
- Jarzab, A.; Kurzawa, N.; Hopf, T.; Moerch, M.; Zecha, J.; Leijten, N.; Bian, Y.; Musiol, E.; Maschberger, M.; Stoehr, G.; et al. Meltome atlas—thermal proteome stability across the tree of life. Nat. Methods 2020, 17, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, P.; Ganscha, S.; Kahraman, A.; Cappelletti, V.; Boersema, P.J.; von Mering, C.; Claassen, M.; Picotti, P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 2017, 355, eaai7825. [Google Scholar] [CrossRef]
- Volkening, J.D.; Stecker, K.E.; Sussman, M.R. Proteome-wide Analysis of Protein Thermal Stability in the Model Higher Plant Arabidopsis thaliana. Mol. Cell. Proteom. 2019, 18, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtyla, Ł.; Garnczarska, M.; Zalewski, T.; Bednarski, W.; Ratajczak, L.; Jurga, S. A comparative study of water distribution, free radical production and activation of antioxidative metabolism in germinating pea seeds. J. Plant Physiol. 2006, 163, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Myers, N.; Moscovitz, O.; Sharon, M.; Prilusky, J.; Shaul, Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol. BioSyst. 2011, 8, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Sun, N.; Tu, C.; Shi, X.; Cheng, H.; Liu, S.; Li, S.; Wang, Y.; Zheng, Y.; et al. Intrinsically Disordered Proteins as Important Players during Desiccation Stress of Soybean Radicles. J. Proteome Res. 2017, 16, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Zamora-Briseño, J.A.; Pereira-Santana, A.; Reyes-Hernández, S.J.; Cerqueda-García, D.; Castaño, E.; Rodríguez-Zapata, L.C. Towards an understanding of the role of intrinsic protein disorder on plant adaptation to environmental challenges. Cell Stress Chaperon 2021, 26, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Dai, L.; Yu, H.; Puspita, B.; Zhao, T.; Li, F.; Tan, J.; Lim, Y.T.; Chen, M.W.; Sobota, R.; et al. Monitoring structural modulation of redox-sensitive proteins in cells with MS-CETSA. Redox Biol. 2019, 24, 101168. [Google Scholar] [CrossRef]
- Azarkovich, M.I. Dehydrins in Orthodox and Recalcitrant Seeds. Russ. J. Plant Physiol. 2020, 67, 221–230. [Google Scholar] [CrossRef]
- DeRocher, A.E.; Vierling, E. Developmental control of small heat shock protein expression during pea seed maturation. Plant J. 1994, 5, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Hundertmark, M.; Buitink, J.; Leprince, O.; Hincha, D.K. The reduction of seed-specific dehydrins reduces seed longevity in Arabidopsis thaliana. Seed Sci. Res. 2011, 21, 165–173. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nam, K.H. Physiological roles of ERD10 in abiotic stresses and seed germination of Arabidopsis. Plant Cell Rep. 2010, 29, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kiyosue, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Characterization of Two cDNAs (ERD10 and ERD14) Corresponding to Genes That Respond Rapidly to Dehydration Stress in Arabidopsis thaliana. Plant Cell Physiol. 1994, 35, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.E.H.; Maruri-López, I.; Graether, S.P.; Jiménez-Bremont, J.F. In vivo evidence for homo- and heterodimeric interactions of Arabidopsis thaliana dehydrins AtCOR47, AtERD10, and AtRAB18. Sci. Rep. 2017, 7, 17036. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Sánchez, I.E.; Maruri-López, I.; Molphe-Balch, E.P.; Becerra-Flora, A.; Jaimes-Miranda, F.; Jiménez-Bremont, J.F. Evidence for in vivo interactions between dehydrins and the aquaporin AtPIP2B. Biochem. Biophys. Res. Commun. 2019, 510, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.L.; Tripathi, P.; Rabara, R.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2011, 10, 2–11. [Google Scholar] [CrossRef]
- Yang, X.; Cheema, J.; Zhang, Y.; Deng, H.; Duncan, S.; Umar, M.I.; Zhao, J.; Liu, Q.; Cao, X.; Kwok, C.K.; et al. RNA G-quadruplex structures exist and function in vivo in plants. Genome Biol. 2020, 21, 226. [Google Scholar] [CrossRef]
- Dang, N.X.; Popova, A.; Hundertmark, M.; Hincha, D.K. Functional characterization of selected LEA proteins from Arabidopsis thaliana in yeast and in vitro. Planta 2014, 240, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.; Rausch, S.; Hundertmark, M.; Gibon, Y.; Hincha, D.K. The intrinsically disordered protein LEA7 from Arabidopsis thaliana protects the isolated enzyme lactate dehydrogenase and enzymes in a soluble leaf proteome during freezing and drying. Biochim. Biophys. Acta BBA Proteins Proteom. 2015, 1854, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.; Hundertmark, M.; Seckler, R.; Hincha, D.K. Structural transitions in the intrinsically disordered plant dehydration stress protein LEA7 upon drying are modulated by the presence of membranes. Biochim. Biophys. Acta BBA Biomembr. 2011, 1808, 1879–1887. [Google Scholar] [CrossRef] [Green Version]
- Zamora-Briseño, J.A.; de Jiménez, E.S. A LEA 4 protein up-regulated by ANA is involved in drought response in maize roots. Mol. Biol. Rep. 2016, 43, 221–228. [Google Scholar] [CrossRef]
- Chauffour, F.; Bailly, M.; Perreau, F.; Cueff, G.; Suzuki, H.; Collet, B.; Frey, A.; Clément, G.; Soubigou-Taconnat, L.; Balliau, T.; et al. Multi-omics Analysis Reveals Sequential Roles for ABA during Seed Maturation. Plant Physiol. 2019, 180, 1198–1218. [Google Scholar] [CrossRef]
- Manfre, A.J.; Lanni, L.M.; Marcotte, W.R. The Arabidopsis Group 1 Late Embryogenesis Abundant Protein ATEM6 Is Required for Normal Seed Development. Plant Physiol. 2006, 140, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Manfre, A.J.; LaHatte, G.A.; Climer, C.R.; Marcotte, W.R. Seed Dehydration and the Establishment of Desiccation Tolerance During Seed Maturation is Altered in the Arabidopsis thaliana Mutant atem6-1. Plant Cell Physiol. 2008, 50, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olvera-Carrillo, Y.; Campos, F.; Reyes, J.L.; Garciarrubio, A.; Covarrubias, A.A. Functional Analysis of the Group 4 Late Embryogenesis Abundant Proteins Reveals Their Relevance in the Adaptive Response during Water Deficit in Arabidopsis. Plant Physiol. 2010, 154, 373–390. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Velazquez, C.; Saab-Rincón, G.; Reyes, J.L.; Covarrubias, A.A. The Unstructured N-terminal Region of Arabidopsis Group 4 Late Embryogenesis Abundant (LEA) Proteins Is Required for Folding and for Chaperone-like Activity under Water Deficit. J. Biol. Chem. 2016, 291, 10893–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhang, F.; Wang, G.; Liu, Y.; Liu, D. Partial deficiency of isoleucine impairs root development and alters transcript levels of the genes involved in branched-chain amino acid and glucosinolate metabolism in Arabidopsis. J. Exp. Bot. 2012, 64, 599–612. [Google Scholar] [CrossRef]
- Erban, A.; Martinez-Seidel, F.; Rajarathinam, Y.; Dethloff, F.; Orf, I.; Fehrle, I.; Alpers, J.; Beine-Golovchuk, O.; Kopka, J. Multiplexed profiling and data processing methods to identify temperature-regulated primary metabolites using gas chromatography coupled to mass spectrometry. Methods Mol. Biol. 2020, 2156, 203–239. [Google Scholar] [PubMed]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography—Mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar] [CrossRef]
- Hummel, J.; Strehmel, N.; Selbig, J.; Walther, D.; Kopka, J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 2010, 6, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dormann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics 2004, 21, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allwood, J.W.; Erban, A.; De Koning, S.; Dunn, W.; Luedemann, A.; Lommen, A.; Kay, L.; Löscher, R.; Kopka, J.; Goodacre, R. Inter-laboratory reproducibility of fast gas chromatography–electron impact–time of flight mass spectrometry (GC–EI–TOF/MS) based plant metabolomics. Metabolomics 2009, 5, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC–MS metabolite profiling. J. Chromatogr. B 2008, 871, 182–190. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2019, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Tohge, T.; Balazadeh, S.; Erban, A.; Giavalisco, P.; Kopka, J.; Mueller-Roeber, B.; Fernie, A.R.; Hoefgen, R. Comprehensive Metabolomics Studies of Plant Developmental Senescence. Adv. Struct. Saf. Stud. 2018, 1744, 339–358. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Vicient, C.M.; Delseny, M. Isolation of Total RNA fromArabidopsis thalianaSeeds. Anal. Biochem. 1999, 268, 412–413. [Google Scholar] [CrossRef]
- Piques, M.; Schulze, W.X.; Höhne, M.; Usadel, B.; Gibon, Y.; Rohwer, J.; Stitt, M. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 2009, 5, 314. [Google Scholar] [CrossRef]
- Flis, A.; Fernández, A.P.; Zielinski, T.; Mengin, V.; Sulpice, R.; Stratford, K.; Hume, A.; Pokhilko, A.; Southern, M.M.; Seaton, D.D.; et al. Defining the robust behaviour of the plant clock gene circuit with absolute RNA timeseries and open infrastructure. Open Biol. 2015, 5, 5. [Google Scholar] [CrossRef]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.-R. Genome-Wide Identification and Testing of Superior Reference Genes for Transcript Normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R; R Studio, Inc.: Boston, MA, USA, 2019. [Google Scholar]
- UniProt, Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. pcaMethods a bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Contreras-López, O.; Moyano, T.C.; Soto, D.; Gutiérrez, R.A. Step-by-Step Construction of Gene Co-expression Networks from High-Throughput Arabidopsis RNA Sequencing Data. Methods Mol. Biol. 2018, 1761, 275–301. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Disorder Content | Counts | Min | Max | Mean |
|---|---|---|---|---|
| (%) | (Residues) | |||
| 0 | 84 | 80 | 843 | 255.6 |
| 0–10 | 144 | 56 | 990 | 342.3 |
| 10–20 | 111 | 82 | 974 | 310.5 |
| 20–30 | 97 | 69 | 976 | 348.9 |
| 30–40 | 62 | 87 | 987 | 341 |
| 40–50 | 50 | 72 | 953 | 309.8 |
| 50–60 | 27 | 98 | 724 | 294.2 |
| 60–70 | 18 | 93 | 772 | 303.4 |
| 70–80 | 9 | 129 | 367 | 199.1 |
| 80–90 | 7 | 111 | 577 | 277 |
| 90–100 | 16 | 131 | 891 | 461.4 |
| 100 | 76 | 62 | 956 | 284.9 |
| LEA ID | Pfam (Hundertmark and Hincha, 2008) | Pearson Correlation | p value |
|---|---|---|---|
| LEA4 | dehydrin | 0.703 ** | 0.010 |
| LEA5 | dehydrin | −0.091 | 0.839 |
| LEA7 | LEA_4 | 0.669 ** | 0.009 |
| LEA8 | dehydrin | 0.052 | 0.873 |
| LEA10 | dehydrin | 0.789 ** | 0.005 |
| LEA14 | dehydrin | 0.212 | 0.577 |
| LEA15 | LEA_6 | 0.581 | 0.061 |
| LEA16 | LEA_6 | −0.169 | 0.664 |
| LEA19 | LEA_4 | 0.461 | 0.131 |
| LEA20 | LEA_5 | 0.250 | 0.507 |
| LEA25 | LEA_4 | 0.409 | 0.191 |
| LEA26 | LEA_2 | 0.050 | 0.873 |
| LEA28 | LEA_4 | 0.070 | 0.839 |
| LEA29 | LEA_4 | 0.695 ** | 0.006 |
| LEA30 | LEA_4 | 0.297 | 0.393 |
| LEA31 | SMP | 0.839 ** | 0.005 |
| LEA32 | SMP | 0.934 *** | 0.0003 |
| LEA34 | dehydrin | 0.813 ** | 0.006 |
| LEA36 | LEA_4 | 0.127 | 0.715 |
| LEA42 | LEA_4 | 0.346 | 0.310 |
| LEA43 | LEA_4 | 0.777 ** | 0.009 |
| LEA46 | LEA_1 | 0.824 *** | 0.0004 |
| LEA48 | LEA_4 | 0.243 | 0.507 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ginsawaeng, O.; Gorka, M.; Erban, A.; Heise, C.; Brueckner, F.; Hoefgen, R.; Kopka, J.; Skirycz, A.; Hincha, D.K.; Zuther, E. Characterization of the Heat-Stable Proteome during Seed Germination in Arabidopsis with Special Focus on LEA Proteins. Int. J. Mol. Sci. 2021, 22, 8172. https://doi.org/10.3390/ijms22158172
Ginsawaeng O, Gorka M, Erban A, Heise C, Brueckner F, Hoefgen R, Kopka J, Skirycz A, Hincha DK, Zuther E. Characterization of the Heat-Stable Proteome during Seed Germination in Arabidopsis with Special Focus on LEA Proteins. International Journal of Molecular Sciences. 2021; 22(15):8172. https://doi.org/10.3390/ijms22158172
Chicago/Turabian StyleGinsawaeng, Orarat, Michal Gorka, Alexander Erban, Carolin Heise, Franziska Brueckner, Rainer Hoefgen, Joachim Kopka, Aleksandra Skirycz, Dirk K. Hincha, and Ellen Zuther. 2021. "Characterization of the Heat-Stable Proteome during Seed Germination in Arabidopsis with Special Focus on LEA Proteins" International Journal of Molecular Sciences 22, no. 15: 8172. https://doi.org/10.3390/ijms22158172
APA StyleGinsawaeng, O., Gorka, M., Erban, A., Heise, C., Brueckner, F., Hoefgen, R., Kopka, J., Skirycz, A., Hincha, D. K., & Zuther, E. (2021). Characterization of the Heat-Stable Proteome during Seed Germination in Arabidopsis with Special Focus on LEA Proteins. International Journal of Molecular Sciences, 22(15), 8172. https://doi.org/10.3390/ijms22158172