
Waterlogging-Stress-Responsive LncRNAs, Their Regulatory Relationships with miRNAs and Target Genes in Cucumber (Cucumis sativus L.)



Abstract
:1. Introduction
2. Results
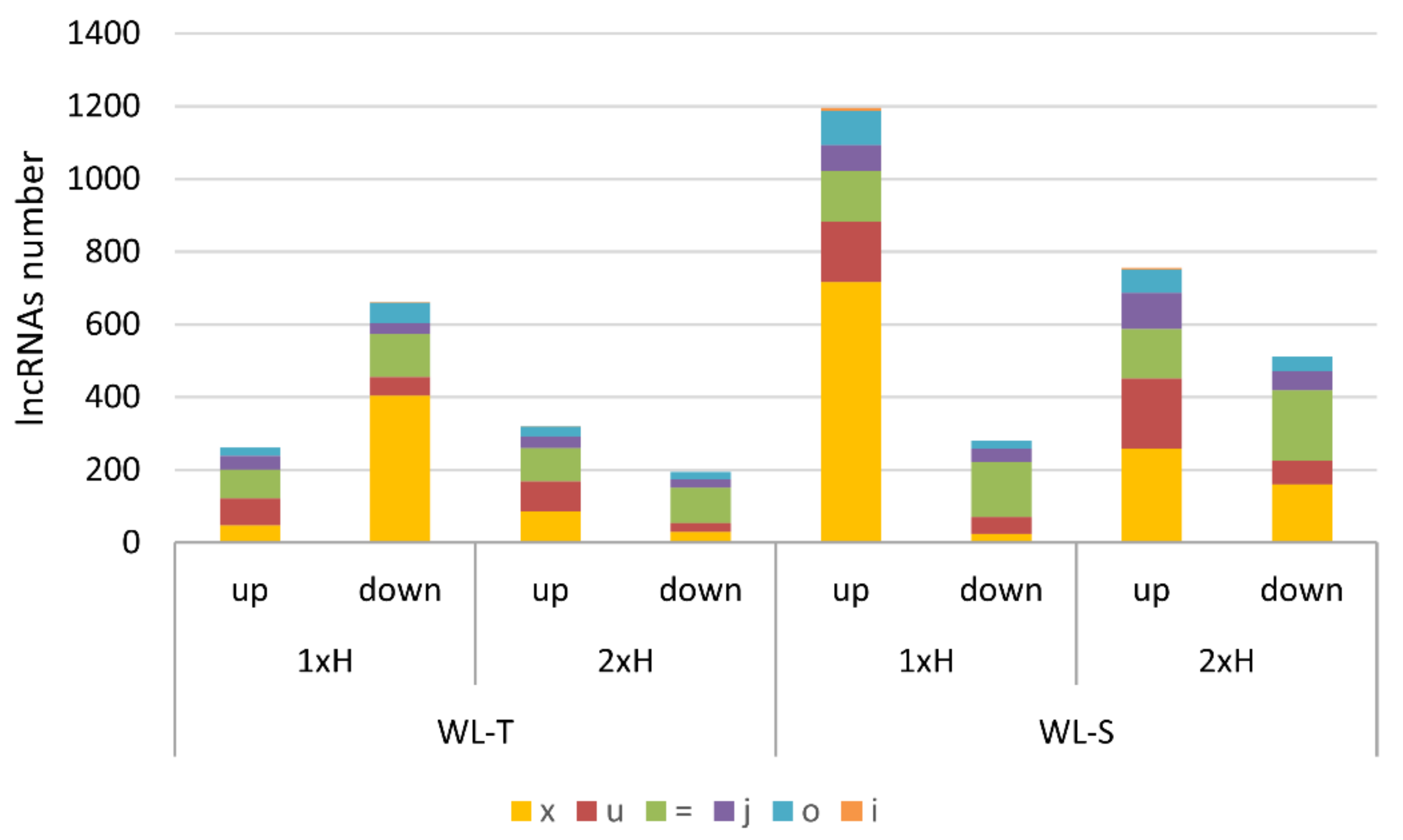
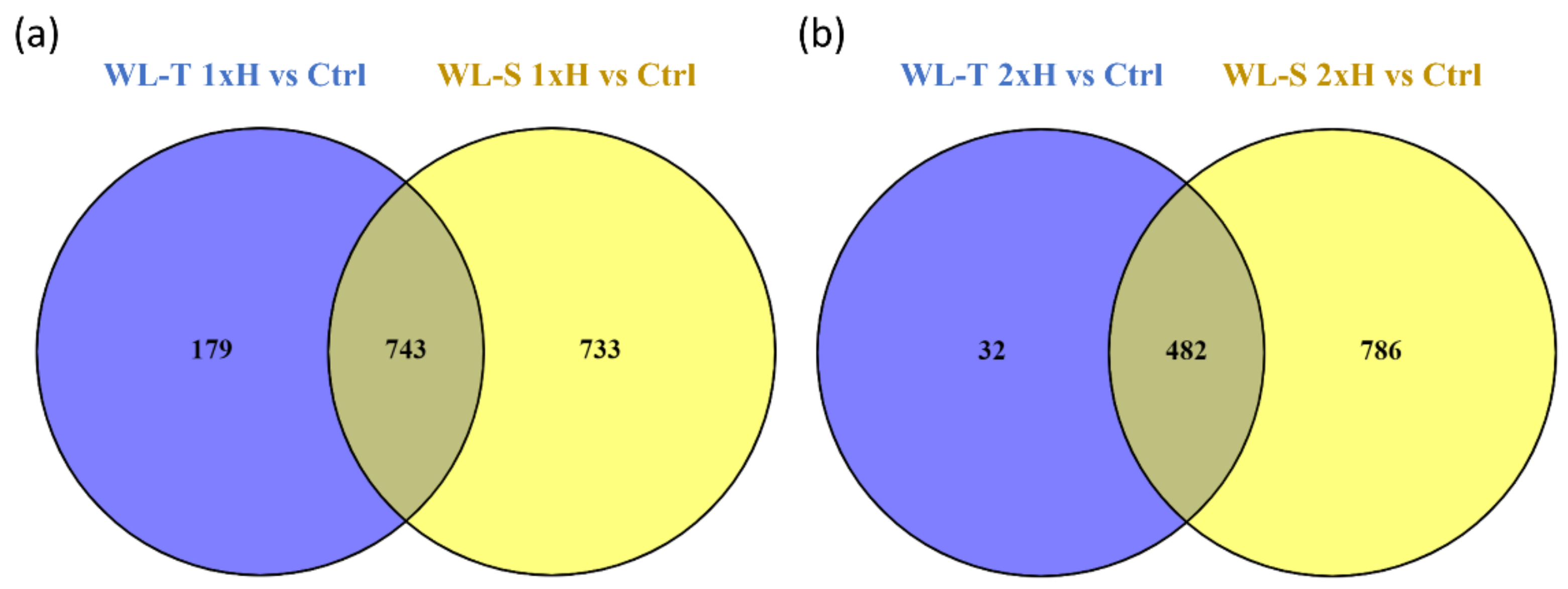
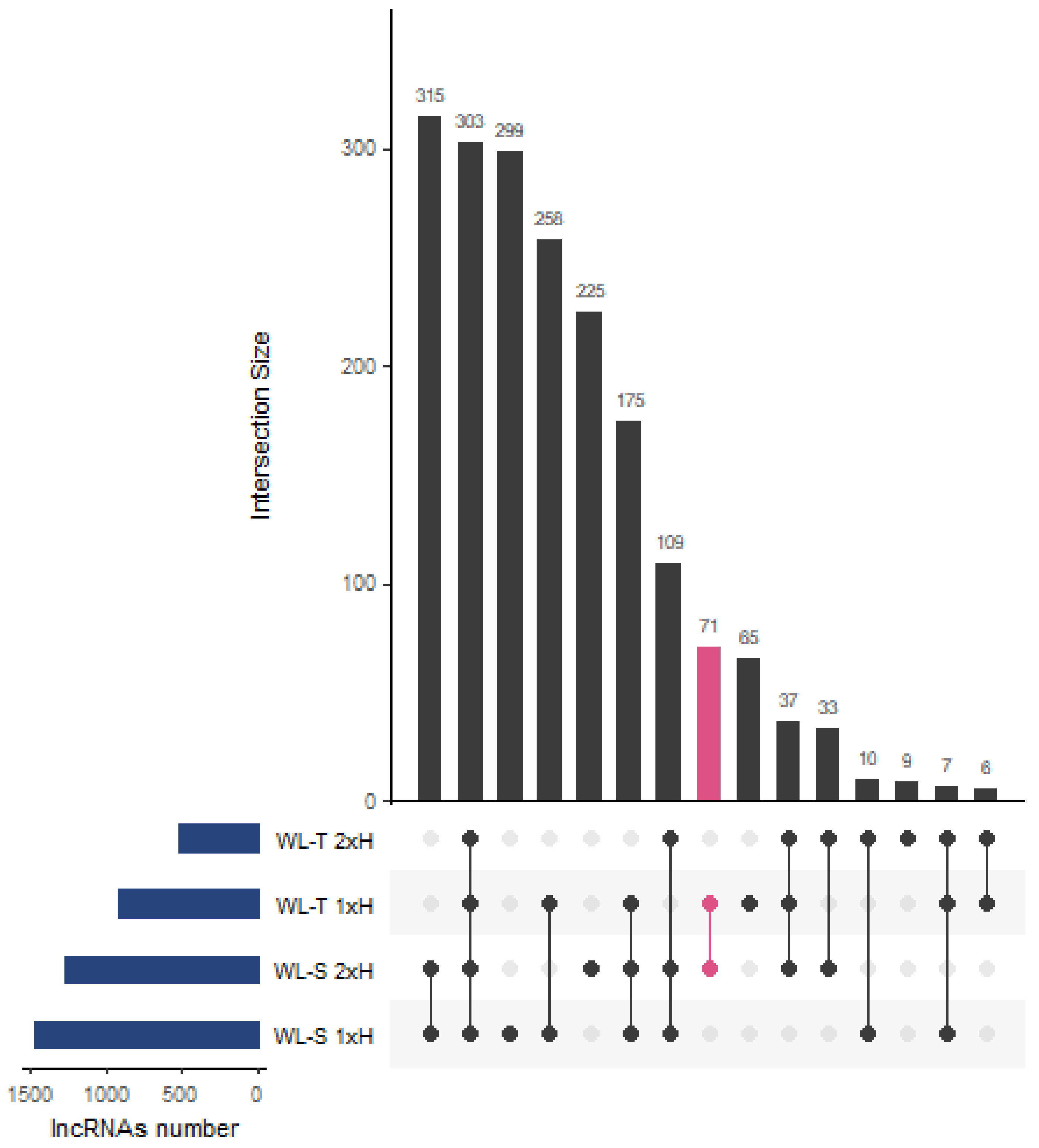
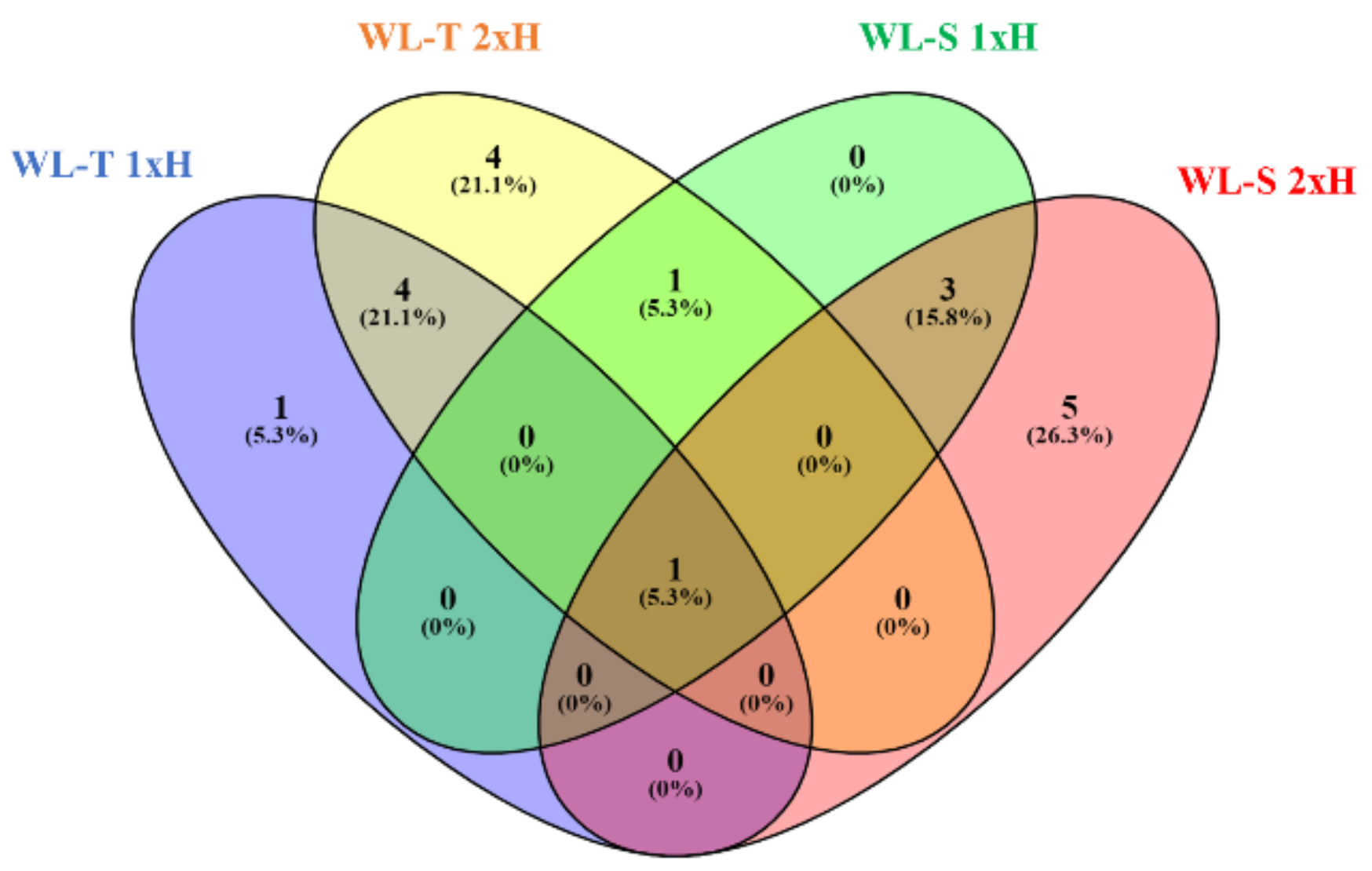
2.1. LncRNAs Identified in Cucumber under Long-Term Waterlogging
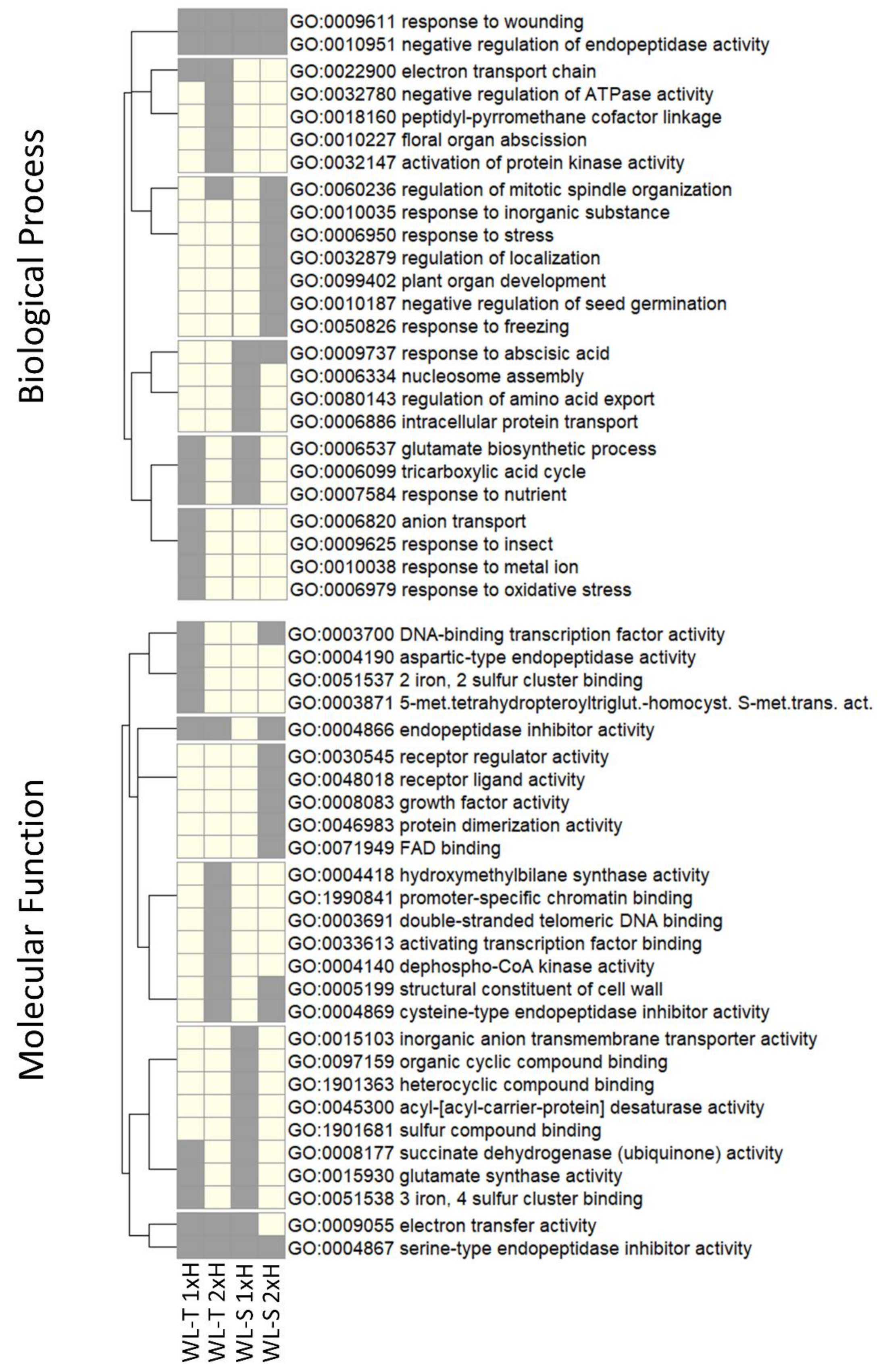
2.2. Target Genes for DE-LncRNAs
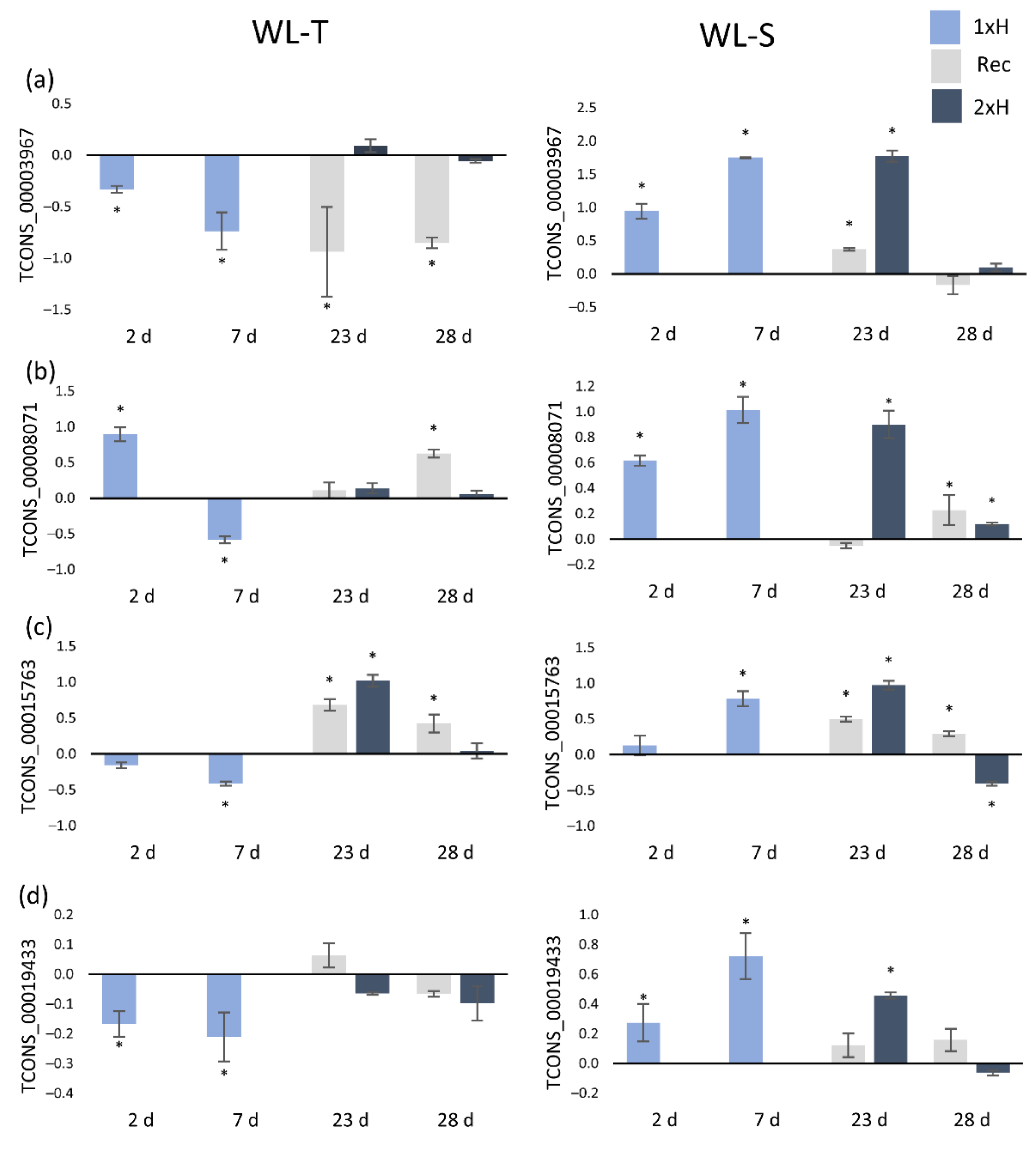
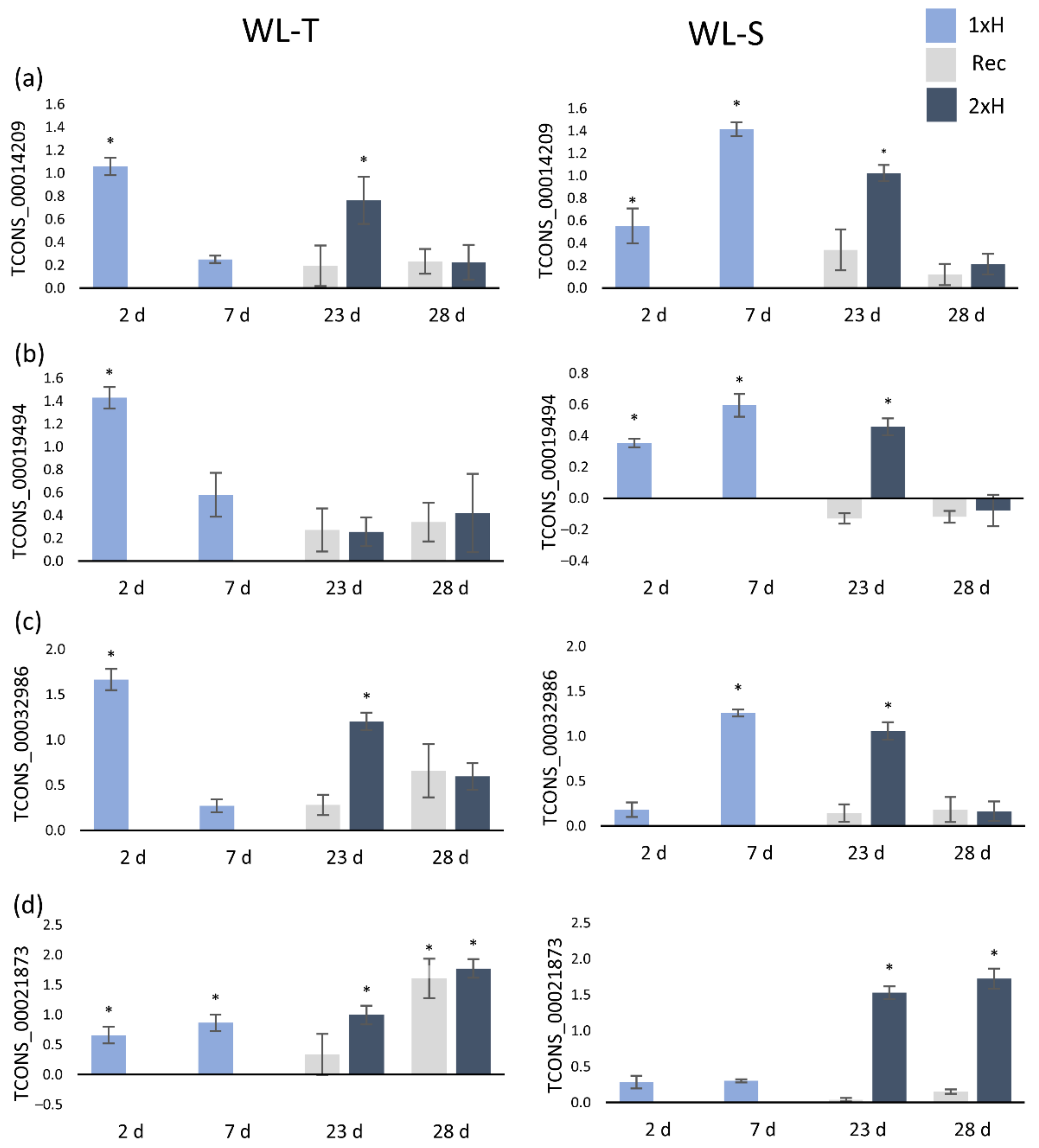
2.3. Validation of lncRNAs with Quantitative Real-Time PCR (QRT-PCR)
2.4. miRNAs Involved in Response to Long-Term Waterlogging Stress
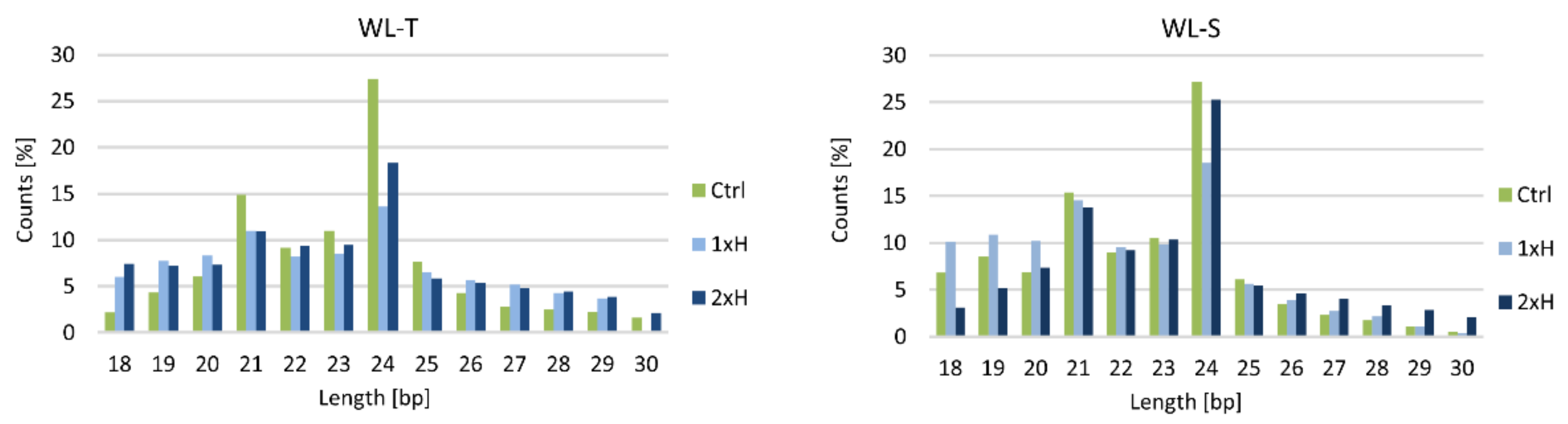
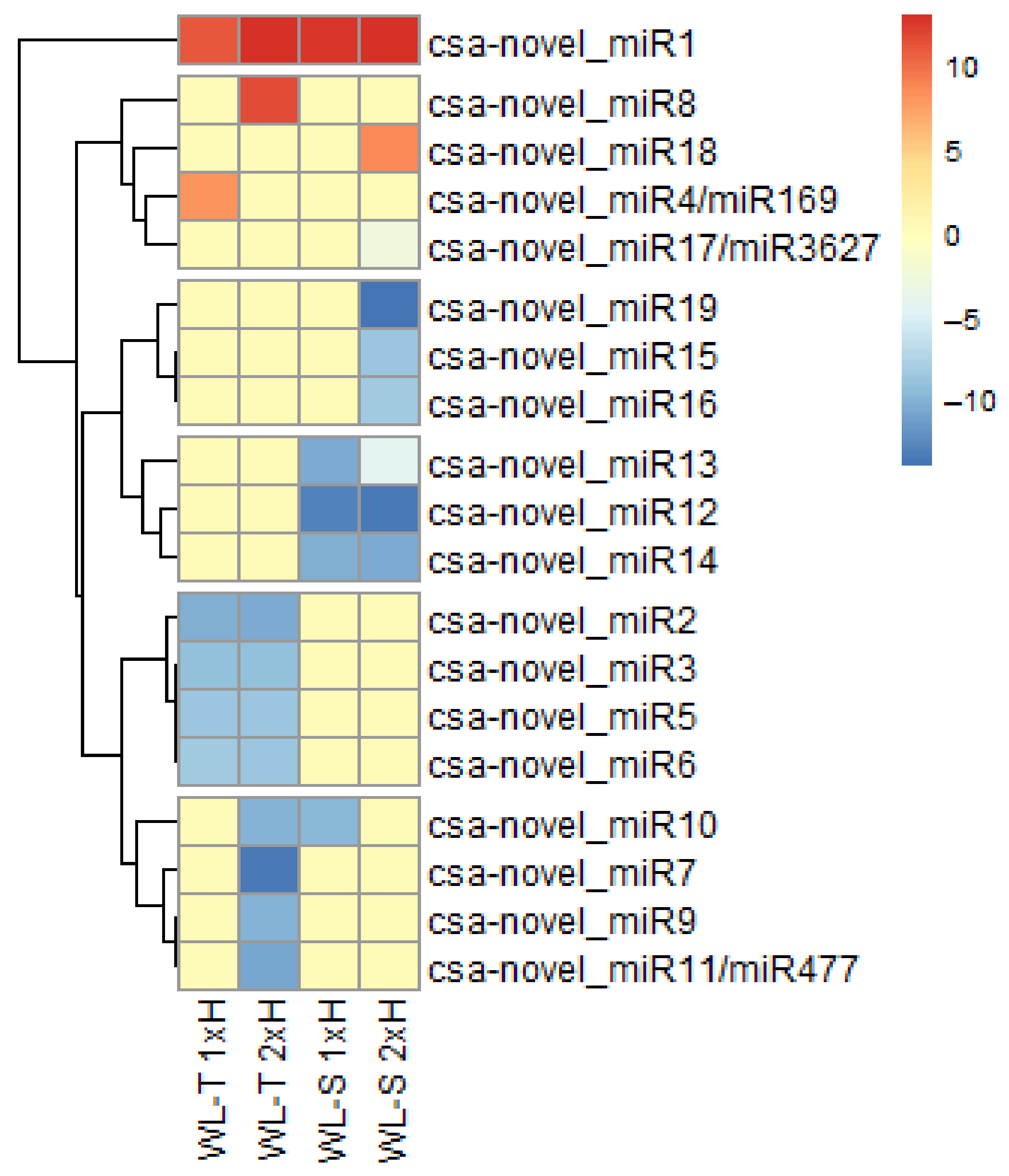
2.5. Long-Term Waterlogging-Responsive Novel miRNAs in Cucumber
2.6. QRT-PCR of miRNAs Involved in Long-Term Waterlogging
2.7. Interaction between LncRNAs and miRNAs
3. Discussion
3.1. LncRNAs Differentially Expressed under Long-Term Waterlogging
3.2. miRNAs Involved in Response to Long-Term Waterlogging
3.3. Interaction between LncRNAs and miRNAs
4. Materials and Methods
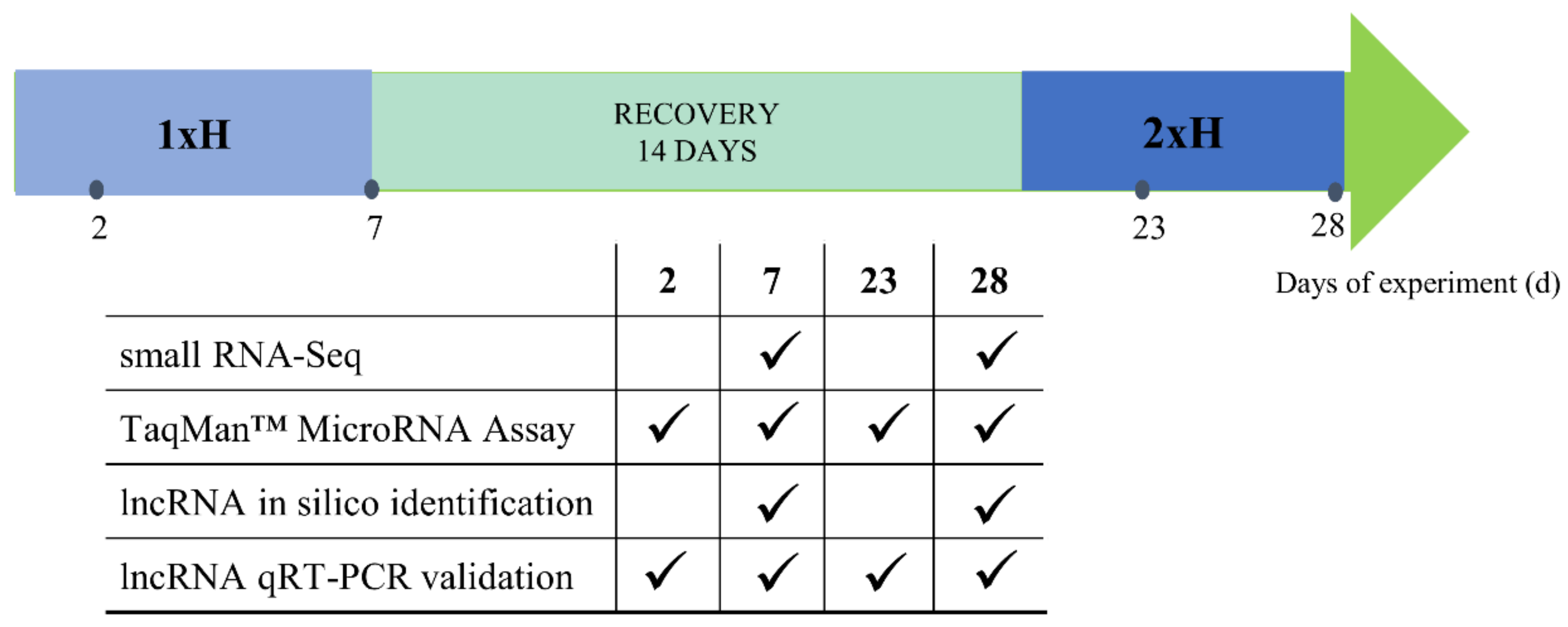
4.1. Plant Growth and Stress Induction
4.2. Sample Collection and RNA Extraction
4.3. Small RNA Library Preparation, Sequencing and Bioinformatic Analysis of Small RNA Sequencing Data
4.4. LncRNA Identification
4.5. Identification of the LncRNA–miRNA Interactions
4.6. Quantitative Real-Time PCR (QRT-PCR) Validation of LncRNAs
4.7. Quantitative Real-Time PCR (QRT-PCR) of miRNAs
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sairam, R.K.; Kumutha, D.; Ezhilmathi, K.; Deshmukh, P.S.; Srivastava, G.C. Physiology and biochemistry of waterlogging tolerance in plants. Biol. Plant. 2008, 52, 401–412. [Google Scholar] [CrossRef]
- Gibbs, D.; Lee, S.C.; Isa, N.M.; Gramuglia, S.; Fukao, T.; Bassel, G.; Correia, C.S.; Corbineau, F.; Theodoulou, F.; Bailey-Serres, J.; et al. Homeostatic response to hypoxia is regulated by the N-end rule pathway in plants. Nat. Cell Biol. 2011, 479, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sui, X.; Guo, J.; Wang, Z.; Cheng, J.; Ma, S.; Li, X.; Zhang, Z. Antisense suppression of cucumber (Cucumis sativus L.) sucrose synthase 3 (CsSUS3) reduces hypoxic stress tolerance. Plant Cell Environ. 2013, 37, 795–810. [Google Scholar] [CrossRef]
- Licausi, F. Regulation of the molecular response to oxygen limitations in plants. New Phytol. 2010, 190, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Kerchev, P.; van der Meer, T.; Sujeeth, N.; Verlee, A.; Stevens, C.V.; Van Breusegem, F.; Gechev, T. Molecular priming as an approach to induce tolerance against abiotic and oxidative stresses in crop plants. Biotechnol. Adv. 2020, 40, 107503. [Google Scholar] [CrossRef]
- Tanou, G.; Fotopoulos, V.; Molassiotis, A. Priming against environmental challenges and proteomics in plants: Update and agricultural perspectives. Front. Plant Sci. 2012, 3, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kęska, K.; Szcześniak, M.; Makałowska, I.; Czernicka, M. Long-Term Waterlogging as Factor Contributing to Hypoxia Stress Tolerance Enhancement in Cucumber: Comparative Transcriptome Analysis of Waterlogging Sensitive and Tolerant Accessions. Genes 2021, 12, 189. [Google Scholar] [CrossRef]
- Barciszewska-Pacak, M.; Milanowska, K.; Knop, K.; Bielewicz, D.; Nuc, P.; Plewka, P.; Pacak, A.M.; Vazquez, F.; Karlowski, W.; Jarmolowski, A.; et al. Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front. Plant Sci. 2015, 6, 410. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Genet. Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef]
- Wang, T.-Z.; Liu, M.; Zhao, M.-G.; Chen, R.; Zhang, W.-H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 2015, 15, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, A.M.L.; Drusin, S.I.; Chorostecki, U.; Mateos, J.L.; Moro, B.; Bologna, N.G.; Bresso, E.G.; Schapire, A.; Rasia, R.M.; Moreno, D.M.; et al. Identification of key sequence features required for microRNA biogenesis in plants. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Stepien, A.; Knop, K.; Dolata, J.; Taube, M.; Bajczyk, M.; Barciszewska-Pacak, M.; Pacak, A.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Posttranscriptional coordination of splicing and miRNA biogenesis in plants. Wiley Interdiscip. Rev. RNA 2016, 8, e1403. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant micro RNA s. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef] [Green Version]
- Iki, T.; Cléry, A.; Bologna, N.; Sarazin, A.; Brosnan, C.A.; Pumplin, N.; Allain, F.H.; Voinnet, O. Structural Flexibility Enables Alternative Maturation, ARGONAUTE Sorting and Activities of miR168, a Global Gene Silencing Regulator in Plants. Mol. Plant 2018, 11, 1008–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Zhang, S.; Yu, B. microRNA biogenesis, degradation and activity in plants. Cell. Mol. Life Sci. 2014, 72, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Mishra, S.S.; Behera, P.K. Drought Tolerance in Rice: Focus on Recent Mechanisms and Approaches. Rice Sci. 2021, 28, 119–132. [Google Scholar] [CrossRef]
- Parmar, S.; Gharat, S.A.; Tagirasa, R.; Chandra, T.; Behera, L.; Dash, S.K.; Shaw, B.P. Identification and expression analysis of miRNAs and elucidation of their role in salt tolerance in rice varieties susceptible and tolerant to salinity. PLoS ONE 2020, 15, e0230958. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Guo, S.; Wang, Y.; Wang, L.; Shu, S.; Sun, J. Systematic identification and analysis of heat-stress-responsive lncRNAs, circRNAs and miRNAs with associated co-expression and ceRNA networks in cucumber (Cucumis sativus L.). Physiol. Plant. 2019, 168, 736–754. [Google Scholar] [CrossRef]
- Fontana, J.E.; Wang, G.; Sun, R.; Xue, H.; Li, Q.; Liu, J.; Davis, K.E.; Thornburg, T.E.; Zhang, B.; Zhang, Z.; et al. Impact of potassium deficiency on cotton growth, development and potential microRNA-mediated mechanism. Plant Physiol. Biochem. 2020, 153, 72–80. [Google Scholar] [CrossRef]
- Noman, A.; Aqeel, M. miRNA-based heavy metal homeostasis and plant growth. Environ. Sci. Pollut. Res. 2017, 24, 10068–10082. [Google Scholar] [CrossRef] [PubMed]
- Barciszewska-Pacak, M.; Knop, K.; Jarmołowski, A.; Szweykowska-Kulińska, Z. Arabidopsis thaliana microRNA162 level is posttranscriptionally regulated via splicing and polyadenylation site selection. Acta Biochim. Pol. 2017, 63, 811–816. [Google Scholar] [CrossRef]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2009, 61, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Liu, Z.; Zou, X.; Jiang, Y.; Qiu, F.; Zheng, Y.; Zhang, Z. Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol. Plant. 2012, 147, 181–193. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, L.; Zhang, Y.; Kang, X.; Zhang, Z.; Wang, Y. Identification of novel and conserved Populus tomentosa microRNA as components of a response to water stress. Funct. Integr. Genom. 2012, 12, 327–339. [Google Scholar] [CrossRef]
- Feyissa, B.A.; Amyot, L.; Nasrollahi, V.; Papadopoulos, Y.; Kohalmi, S.E.; Hannoufa, A. Involvement of the miR156/SPL module in flooding response in Medicago sativa. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Hou, Y.; Jiang, F.; Zheng, X.; Wu, Z. Identification and analysis of oxygen responsive microRNAs in the root of wild tomato (S. habrochaites). BMC Plant Biol. 2019, 19, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Wang, K.; Pan, J.; Chen, X. Small RNA sequencing identifies cucumber miRNA roles in waterlogging-triggered adventitious root primordia formation. Mol. Biol. Rep. 2019, 46, 6381–6389. [Google Scholar] [CrossRef] [PubMed]
- Nejat, N.; Mantri, N. Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 2018, 38, 93–105. [Google Scholar] [CrossRef]
- Simopoulos, C.M.A.; Weretilnyk, E.A.; Golding, G.B. Prediction of plant lncRNA by ensemble machine learning classifiers. BMC Genom. 2018, 19, 316. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Bajic, V.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Q.-H.; Kaufmann, K. Long non-coding RNAs in plants: Emerging modulators of gene activity in development and stress responses. Planta 2020, 252, 1–14. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long Non-coding RNA in Plants in the Era of Reference Sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Melissari, M.T.; Grote, P. Roles for long non-coding RNAs in physiology and disease. Pflügers Archiv. Eur. J. Physiol. 2016, 468, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Petri, A.; Dybkær, K.; Bøgsted, M.; Thrue, C.A.; Hagedorn, P.H.; Schmitz, A.; Bødker, J.S.; Johnsen, H.E.; Kauppinen, S. Long Noncoding RNA Expression during Human B-Cell Development. PLoS ONE 2015, 10, e0138236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, P.; Yang, W.; Ruan, X.; Kiesewetter, K.; Zhu, J.; Cao, H. Integrative Transcriptome Analyses of Metabolic Responses in Mice Define Pivotal LncRNA Metabolic Regulators. Cell Metab. 2016, 24, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xia, X.; Jiang, B.; Ma, K.; Zhu, L.; Wang, L.; Jin, B. Identification and characterization of novel lncRNAs in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2017, 488, 348–354. [Google Scholar] [CrossRef]
- Campalans, A.; Kondorosi, A.; Crespi, M. Enod40, a Short Open Reading Frame–Containing mRNA, Induces Cytoplasmic Localization of a Nuclear RNA Binding Protein in Medicago truncatula. Plant Cell 2004, 16, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wu, P.; Wang, Q.; Wang, W.; Zhang, C.; Sun, F.; Liu, Z.; Li, Y.; Hou, X. Comparative transcriptome discovery and elucidation of the mechanism of long noncoding RNAs during vernalization in Brassica rapa. Plant Growth Regul. 2018, 85, 27–39. [Google Scholar] [CrossRef]
- Chekanova, J. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sun, Z.; Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crop. Prod. 2018, 118, 334–346. [Google Scholar] [CrossRef]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 2017, 89, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.-Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of Maize Long Non-Coding RNAs Responsive to Drought Stress. PLoS ONE 2014, 9, e98958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.-H.; Xu, X.-W.; Lin, X.-J.; Zhang, W.-J.; Chen, X.-H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics 2012, 99, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chen, M.; Ji, J.; Xu, Q.; Qi, X.; Chen, X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017, 17, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.K.; Singh, A.K.; Tripathi, N.; Yadav, D.; Hemantaranjan, A. Flooding: Abiotic Constraint Limiting Vegetable Productivity. Adv. Plants Agric. Res. 2014, 1, 00016. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Hao, J.; Meng, Y.; Luo, L.; Li, J. Identifying optimal reference genes for the normalization of microRNA expression in cucumber under viral stress. PLoS ONE 2018, 13, e0194436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floris, M.; Mahgoub, H.; Lanet, E.; Robaglia, C.; Menand, B. Post-transcriptional Regulation of Gene Expression in Plants during Abiotic Stress. Int. J. Mol. Sci. 2009, 10, 3168–3185. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, J.; Yang, Y.; Tan, C.; Zhu, Y.; Hu, L.; Qi, Y.; Lu, Z.J. Stress-responsive regulation of long non-coding RNA polyadenylation in Oryza sativa. Plant J. 2018, 93, 814–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjo, Y.; Maruyama, K.; Yasue, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Komatsu, S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol. 2011, 77, 129–144. [Google Scholar] [CrossRef]
- Kołton, A.; Kęska, K.; Czernicka, M. Selection of Tomato and Cucumber Accessions for Waterlogging Sensitivity through Morpho-Physiological Assessment at an Early Vegetative Stage. Agronomy 2020, 10, 1490. [Google Scholar] [CrossRef]
- Mao, W.; Li, Z.; Xia, X.; Li, Y.; Yu, J. A Combined Approach of High-Throughput Sequencing and Degradome Analysis Reveals Tissue Specific Expression of MicroRNAs and Their Targets in Cucumber. PLoS ONE 2012, 7, e33040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Li, S.; Hu, L.; Zhang, C. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (Brassica napus L.) genotypes subjected to drought stress and re-watering. BMC Plant Biol. 2020, 20, 81. [Google Scholar] [CrossRef] [Green Version]
- Waseem, M.; Liu, Y.; Xia, R. Long Non-Coding RNAs, the Dark Matter: An Emerging Regulatory Component in Plants. Int. J. Mol. Sci. 2020, 22, 86. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Bhuyan, M.; Zulfiqar, F.; Raza, A.; Mohsin, S.; Mahmud, J.; Fujita, M.; Fotopoulos, V. Reactive Oxygen Species and Antioxidant Defense in Plants under Abiotic Stress: Revisiting the Crucial Role of a Universal Defense Regulator. Antioxidants 2020, 9, 681. [Google Scholar] [CrossRef]
- Shabala, S.; Shabala, L.; Barceló, J.; Poschenrieder, C. Membrane transporters mediating root signalling and adaptive responses to oxygen deprivation and soil flooding. Plant Cell Environ. 2014, 37, 2216–2233. [Google Scholar] [CrossRef]
- Xu, X.; Ji, J.; Ma, X.; Xu, Q.; Qi, X.; Chen, X. Comparative Proteomic Analysis Provides Insight into the Key Proteins Involved in Cucumber (Cucumis sativus L.) Adventitious Root Emergence under Waterlogging Stress. Front. Plant Sci. 2016, 7, 1515. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-Q.; Gao, J.; Dong, A.-W.; Shen, W.-H. A Truncated Arabidopsis NUCLEOSOME ASSEMBLY PROTEIN 1, AtNAP1;3T, Alters Plant Growth Responses to Abscisic Acid and Salt in the Atnap1;3-2 Mutant. Mol. Plant 2009, 2, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Bäurle, I.; Trindade, I. Chromatin regulation of somatic abiotic stress memory. J. Exp. Bot. 2020, 71, 5269–5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mironov, A.A.; Beznoussenko, G.V. Models of Intracellular Transport: Pros and Cons. Front. Cell Dev. Biol. 2019, 7, 146. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wei, Q.; Huang, H.; Xia, J. Amino Acid Transporters in Plant Cells: A Brief Review. Plants 2020, 9, 967. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapa ssp.chinensis). Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Valliyodan, B.; Prince, S.; Wan, J.; Nguyen, H.T. Characterization of the XTH Gene Family: New Insight to the Roles in Soybean Flooding Tolerance. Int. J. Mol. Sci. 2018, 19, 2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, H.; Philippe, F.; Domon, J.-M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell Wall Metabolism in Response to Abiotic Stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Luan, H.; Shen, H.; Pan, Y.; Guo, B.; Lv, C.; Xu, R. Elucidating the hypoxic stress response in barley (Hordeum vulgare L.) during waterlogging: A proteomics approach. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Wang, Q.-J.; Sun, H.; Dong, Q.-L.; Sun, T.-Y.; Jin, Z.-X.; Hao, Y.-J.; Yao, Y.-X. The enhancement of tolerance to salt and cold stresses by modifying the redox state and salicylic acid content via thecytosolic malate dehydrogenasegene in transgenic apple plants. Plant Biotechnol. J. 2016, 14, 1986–1997. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-M.; Fang, Y.-S.; Zhang, C.-J.; Hao, Q.-N.; Cao, D.; Yuan, S.-L.; Chen, H.-F.; Yang, Z.-L.; Chen, S.-L.; Shan, Z.-H.; et al. GmSYP24, a putative syntaxin gene, confers osmotic/drought, salt stress tolerances and ABA signal pathway. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mangrauthia, S.K.; Maliha, A.; Prathi, N.B.; Marathi, B. MicroRNAs: Potential target for genome editing in plants for traits improvement. Indian J. Plant Physiol. 2017, 22, 530–548. [Google Scholar] [CrossRef]
- Xu, X.; Zhong, C.; Tan, M.; Song, Y.; Qi, X.; Xu, Q.; Chen, X. Identification of MicroRNAs and Their Targets That Respond to Powdery Mildew Infection in Cucumber by Small RNA and Degradome Sequencing. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Barrera-Figueroa, B.E.; Juntawong, P.; Peña-Castro, J.M. Submergence and Waterlogging Stress in Plants: A Review Highlighting Research Opportunities and Understudied Aspects. Front. Plant Sci. 2019, 10, 340. [Google Scholar] [CrossRef]
- Tamang, B.G.; Li, S.; Rajasundaram, D.; Lamichhane, S.; Fukao, T. Overlapping and stress-specific transcriptomic and hormonal responses to flooding and drought in soybean. Plant J. 2021. [Google Scholar] [CrossRef]
- Bouzroud, S.; Gouiaa, S.; Hu, N.; Bernadac, A.; Mila, I.; Bendaou, N.; Smouni, A.; Bouzayen, M.; Zouine, M. Auxin Response Factors (ARFs) are potential mediators of auxin action in tomato response to biotic and abiotic stress (Solanum lycopersicum). PLoS ONE 2018, 13, e0193517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Chen, J.; Kuang, L.; Wang, N.; Zhang, G.; Jiang, L.; Wu, D. Effects of waterlogging stress on early seedling development and transcriptomic responses in Brassica napus. Mol. Breed. 2020, 40, 1–14. [Google Scholar] [CrossRef]
- Licciardello, C.; Tononi, P.; Rossato, M.; Delledonne, M.; Caruso, P. A transcriptional analysis reveals an extensive range of genes responsible for increasing the tolerance of Carrizo citrange to oxygen deficiency. Tree Genet. Genomes 2019, 15, 19. [Google Scholar] [CrossRef]
- Wani, S.H.; Kumar, V.; Khare, T.; Tripathi, P.; Shah, T.; Ramakrishna, C.; Aglawe, S.; Mangrauthia, S.K. miRNA applications for engineering abiotic stress tolerance in plants. Biologia 2020, 75, 1063–1081. [Google Scholar] [CrossRef]
- Eom, S.H.; Lee, H.J.; Lee, J.H.; Wi, S.H.; Kim, S.K.; Hyun, T.K. Identification and Functional Prediction of Drought-Responsive Long Non-Coding RNA in Tomato. Agronomy 2019, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Song, Y.; Du, Q.; Yang, X.; Ci, D.; Chen, J.; Xie, J.; Li, B.; Zhang, D. Population genomic analysis of gibberellin-responsive long non-coding RNAs in Populus. J. Exp. Bot. 2016, 67, 2467–2482. [Google Scholar] [CrossRef] [Green Version]
- Khemka, N.; Singh, V.K.; Garg, R.; Jain, M. Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development. Sci. Rep. 2016, 6, 33297. [Google Scholar] [CrossRef] [Green Version]
- Karakülah, G.; Kurtoğlu, K.Y.; Unver, T. PeTMbase: A Database of Plant Endogenous Target Mimics (eTMs). PLoS ONE 2016, 11, e0167698. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, A.; Yu, B.; Li, S. Interplay between miRNAs and lncRNAs: Mode of action and biological roles in plant development and stress adaptation. Comput. Struct. Biotechnol. J. 2021, 19, 2567–2574. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Somoza, I.R.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Sun, J.; Dong, Y.; Wang, C.; Xiao, S.; Mo, L.; Jiao, Z. Comparative transcriptome analysis uncovers regulatory roles of long non-coding RNAs involved in resistance to powdery mildew in melon. BMC Genom. 2020, 21, 125. [Google Scholar] [CrossRef]
- Sun, Z.; Evans, J.; Bhagwate, A.; Middha, S.; Bockol, M.; Yan, H.; Kocher, J.-P. CAP-miRSeq: A comprehensive analysis pipeline for microRNA sequencing data. BMC Genom. 2014, 15, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, M.; Mackowiak, S.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.E.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Negri, T.D.C.; Alves, W.A.L.; Bugatti, P.H.; Saito, P.T.M.; Domingues, D.; Paschoal, A.R. Pattern recognition analysis on long noncoding RNAs: A tool for prediction in plants. Briefings Bioinform. 2018, 20, 682–689. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.; Bateman, A.; Petrov, A.I. Non-Coding RNA Analysis Using the Rfam Database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. TopGo: Enrichment Analysis for Gene Ontology. R Package Version 2.38. Available online: https://bioconductor.riken.jp/packages/3.3/bioc/html/topGO.html (accessed on 27 November 2020).
- Bonnet, E.; He, Y.; Billiau, K.; Van De Peer, Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Bouba, I.; Kang, Q.; Luan, Y.-S.; Meng, J. Predicting miRNA-lncRNA interactions and recognizing their regulatory roles in stress response of plants. Math. Biosci. 2019, 312, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA | WL-T | WL-S | Class Code | Nearest Gene | Gene Description | Gene Ontology | ||
|---|---|---|---|---|---|---|---|---|
| 1xH * | 2xH ** | 1xH | 2xH | |||||
| TCONS_00003967 | −4.57 | ns *** | 6.93 | ns | x | Csa1M422990.1 | Xyloglucan endotransglucosylase/hydrolase | - |
| TCONS_00008071 | −4.66 | ns | 7.44 | ns | x | Csa2M174150.1 | Malate dehydrogenase | BP: GO:0006099 |
| TCONS_00015763 | −8.06 | ns | 6.62 | −4.41 | x | Csa3M782680.1 | Syntaxin, putative | BP: GO:0009737 |
| TCONS_00019433 | −7.42 | ns | 7.13 | ns | x | Csa4M054300.1 | 26S proteasome non-ATPase regulatory subunit | - |
| TCONS_00014209 | ns | ns | 5.51 | ns | u | - | - | - |
| TCONS_00019494 | ns | ns | 8.87 | ns | x | Csa4M063450.1 | ATP-dependent RNA helicase, putative | MF: GO:0097159, GO:1901363 |
| TCONS_00032986 | ns | ns | 6.07 | ns | x | Csa7M357030.1 | Transcription initiation factor TFIID subunit 1-A | - |
| TCONS_00021873 | 5.23 | 7.34 | ns | 3.50 | u | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kęska, K.; Szcześniak, M.W.; Adamus, A.; Czernicka, M. Waterlogging-Stress-Responsive LncRNAs, Their Regulatory Relationships with miRNAs and Target Genes in Cucumber (Cucumis sativus L.). Int. J. Mol. Sci. 2021, 22, 8197. https://doi.org/10.3390/ijms22158197
Kęska K, Szcześniak MW, Adamus A, Czernicka M. Waterlogging-Stress-Responsive LncRNAs, Their Regulatory Relationships with miRNAs and Target Genes in Cucumber (Cucumis sativus L.). International Journal of Molecular Sciences. 2021; 22(15):8197. https://doi.org/10.3390/ijms22158197
Chicago/Turabian StyleKęska, Kinga, Michał Wojciech Szcześniak, Adela Adamus, and Małgorzata Czernicka. 2021. "Waterlogging-Stress-Responsive LncRNAs, Their Regulatory Relationships with miRNAs and Target Genes in Cucumber (Cucumis sativus L.)" International Journal of Molecular Sciences 22, no. 15: 8197. https://doi.org/10.3390/ijms22158197