
Nuclear Transfer Arrest Embryos Show Massive Dysregulation of Genes Involved in Transcription Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
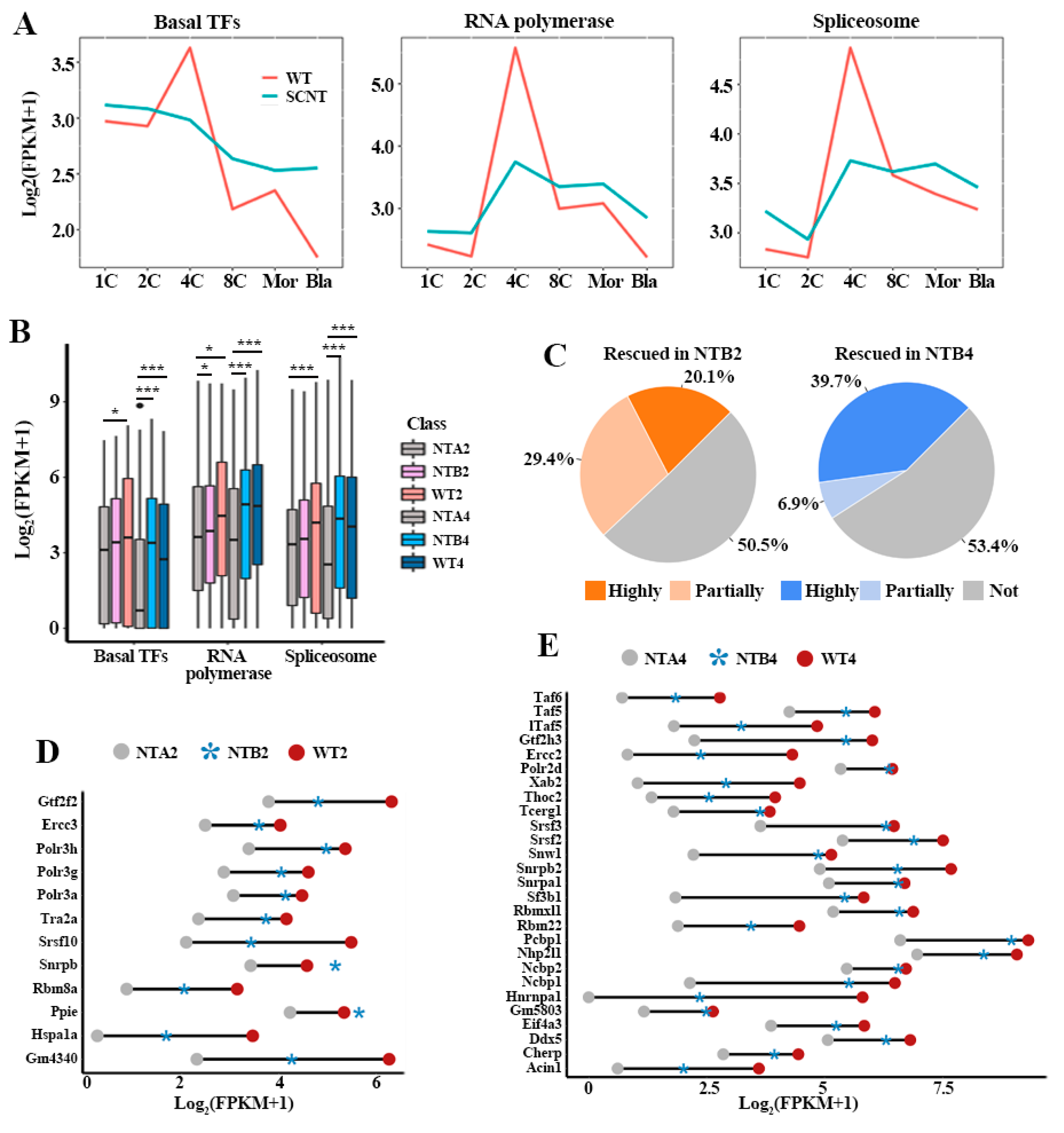
2.1. Incomplete Activation of Transcription Pathways in SCNT Embryos
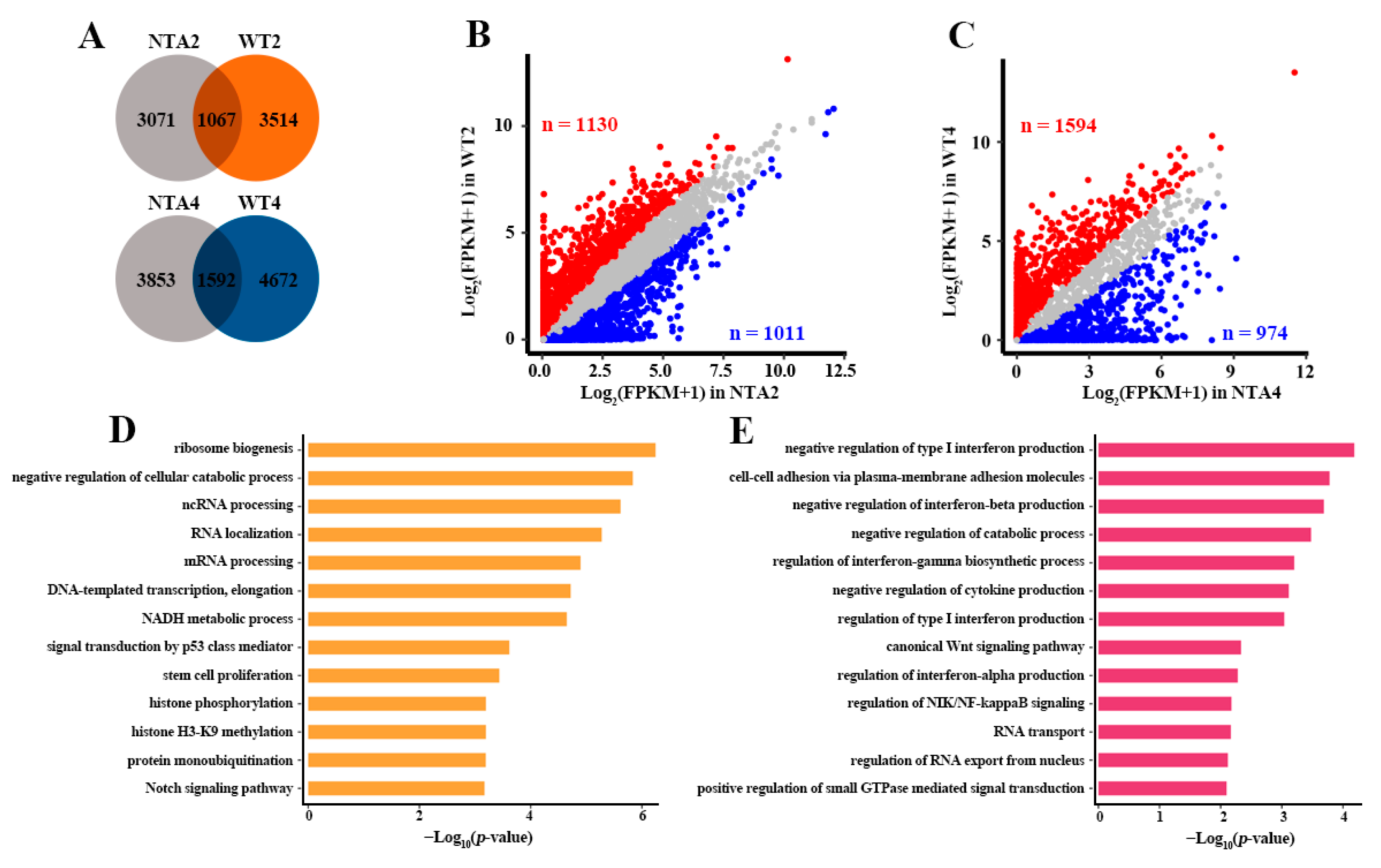
2.2. Abnormal Transcription Pathway was Related to Massive Dysregulation of Genes in NTA Embryos
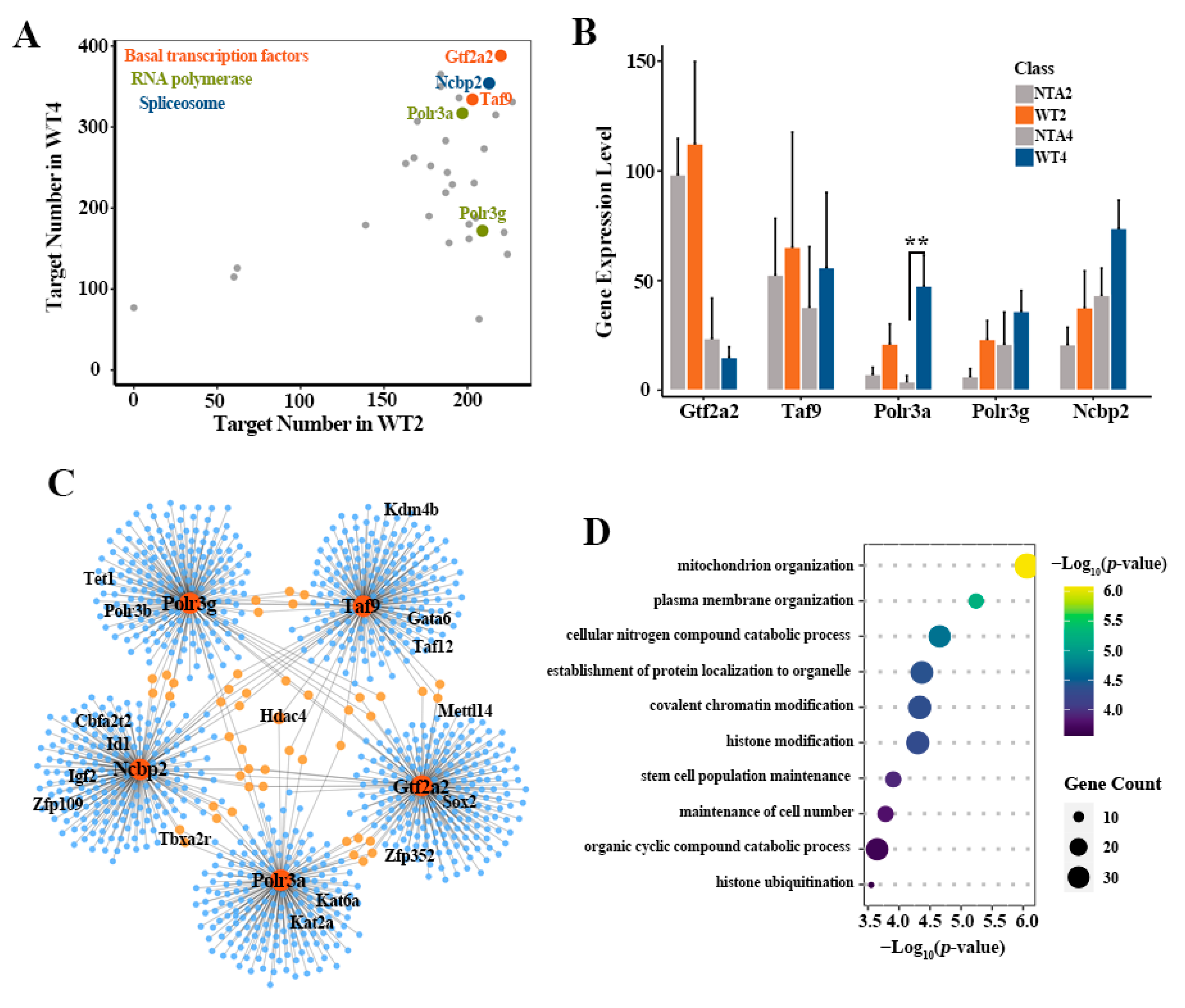
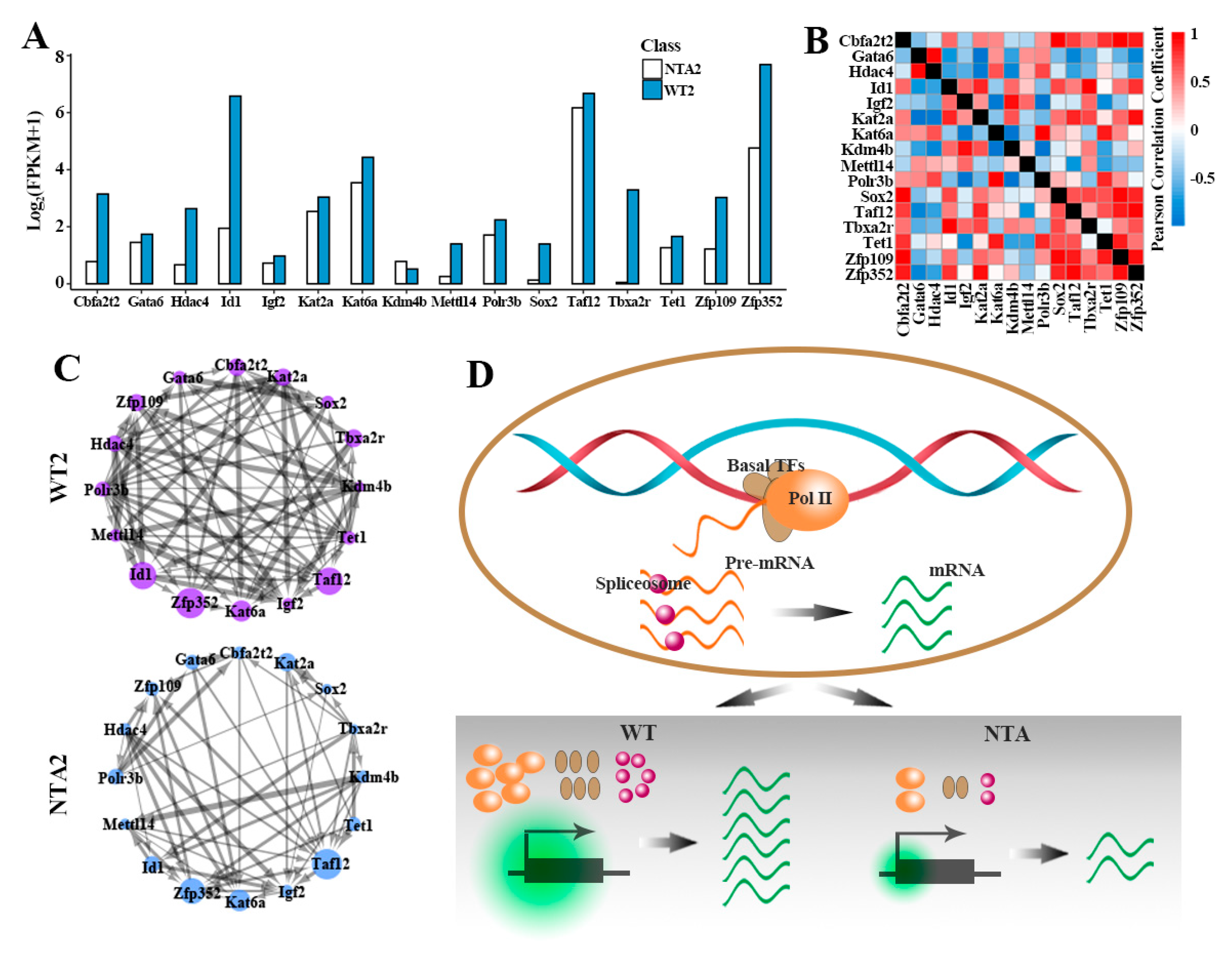
2.3. Defective Activation of Transcription Pathways Downstream GRNs in NTA Embryos
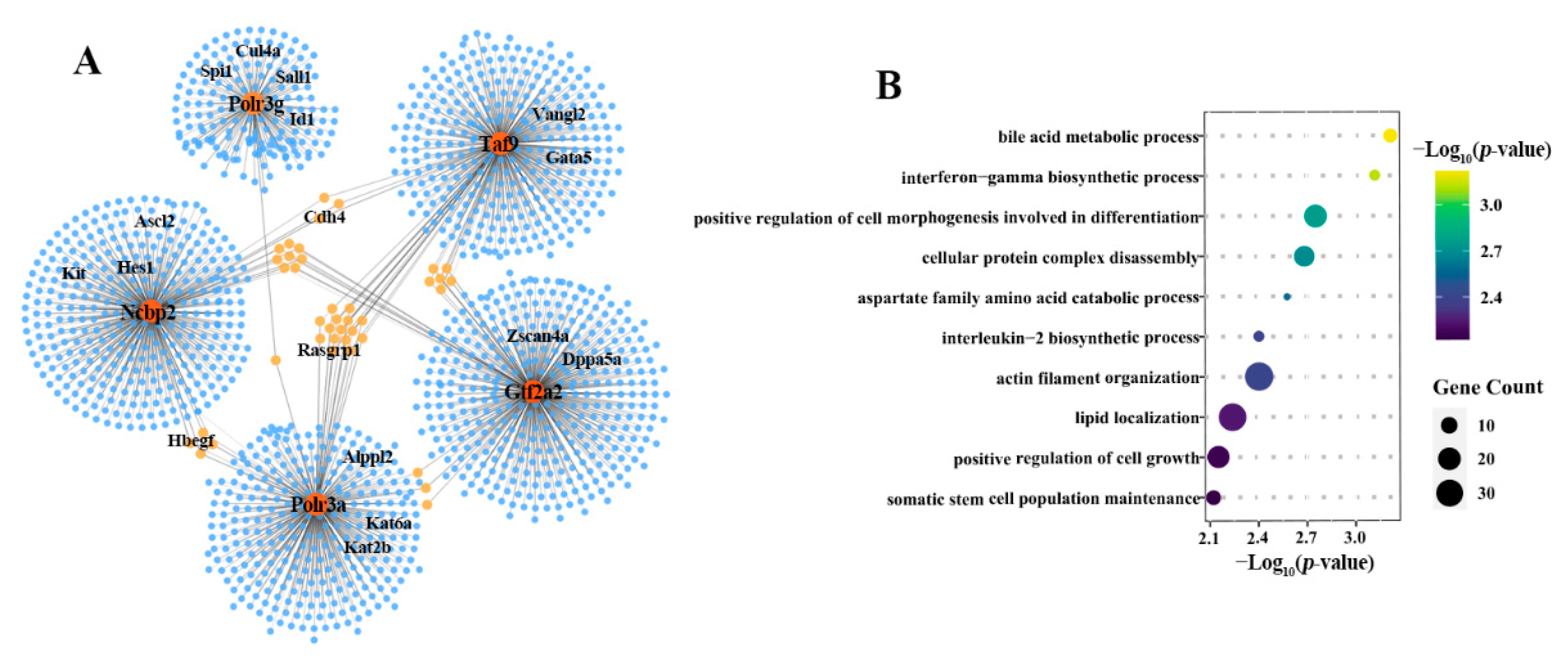
2.4. NTA Embryos Showed Weak Coordination of Key Predicted Target Genes
3. Discussion
4. Materials and Methods
4.1. Dataset Collection
4.2. RNA-seq Data Processing
4.3. Transcription-Related Pathways Selection
4.4. Differential Genes Expression Analysis
4.5. Definition of Transcription Related Gene Rescue in NTB Embryos
4.6. Single-Cell Gene Regulatory Network Inference
4.7. Functional Pathways Enrichment and Statistical Analysis
4.8. Data Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Qu, J.; Li, J.; He, H.; Liu, Z.; Huan, Y. Epigenetic reprogramming during somatic cell nuclear transfer: Recent progress and future directions. Front. Genet. 2020, 11, 205. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.H.S.; Fisher, P.; Chen, W.C.; Choi, I.; Kelly, R.D.W.; Lee, J.H.; Xhu, J. Somatic cell nuclear transfer: Past, present and future perspectives. Theriogenology 2007, 68, S214–S231. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Yang, X.; Smith, S.L.; Tian, X.C.; Lewin, H.A.; Renard, J.P.; Wakayama, T. Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat. Genet. 2007, 39, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Lanza, R.P.; Cibelli, J.B.; West, M.D. Prospects for the use of nuclear transfer in human transplantation. Nat. Biotechnol. 1999, 17, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Ogura, A.; Inoue, K.; Wakayama, T. Recent advancements in cloning by somatic cell nuclear transfer. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2013, 368, 20110329. [Google Scholar] [CrossRef] [Green Version]
- Loi, P.; Iuso, D.; Czernik, M.; Ogura, A. A new, dynamic era for somatic cell nuclear transfer? Trends Biotechnol. 2016, 34, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wang, C.F.; Gao, Y.W.; Xiu, W.C.; Chen, J.Y.; Kou, X.C.; Zhao, Y.H.; Liao, Y.H.; Bai, D.D.; Qiao, Z.B.; et al. Inhibition of aberrant dna re-methylation improves post-implantation development of somatic cell nuclear transfer embryos. Cell Stem Cell 2018, 23, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, X.; Wang, C.; Gao, Y.; Gao, R.; Kou, X.; Zhao, Y.; Li, J.; Wu, Y.; Xiu, W.; et al. Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov. 2016, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Liu, Y.; Lu, F.; Iwabuchi, K.A.; Shen, L.; Inoue, A.; Zhang, Y. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 2014, 159, 884–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, S.; Wang, H.; Jiang, L.; Lu, F.; Iwabuchi, K.A.; Wu, X.; Inoue, K.; Yang, L.; Press, W.; Lee, J.T.; et al. Loss of H3K27me3 imprinting in somatic cell nuclear transfer embryos disrupts post-implantation development. Cell Stem Cell 2018, 23, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Y.; Gao, Y.; Su, J.; Zhang, J.; Xing, X.; Zhou, C.; Yao, K.; An, Q.; Zhang, Y. H3K9 demethylase KDM4E is an epigenetic regulator for bovine embryonic development and a defective factor for nuclear reprogramming. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Zhang, H.; Wei, R.; Li, Q.; Weng, X.; Kong, Q.; Liu, Z. Histone H3 lysine 27 trimethylation acts as an epigenetic barrier in porcine nuclear reprogramming. Reproduction 2016, 151, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, F.; Worrad, D.M.; Schultz, R.M. Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo. Dev. Biol. 1997, 181, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, C.; Li, W.; Liang, P.; Liu, S.; Zuo, Y. Transcriptome comparisons of multi-species identify differential genome activation of mammals embryogenesis. IEEE Access 2019, 7, 7794–7802. [Google Scholar] [CrossRef]
- Schultz, R.M. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum. Reprod. Update 2002, 8, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurita, M.; Reynaud, E.; Aguilar-Fuentes, J. From the beginning: The basal transcription machinery and onset of transcription in the early animal embryo. Cell. Mol. Life Sci. CMLS 2008, 65, 212–227. [Google Scholar] [CrossRef]
- Li, H.; Ta, N.; Long, C.; Zhang, Q.; Li, S.; Liu, S.; Yang, L.; Zuo, Y. The spatial binding model of the pioneer factor Oct4 with its target genes during cell reprogramming. Comput. Struct. Biotechnol. J. 2019, 17, 1226–1233. [Google Scholar] [CrossRef]
- Liu, B.; Xu, Q.; Wang, Q.; Feng, S.; Lai, F.; Wang, P.; Zheng, F.; Xiang, Y.; Wu, J.; Nie, J.; et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 2020, 587, 139–144. [Google Scholar] [CrossRef]
- Zuo, Y.; Su, G.; Cheng, L.; Liu, K.; Feng, Y.; Wei, Z.; Bai, C.; Cao, G.; Li, G. Coexpression analysis identifies nuclear reprogramming barriers of somatic cell nuclear transfer embryos. Oncotarget 2017, 8, 65847–65859. [Google Scholar] [CrossRef]
- Zuo, Y.; Gao, Y.; Su, G.; Bai, C.; Wei, Z.; Liu, K.; Li, Q.; Bou, S.; Li, G. Irregular transcriptome reprogramming probably causes thec developmental failure of embryos produced by interspecies somatic cell nuclear transfer between the Przewalski’s gazelle and the bovine. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Aibara, S.; Schilbach, S.; Cramer, P. Structures of mammalian RNA polymerase II pre-initiation complexes. Nature 2021, 594, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Su, G.; Wang, S.; Lei, Y.; Li, G. Exploring timing activation of functional pathway based on differential co-expression analysis in preimplantation embryogenesis. Oncotarget 2016, 7, 74120–74131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossant, J.; Tam, P.P.L. New insights into early human development: Lessons for stem cell derivation and differentiation. Cell Stem Cell 2017, 20, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Bushnell, D.A.; Kornberg, R.D. RNA polymerase II transcription: Structure and mechanism. Biochim. Biophys. Acta 2013, 1829, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, Y.; Wu, Z.; Zhang, J.; Zhu, J.; Liu, Z.; Song, X. Epigenetic modification agents improve gene-specific methylation reprogramming in porcine cloned embryos. PLoS ONE 2015, 10, e0129803. [Google Scholar] [CrossRef]
- Kiefer, H.; Jouneau, L.; Campion, É.; Rousseau-Ralliard, D.; Larcher, T.; Martin-Magniette, M.L.; Balzergue, S.; Ledevin, M.; Prézelin, A.; Chavatte-Palmer, P.; et al. Altered DNA methylation associated with an abnormal liver phenotype in a cattle model with a high incidence of perinatal pathologies. Sci. Rep. 2016, 6, 38869. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Cai, Y.; Wang, Y.; Nie, Y.; Zhang, C.; Xu, Y.; Zhang, X.; Lu, Y.; Wang, Z.; Poo, M.; et al. Cloning of macaque monkeys by somatic cell nuclear transfer. Cell 2018, 172, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gao, S.; Liu, X. Advance in the role of epigenetic reprogramming in somatic cell nuclear transfer-mediated embryonic development. Stem Cells Int. 2021, 2021, 6681337. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, M.; Yang, W.; Cao, P.; Zheng, L.; Zuo, Y. A comparative analysis of single-cell transcriptome identifies reprogramming driver factors for efficiency improvement. Mol. Ther. Nucleic Acids 2020, 19, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lin, L.; Wang, X.; Gao, M.; Cao, S.; Mai, Y.; Wu, F.; Kuang, J.; Liu, H.; Yang, J.; et al. Resolving cell fate decisions during somatic cell reprogramming by single-cell rna-seq. Mol. Cell 2019, 73, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Aloia, L.; Gutierrez, A.; Caballero, J.M.; Di Croce, L. Direct interaction between Id1 and Zrf1 controls neural differentiation of embryonic stem cells. EMBO Rep. 2015, 16, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.S.; Volk, C.; Marga, C.; Navarrete Santos, A.; Jung, M.; Rujescu, D.; Navarrete Santos, A. Adipose-derived stem/stromal cells recapitulate aging biomarkers and show reduced stem cell plasticity affecting their adipogenic differentiation capacity. Cell Reprogram. 2019, 21, 187–199. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, Z.; Chen, J.; Huang, H.; Xu, Y.; Ding, X.; Zheng, Y.; Nishinakamura, R.; Xu, G.L.; Wang, H.; et al. Cooperative action between SALL4A and TET proteins in stepwise oxidation of 5-methylcytosine. Mol. Cell 2016, 64, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Gurdon, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar]
- Czernik, M.; Anzalone, D.A.; Palazzese, L.; Oikawa, M.; Loi, P. Somatic cell nuclear transfer: Failures, successes and the challenges ahead. Int. J. Dev. Biol. 2019, 63, 123–130. [Google Scholar] [CrossRef]
- Cramer, P.; Bushnell, D.A.; Fu, J.; Gnatt, A.L.; Maier-Davis, B.; Thompson, N.E.; Burgess, R.R.; Edwards, A.M.; David, P.R.; Kornberg, R.D. Architecture of RNA polymerase II and implications for the transcription mechanism. Science 2000, 288, 640–649. [Google Scholar] [CrossRef] [Green Version]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef] [Green Version]
- Weinmann, R. The basic RNA polymerase II transcriptional machinery. Gene Expr. J. Liver Res. 1992, 2, 81–91. [Google Scholar]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.F.; Liu, D.Y.; Wang, Z.R.; Tian, R.X.; Zuo, Y.C. Multi-substrate selectivity based on key loops and non-homologous domains: New insight into ALKBH family. Cell. Mol. Life Sci. 2021, 78, 129–141. [Google Scholar] [CrossRef]
- Zuo, Y.C.; Song, M.M.; Li, H.S.; Chen, X.; Cao, P.B.; Zheng, L.; Cao, G.F. Analysis of the epigenetic signature of cell reprogramming by computational DNA methylation profiles. Curr. Bioinform. 2020, 15, 589–599. [Google Scholar] [CrossRef]
- Liu, D.; Li, G.; Zuo, Y. Function determinants of TET proteins: The arrangements of sequence motifs with specific codes. Brief. Bioinform. 2019, 20, 1826–1835. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Xu, B.; Tian, R.; Zuo, Y. Modular arrangements of sequence motifs determine the functional diversity of KDM proteins. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, F.; Zhang, L.; Wu, X.; Li, D.; Xin, J.; Xie, J.; Kong, F.; Wang, W.; Wu, Q.; et al. Transcriptional defects and reprogramming barriers in somatic cell nuclear reprogramming as revealed by single-embryo RNA sequencing. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varet, H.; Brillet-Gueguen, L.; Coppee, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Long, C.; Xiang, J.; Liang, P.; Li, X.; Zuo, Y. Dppa2/4 as a trigger of signaling pathways to promote zygote genome activation by binding to CG-rich region. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Janky, R.; Aerts, S. iRegulon and i-cisTarget: Reconstructing Regulatory Networks Using Motif and Track Enrichment. Curr. Protoc. Bioinform. 2015, 52, 2–16. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, C.; Li, H.; Li, X.; Yang, W.; Zuo, Y. Nuclear Transfer Arrest Embryos Show Massive Dysregulation of Genes Involved in Transcription Pathways. Int. J. Mol. Sci. 2021, 22, 8187. https://doi.org/10.3390/ijms22158187
Long C, Li H, Li X, Yang W, Zuo Y. Nuclear Transfer Arrest Embryos Show Massive Dysregulation of Genes Involved in Transcription Pathways. International Journal of Molecular Sciences. 2021; 22(15):8187. https://doi.org/10.3390/ijms22158187
Chicago/Turabian StyleLong, Chunshen, Hanshuang Li, Xinru Li, Wuritu Yang, and Yongchun Zuo. 2021. "Nuclear Transfer Arrest Embryos Show Massive Dysregulation of Genes Involved in Transcription Pathways" International Journal of Molecular Sciences 22, no. 15: 8187. https://doi.org/10.3390/ijms22158187
APA StyleLong, C., Li, H., Li, X., Yang, W., & Zuo, Y. (2021). Nuclear Transfer Arrest Embryos Show Massive Dysregulation of Genes Involved in Transcription Pathways. International Journal of Molecular Sciences, 22(15), 8187. https://doi.org/10.3390/ijms22158187