
Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases

 , , , , ,
, , , , ,  and
and
Abstract
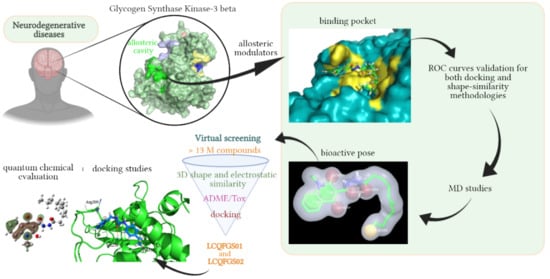
:
1. Introduction
2. Results
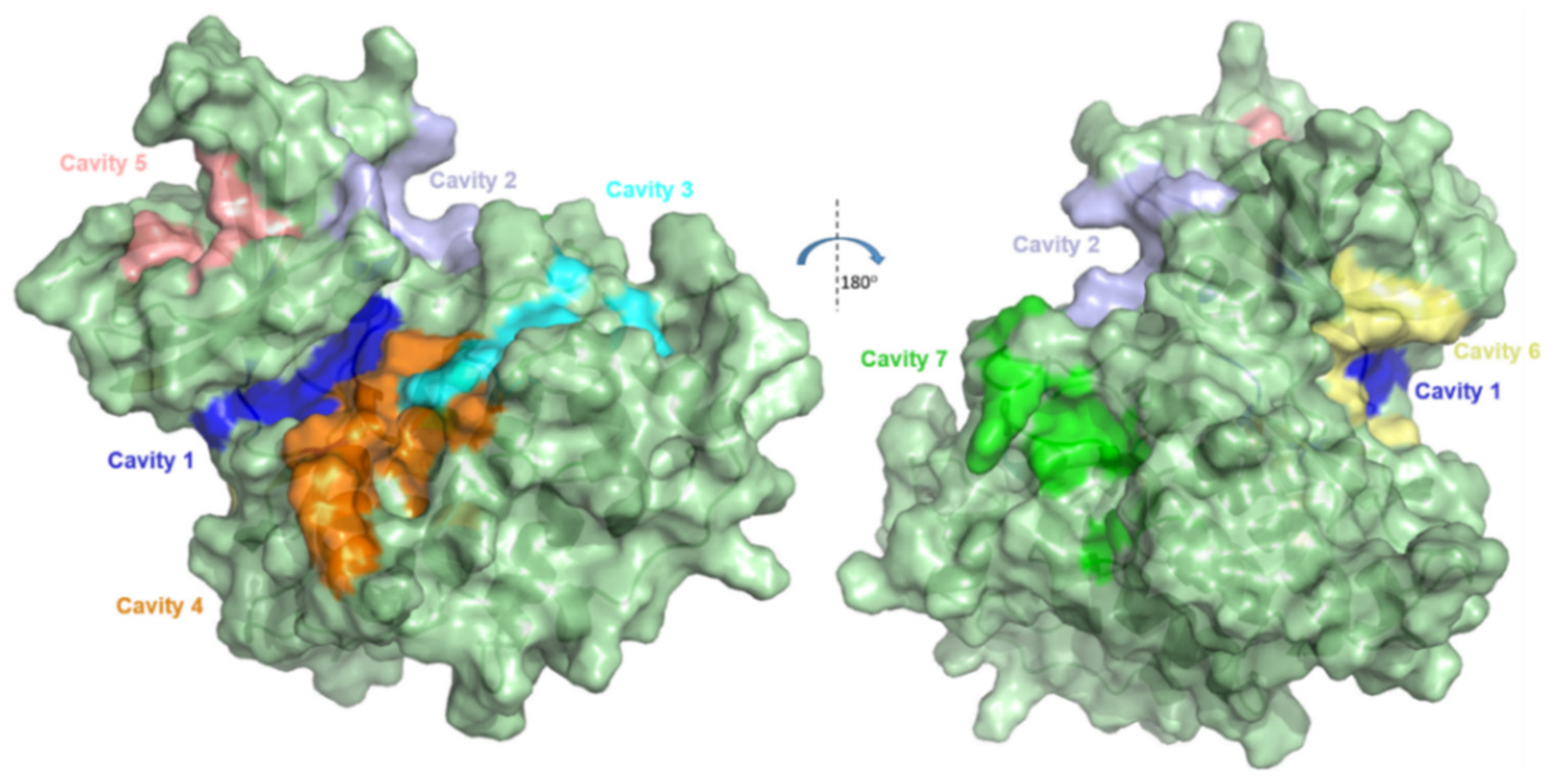
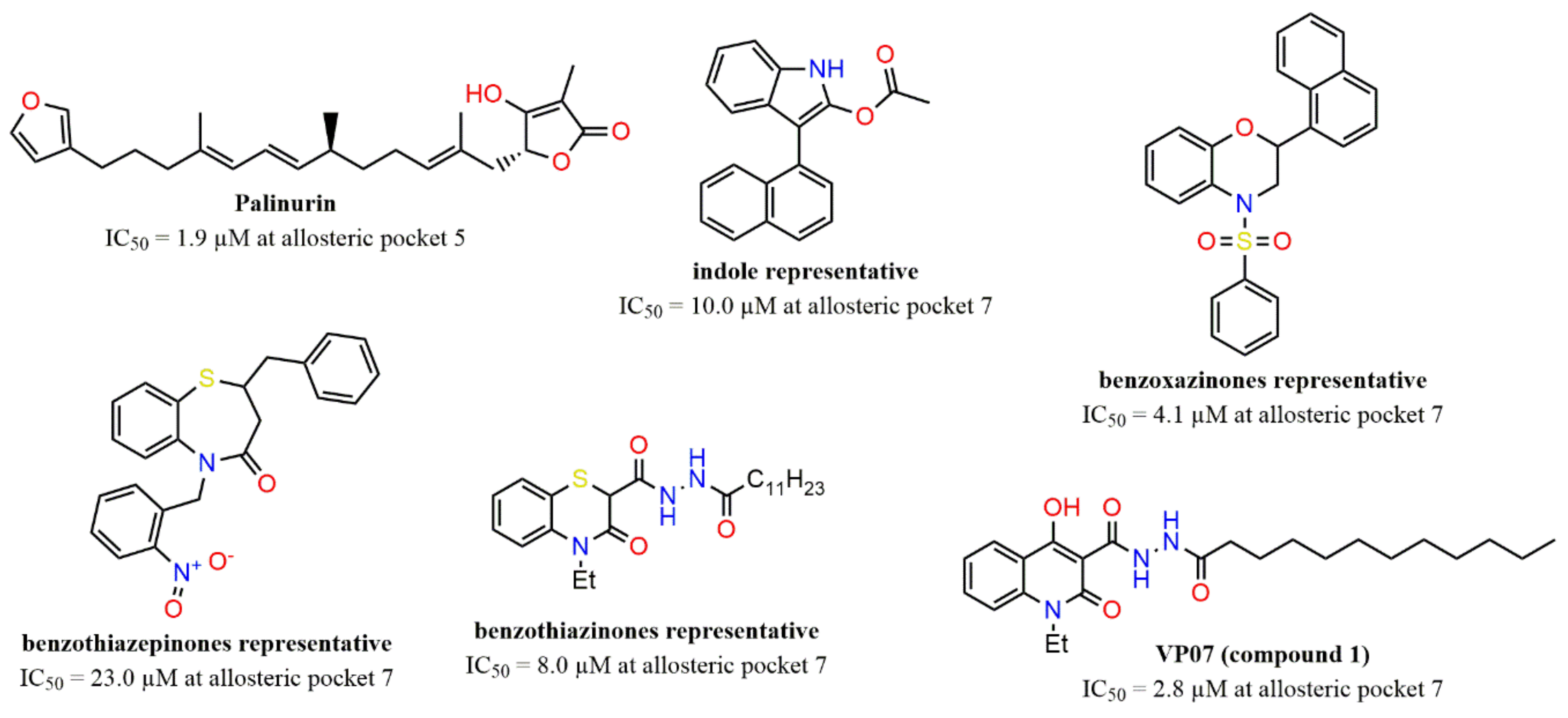
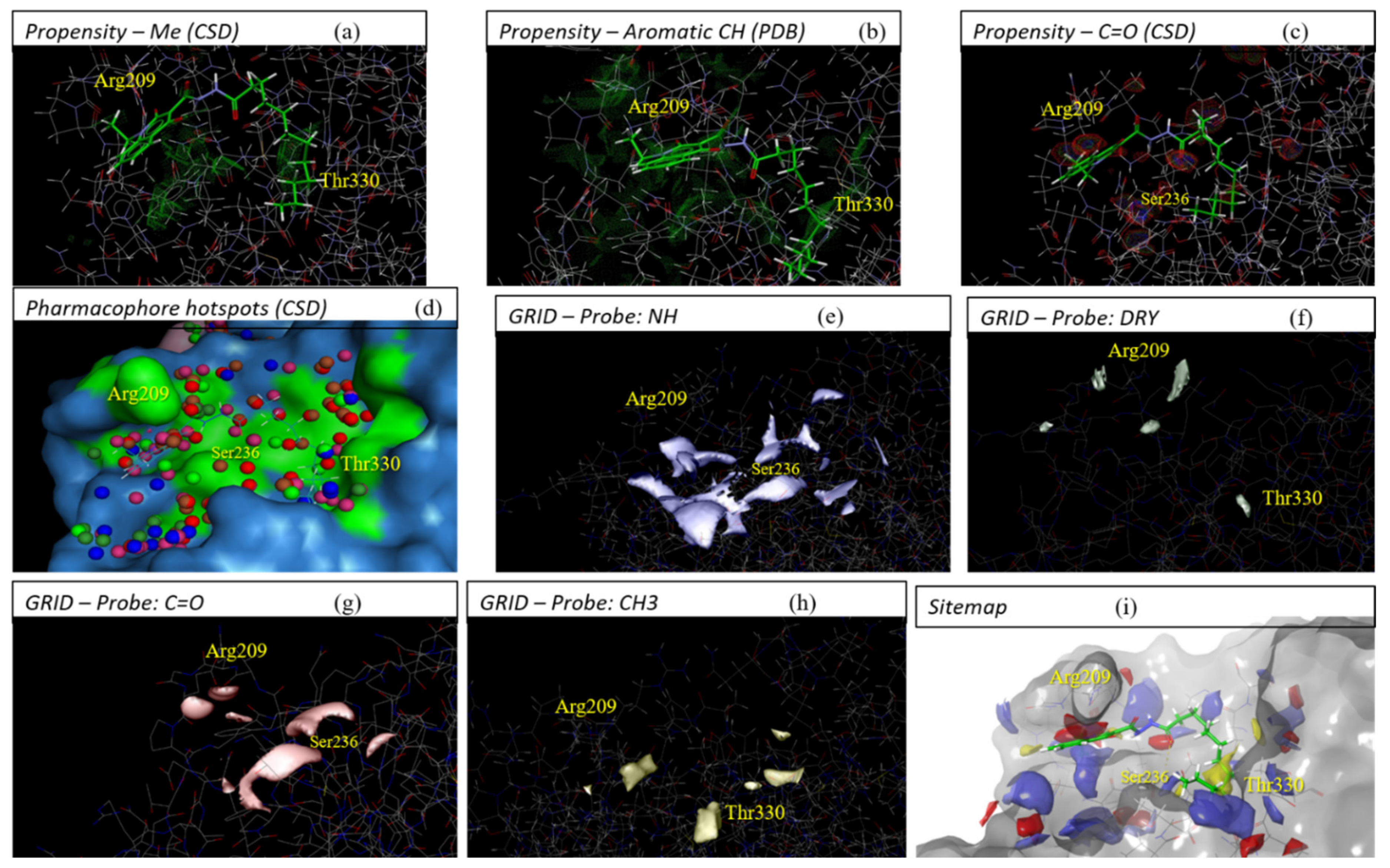
2.1. Detection and Prediction of the Potential of Allosteric Pockets
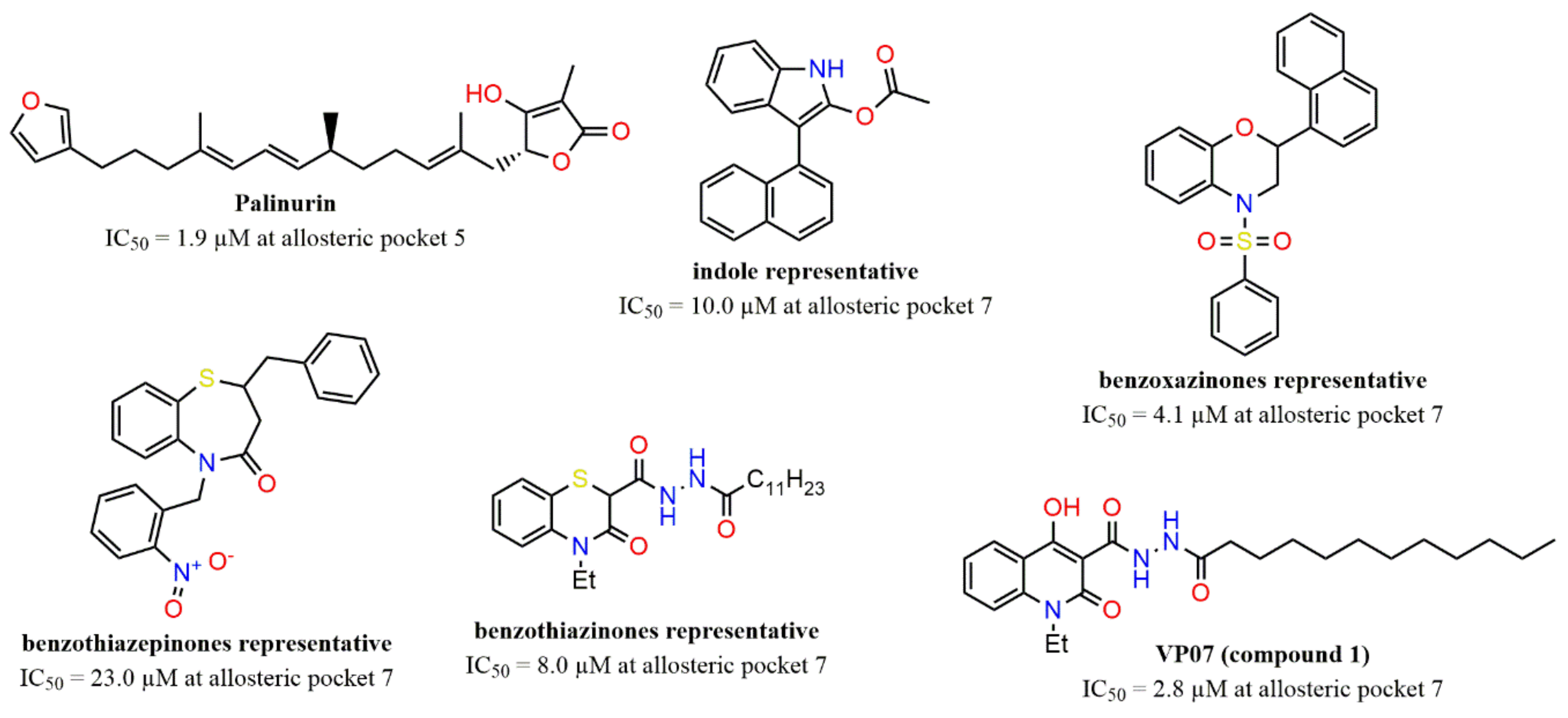
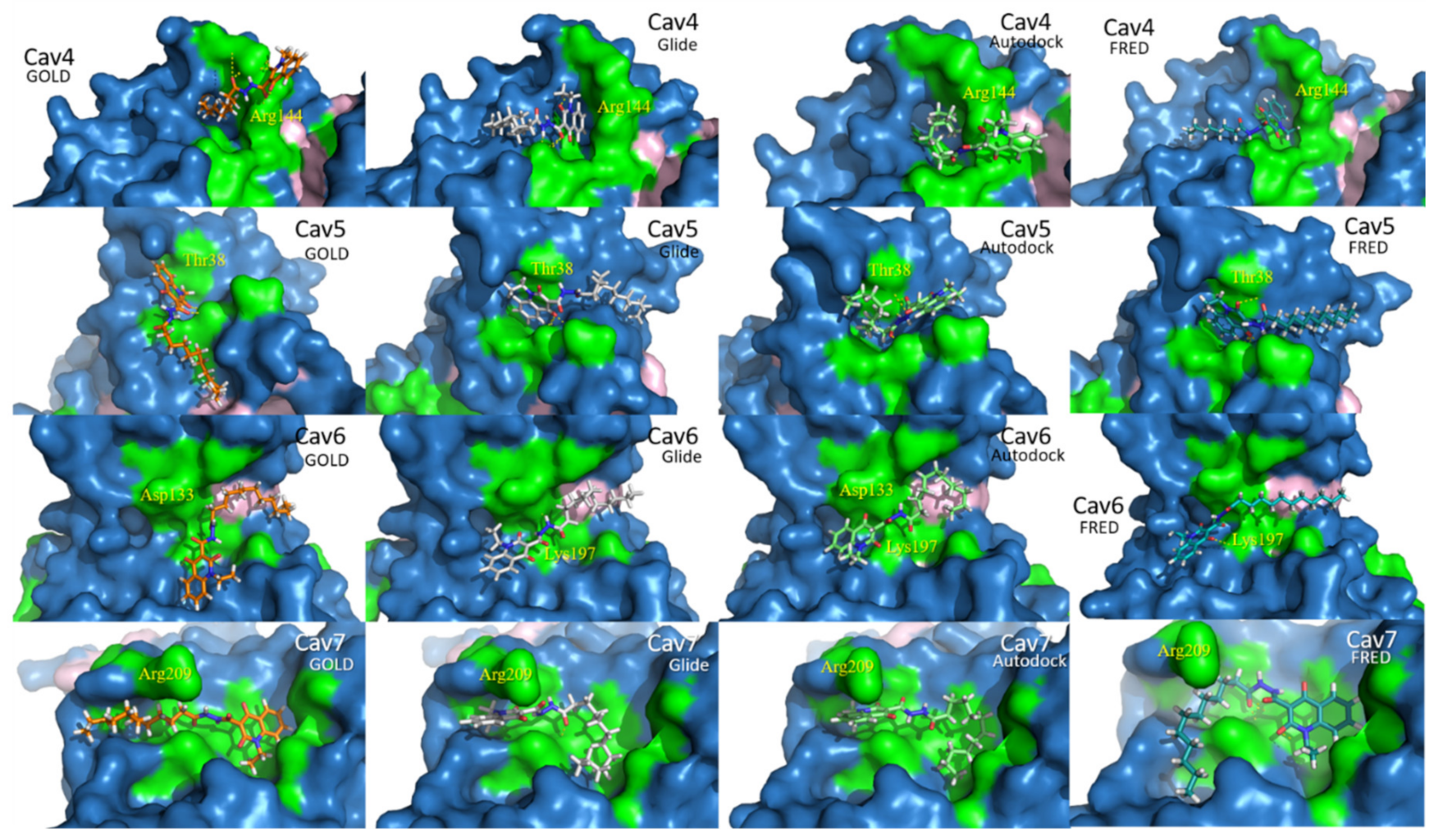
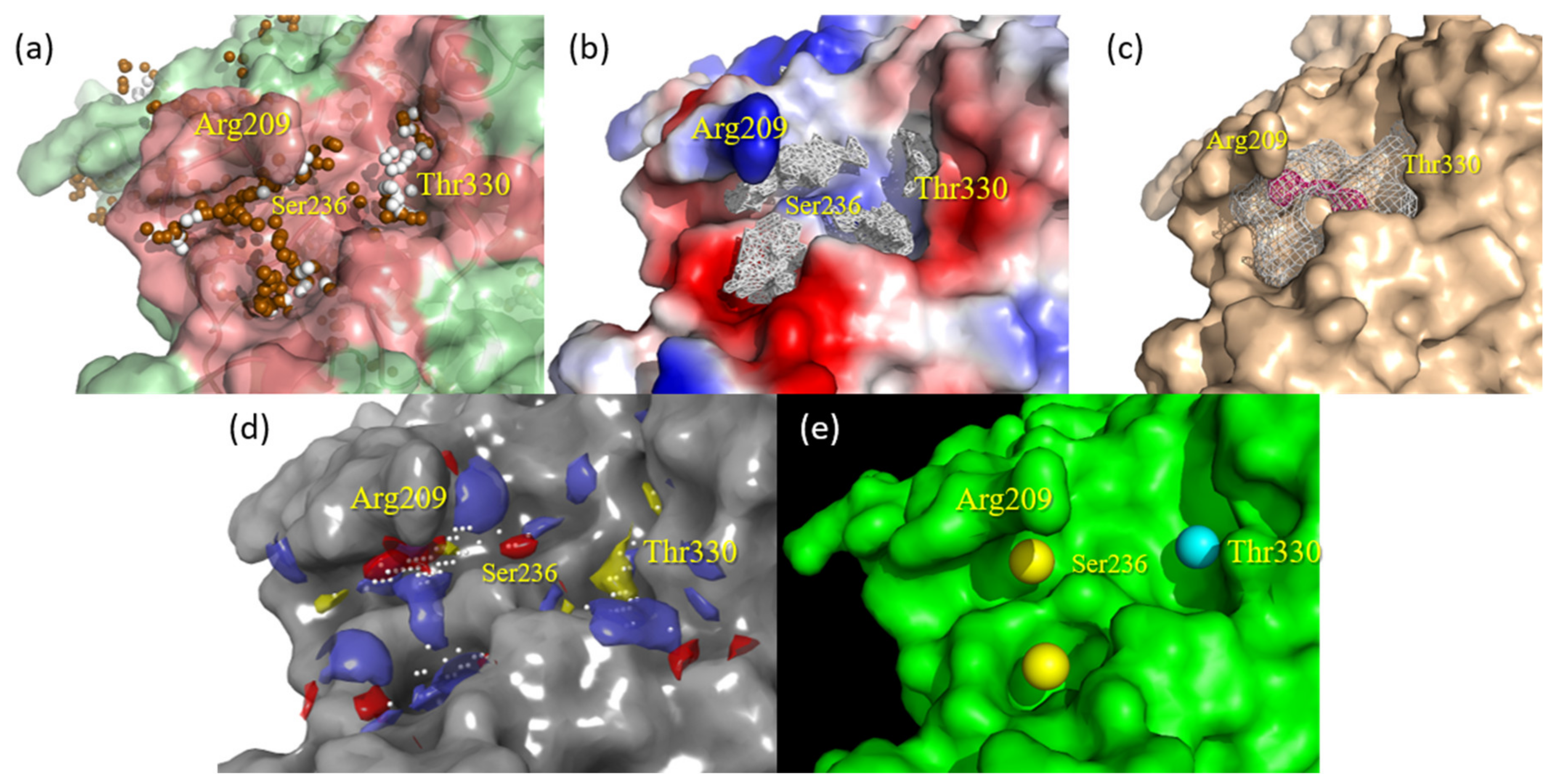
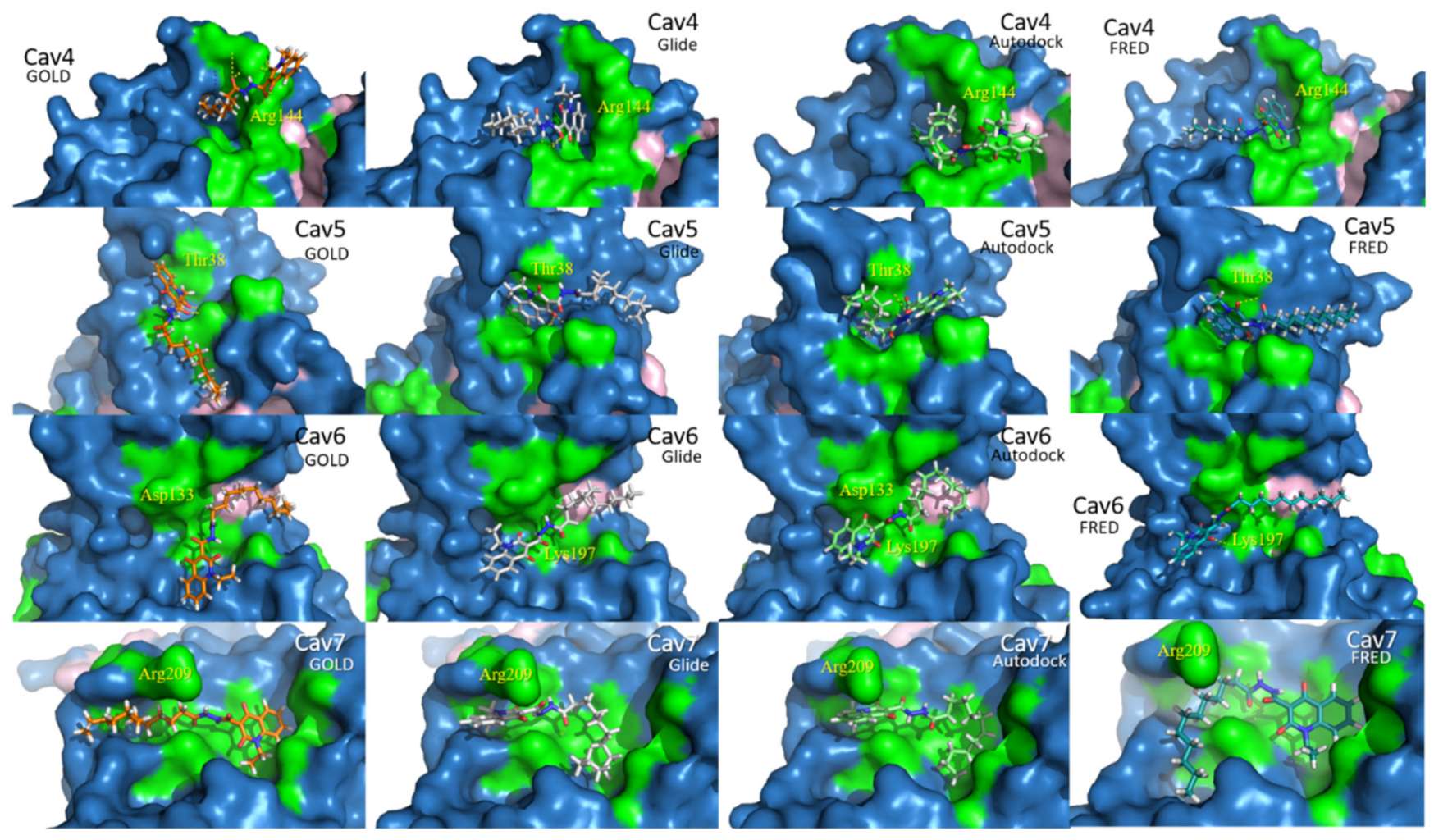
2.2. Docking Assessment—Pocket Perspective
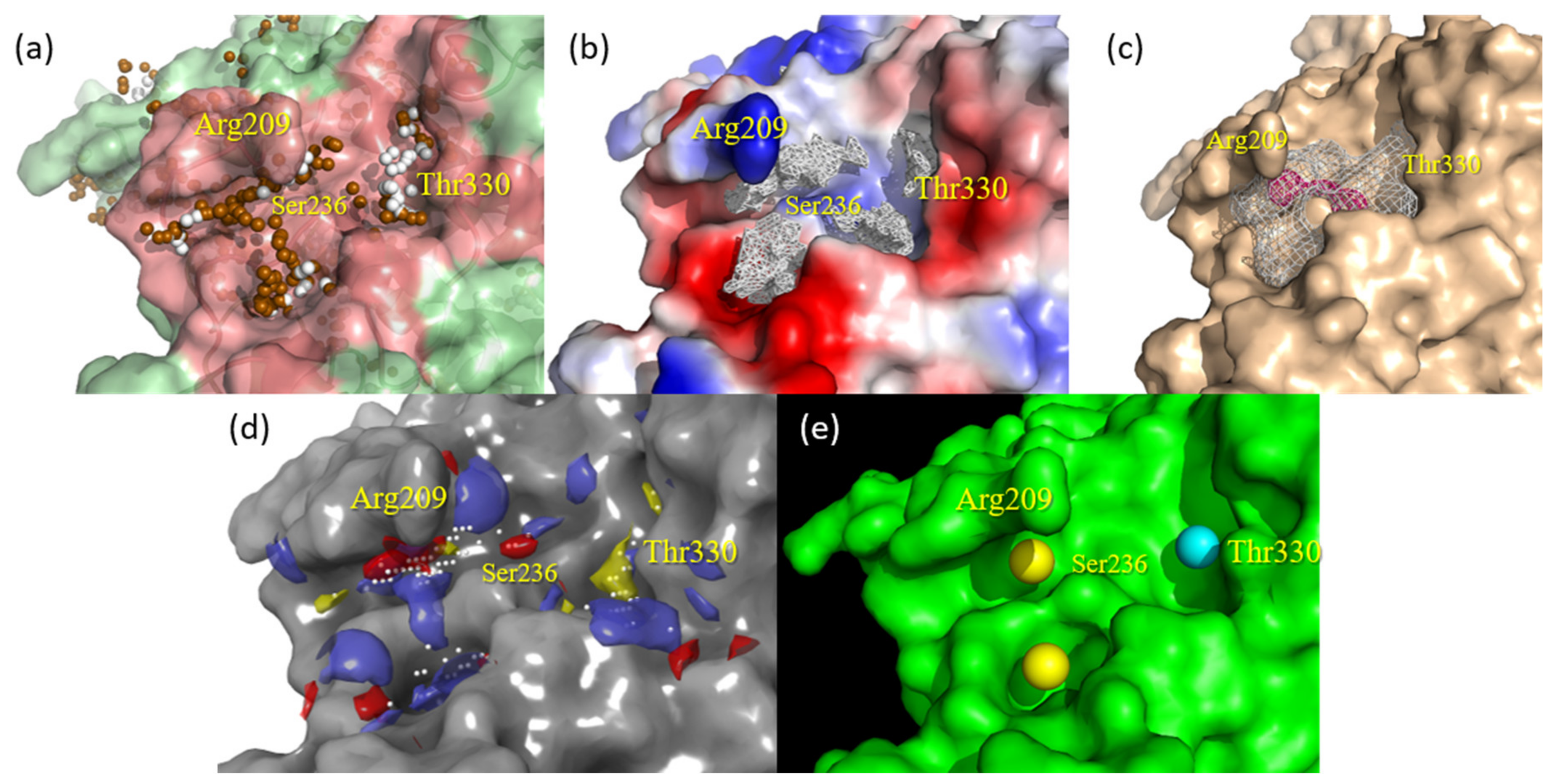
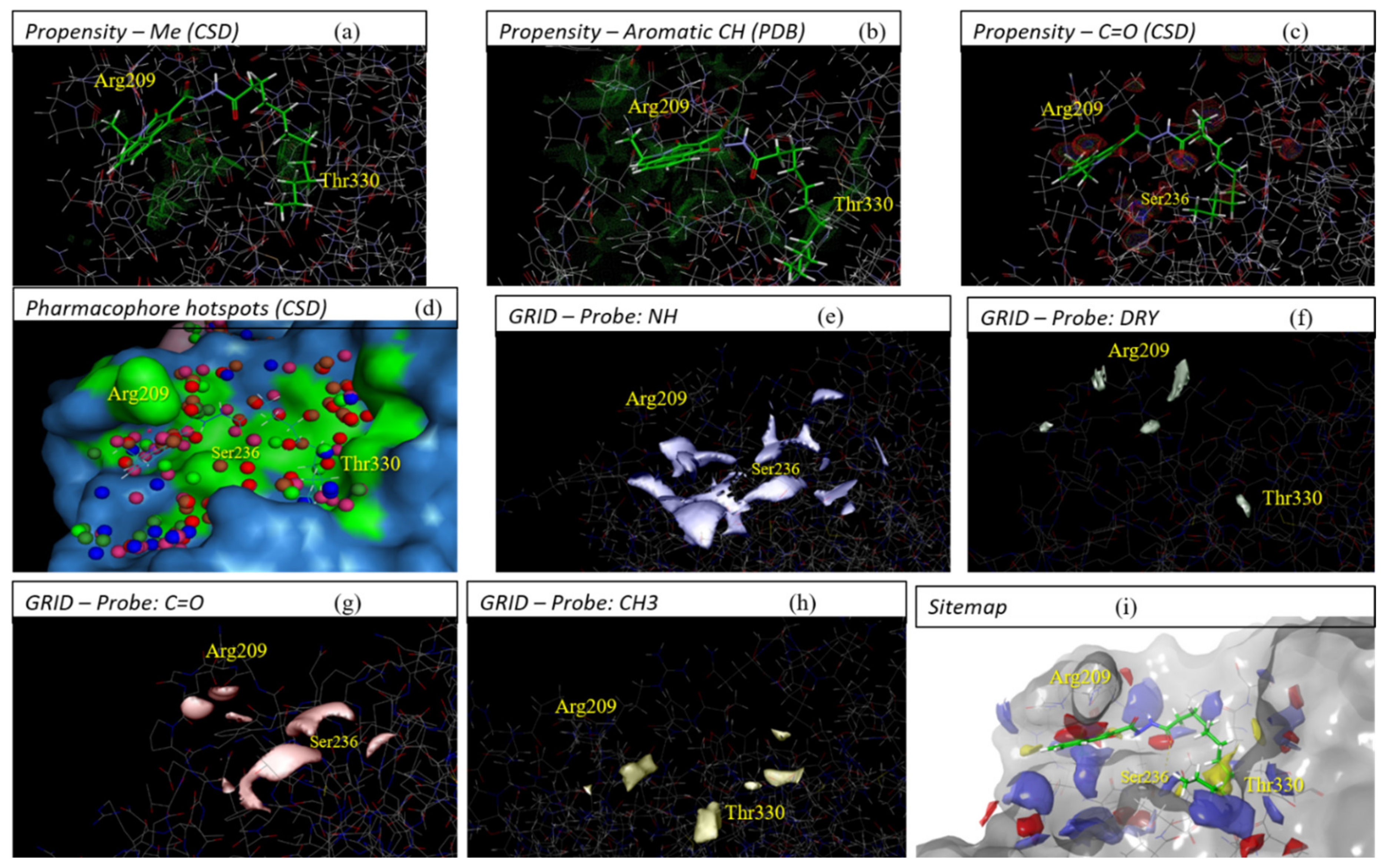
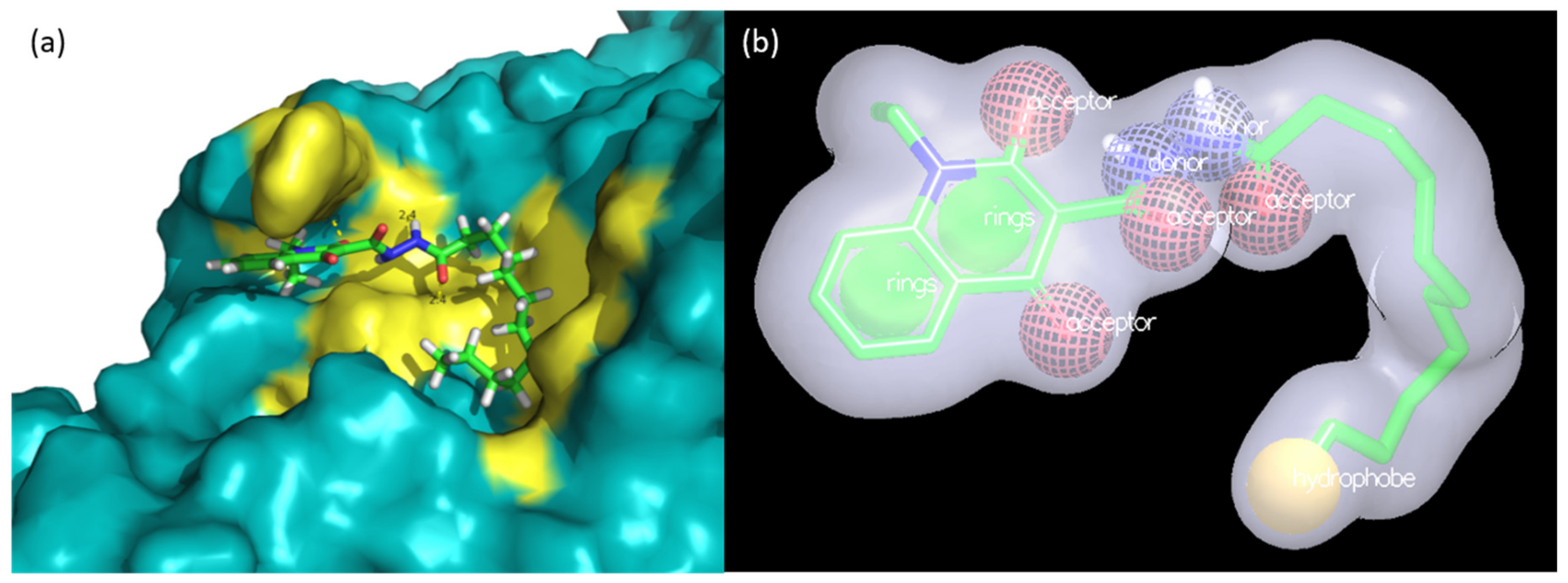
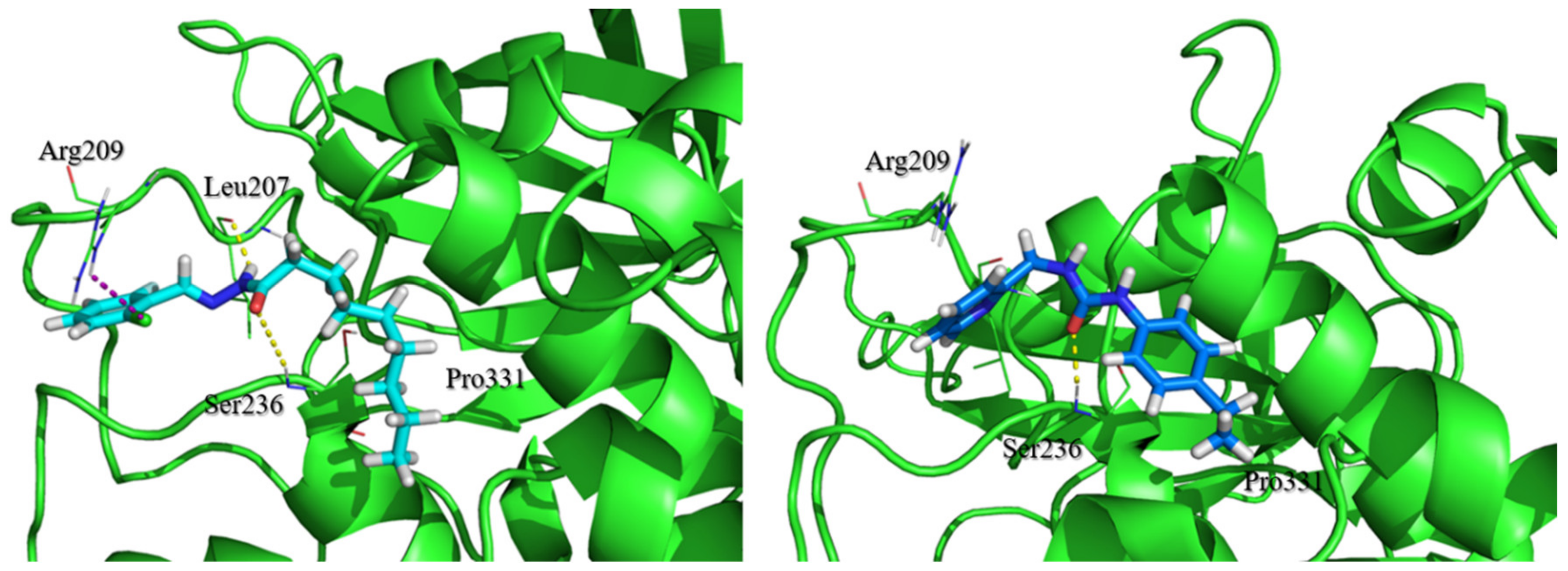
2.3. Evaluation of Compound 1 Pose within Allosteric Pocket 7
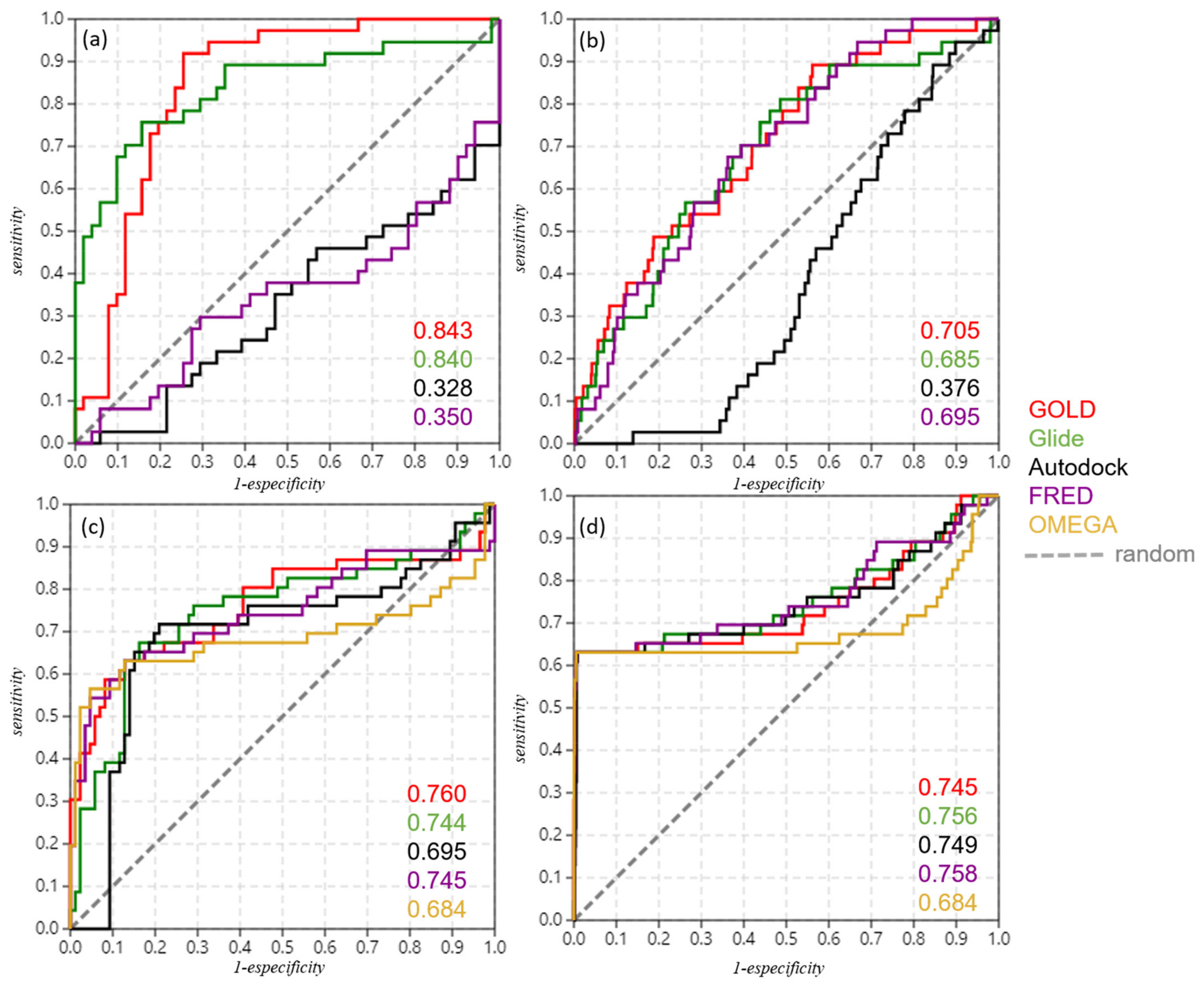
2.4. Refinement and Validation of Docking Protocols—Pose Perspective
2.5. Optimizing Docking Validation
2.6. Shape Similarity and Query Validation
2.7. Molecular Dynamics Study
2.8. Virtual Screening
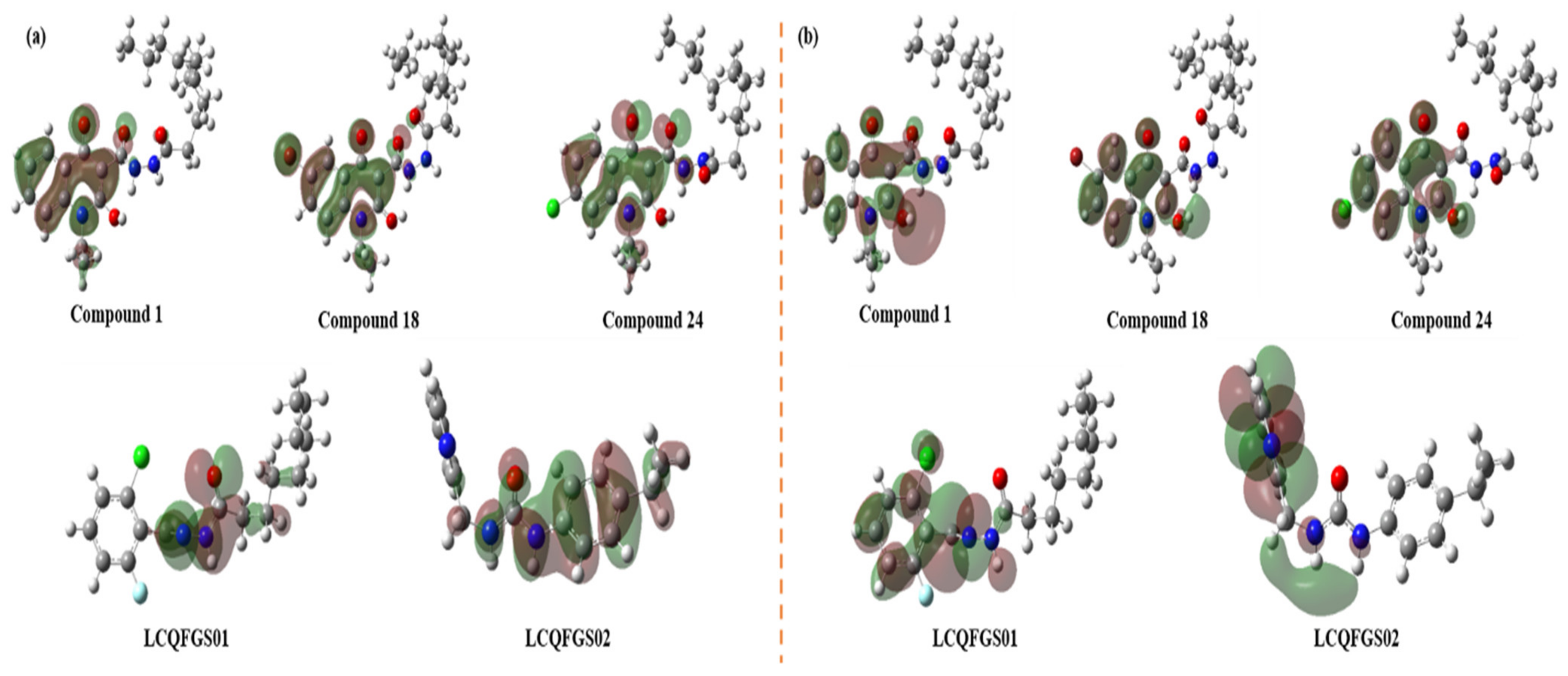
2.9. Quantum Chemical Studies
3. Discussion
4. Materials and Methods
4.1. Pocket Detectors/Predictors
4.2. Docking Simulations
4.3. Generation of Contour/Surface Maps
4.4. Dataset Compilation
4.5. Refinement of Docking Protocols
4.6. Validation of Docking Protocols
4.7. Shape Similarity and Query Validation
4.8. Molecular Dynamics Studies
4.9. Virtual Screening Campaign
4.10. Quantum Chemical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADME/Tox | Absorption, distribution, metabolism, and excretion/toxicity |
| APP | Amyloid precursor protein |
| ATP | Adenosine triphosphate |
| AUC | Area under the curve |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| CNS | Central nervous system |
| DFT | Density function theory |
| EONTC | EON TanimotoCombo indices |
| FMO | Frontier molecular orbital |
| GSK-3β | Glycogen synthase kinase 3 beta |
| HOA% | Human oral absorption in % |
| HOMO | Highest occupied molecular orbital |
| IP | Ionization potential |
| LBVS | Ligand-based virtual screening |
| LUMO | Lowest unoccupied molecular orbital |
| MAPT | Microtubule-associated protein tau |
| MD | Molecular dynamics |
| MIFs | Molecular interaction fields |
| MW | Molecular weight |
| NFTs | Neurofibrillary tangles |
| PD | Parkinson’s disease |
| PDB | Protein Data Bank |
| PSA | Polar surface area |
| (QP)logBB | Logarithm of blood-brain barrier predicted by QikProp |
| (QP)logPo/w | Logarithm of partition coefficient in 1-octanol/water predicted by QikProp |
| (QP)PCaco | Permeability across Caco-2 cells predicted by QikProp |
| (QP)PMDCK | Permeability across Madin-Darby Canine Kidney cells predicted by QikProp |
| Rg | Radius of gyrate |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| ROC | Receiver operating characteristic |
| ROCSTC | ROCS TanimotoCombo indices |
| SASA | Solvent-accessible surface area |
| VS | Virtual screening |
| XPscore | Glide extra precision score values |
References
- Dumurgier, J.; Tzourio, C. Epidemiology of neurological diseases in older adults. Rev. Neurol. 2020, 176, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, R.; Munjal, N.S.; Singh, T.R. Identification of novel small molecules against GSK3β for Alzheimer’s disease using chemoinformatics approach. J. Mol. Graph. Model. 2019, 91, 91–104. [Google Scholar] [CrossRef]
- Gao, C.; Hlscher, C.; Liu, Y.; Li, L. GSK3: A key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev. Neurosci. 2012, 23, 1–11. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fujishiro, H.; Takechi, H. Efficacy and safety of glycogen synthase kinase 3 inhibitors for Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2019, 69, 1031–1039. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Ilouz, R.; Kaidanovich, O.; Gurwitz, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3β by bivalent zinc ions: Insight into the insulin-mimetic action of zinc. Biochem. Biophys. Res. Commun. 2002, 295, 102–106. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget 2017, 8, 14221–14250. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Lupo, G.; Pennisi, M.; Basile, M.; Anfuso, C.; Petralia, M.; Gattuso, G.; Vivarelli, S.; Spandidos, D.; Libra, M.; et al. The analysis of miRNA expression profiling datasets reveals inverse microRNA patterns in glioblastoma and Alzheimer’s disease. Oncol. Rep. 2019, 42, 911–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snitow, M.E.; Bhansali, R.S.; Klein, P.S. Lithium and Therapeutic Targeting of GSK-3. Cells 2021, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Hernández, F. GSK-3 inhibitors for Alzheimer’ s disease. Expert Rev. Neurother. 2007, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.H. Magnesium ion catalyzed ATP hydrolysis. J. Am. Chem. Soc. 2000, 122, 12023–12024. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.-Z.; Mandelkow, E.-M.; et al. Indirubins Inhibit Glycogen Synthase Kinase-3β and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease. J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Meijer, L.; Thunnissen, A.M.W.H.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Silva, G.M.; Barcelos, M.P.; Poiani, J.G.C.; Hage-Melim, L.I.D.S.; da Silva, C.H.T.D.P. Allosteric Modulators of Potential Targets Related to Alzheimer’s Disease: A Review. ChemMedChem 2019, 14, 1467–1483. [Google Scholar] [CrossRef] [Green Version]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A phase II trial of tideglusib in alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the binding sites of glycogen synthase kinase 3. identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef] [Green Version]
- The Pymol Molecular Graphics System; 1.3; Schrödinger LLC: New York, NY, USA, 2010.
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Ramunno, A.; Savi, L.; Chemi, G.; Alfano, G.; Pecorelli, A.; Pambianchi, E.; Galatello, P.; Compagnoni, G.; Focher, F.; et al. First dual AK/GSK-3β inhibitors endowed with antioxidant properties as multifunctional, potential neuroprotective agents. Eur. J. Med. Chem. 2017, 138, 438–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, H.-R.; Bian, S.-H.; Huang, Z.-H.; Chu, Y.; Ye, D.-Y. Design, synthesis and biological evaluation of benzothiazepinones (BTZs) as novel non-ATP competitive inhibitors of glycogen synthase kinase-3β (GSK-3β). Eur. J. Med. Chem. 2013, 61, 95–103. [Google Scholar] [CrossRef]
- Zhang, P.; Li, S.; Gao, Y.; Lu, W.; Huang, K.; Ye, D.; Li, X.; Chu, Y. Novel benzothiazinones (BTOs) as allosteric modulator or substrate competitive inhibitor of glycogen synthase kinase 3β (GSK-3β) with cellular activity of promoting glucose uptake. Bioorganic Med. Chem. Lett. 2014, 24, 5639–5643. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, P.; Cui, A.; Ye, D.-Y.; Xiang, M.; Chu, Y. Discovery and anti-inflammatory evaluation of benzothiazepinones (BTZs) as novel non-ATP competitive inhibitors of glycogen synthase kinase-3β (GSK-3β). Bioorg. Med. Chem. 2018, 26, 5479–5493. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y.; Koh, S.-H.; Noh, M.Y.; Park, K.-W.; Lee, Y.J.; Kim, S.H. Glycogen synthase kinase-3β activity plays very important roles in determining the fate of oxidative stress-inflicted neuronal cells. Brain Res. 2007, 1129, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SuperStar Cambridge Crystallographic Data Centre. SuperStar User Guide and Tutorials—Release Update 3. 2018, p. 82. Available online: https://www.ccdc.cam.ac.uk/support-and-resources/ccdcresources/34351740a7f346c980d01ec3324e9328.pdf (accessed on 18 May 2021).
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 2011. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009. [Google Scholar] [CrossRef] [PubMed]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CCDC GOLD—Protein Ligand Docking Software. Available online: https://www.ccdc.cam.ac.uk/solutions/csd-discovery/Components/Gold/ (accessed on 18 May 2021).
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein—Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 623, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Glide Schrödinger Suite; 2019-2; Schrödinger: New York, NY, USA, 2019.
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Lindstrom, W.; Huey, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- The Scripps Research Institute Autodock 4. Available online: http://autodock.scripps.edu/ (accessed on 18 May 2021).
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011. [Google Scholar] [CrossRef]
- FRED; 3.5.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- vROCS; 3.3.2.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- OMEGA; 4.0.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- Kurt, M.; Babu, P.C.; Sundaraganesan, N.; Cinar, M.; Karabacak, M. Molecular structure, vibrational, UV and NBO analysis of 4-chloro-7-nitrobenzofurazan by DFT calculations. Spectrochim. Acta. Part A Mol. Biomol. Spectrosc. 2011, 79, 1162–1170. [Google Scholar] [CrossRef]
- Breno Rodrigues dos Santos, C.; Carvalho Lobato, C.; Alexandre Costa de Sousa, M.; Jorge da Cruz Macêdo, W.; Carlos Tavares Carvalho, J. Molecular Modeling: Origin, Fundamental Concepts and Applications Using Structure-Activity Relationship and Quantitative Structure-Activity Relationship. Rev. Theor. Sci. 2013, 2, 1–25. [Google Scholar] [CrossRef]
- Aihara, J.I. Weighted HOMO-LUMO energy separation as an index of kinetic stability for fullerenes. Theor. Chem. Acc. 1999, 102, 134–138. [Google Scholar] [CrossRef]
- Mendes, A.P.S.; Borges, R.S.; Neto, A.M.J.C.; De Macedo, L.G.M.; Da Silva, A.B.F. The basic antioxidant structure for flavonoid derivatives. J. Mol. Model. 2012, 18, 4073–4080. [Google Scholar] [CrossRef] [PubMed]
- Santos, K.L.B.; Queiroz, A.N.; Lobato, C.C.; Vale, J.K.L.; Santos, C.B.R.; Borges, R.S. A comparative theoretical mechanism on simplified flavonoid derivatives and isoxazolone analogous as Michael system inhibitor. J. Mol. Model. 2021, 27, 1–7. [Google Scholar] [CrossRef]
- Kaya, S.; Tüzün, B.; Kaya, C.; Obot, I.B. Determination of corrosion inhibition effects of amino acids: Quantum chemical and molecular dynamic simulation study. J. Taiwan Inst. Chem. Eng. 2016, 58, 528–535. [Google Scholar] [CrossRef]
- OpenEye Scientific Software. Documentation—Applications 2020.0.4. Available online: https://docs.eyesopen.com/applications/index.html (accessed on 7 February 2020).
- Ferreira, L.G.; Santos, R.N.D.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421, ISBN 5516337380. [Google Scholar] [CrossRef] [PubMed]
- Maestro Schrödinger Suite; 2020-1; Schrödinger: New York, NY, USA, 2020.
- Chemdraw Professional; 18.2.0.48; PerkinElmer Informatics Inc.: Waltham, MA, USA, 2021.
- LigPrep—Suite; 2015-2; Schrödinger: New York, NY, USA, 2018.
- Protein Preparation Wizard—Suite; 2015-2; Schrödinger: New York, NY, USA, 2018.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Westbrook, J.D.; Shao, C.; Feng, Z.; Zhuravleva, M.; Velankar, S.; Young, J. The chemical component dictionary: Complete descriptions of constituent molecules in experimentally determined 3D macromolecules in the Protein Data Bank. Bioinformatics 2015. [Google Scholar] [CrossRef] [Green Version]
- OpenEye Scientific Software. 2020. Available online: https://www.eyesopen.com/ (accessed on 18 May 2021).
- Krishnan, N.; Bonham, C.A.; Rus, I.A.; Shrestha, O.K.; Gauss, C.M.; Haque, A.; Tocilj, A.; Joshua-Tor, L.; Tonks, N.K. Harnessing insulin-and leptin-induced oxidation of PTP1B for therapeutic development. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer-An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Nicholls, A. What do we know and when do we know it? J. Comput. Aided. Mol. Des. 2008, 22, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Chávez Thielemann, H.; Cardellini, A.; Fasano, M.; Bergamasco, L.; Alberghini, M.; Ciorra, G.; Chiavazzo, E.; Asinari, P. From GROMACS to LAMMPS: GRO2LAM. J. Mol. Model. 2019. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.B.; Silva, G.M.; de Fraga Dias, A.; Figueiró, F.; Battastini, A.M.O.; dos Santos, C.B.R.; Costa, L.T.; Rosa, J.M.C.; de Paula da Silva, C.H.T. Identification of novel αβ-tubulin modulators with antiproliferative activity directed to cancer therapy using ligand and structure-based virtual screening. Int. J. Biol. Macromol. 2020, 165, 3040–3050. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Mackerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- De Oliveira, O.V.; Rocha, G.B.; Paluch, A.S.; Costa, L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- ChemBridge The Gold Standard in Small Molecule Screening Libraries and Building Blocks. Available online: https://www.chembridge.com/screening_libraries/index.php (accessed on 18 May 2021).
- Princeton Chemistry Princeton University Library. Available online: https://library.princeton.edu/databases/subject/chemistry (accessed on 18 May 2021).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.H.T.D.P.; Taft, C.A. 3D descriptors calculation and conformational search to investigate potential bioactive conformations, with application in 3D-QSAR and virtual screening in drug design. J. Biomol. Struct. Dyn. 2017, 35, 2966–2974. [Google Scholar] [CrossRef] [PubMed]
- ROCS; 3.4.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- EON; 2.3.3.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, Z. CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain. J. Med. Chem. 2017, 60, 5943–5954. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry; Springer: Dordrecht, The Netherlands, 1980; pp. 5–15. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, R.S.; Oliveira, J.P.; Matos, R.F.; Chaves Neto, A.M.J.; Carneiro, A.S.; Monteiro, M.C. Involvement of electron and hydrogen transfers through redox metabolism on activity and toxicity of the nimesulide. J. Mol. Model. 2015, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GOLD-CHEMPLP | Glide-XP (Kcal/Mol) | Autodock-Binding Energy (Kcal/Mol) | FRED-Chemgauss4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cavity | Cavity | Cavity | Cavity | ||||||||||||
| 4 | 5 | 6 | 7 | 4 | 5 | 6 | 7 | 4 | 5 | 6 | 7 | 4 | 5 | 6 | 7 |
| 54.88 | 71.20 | 56.07 | 88.36 | −4.47 | −5.85 | −2.92 | −6.13 | −2.45 | −4.12 | −3.32 | −6.98 | −3.99 | −3.46 | −4.36 | −5.53 |
| Docking Validation | 88 Compounds | 88 Compounds + Decoys | vROCS Validation | 88 Compounds | 88 Compounds + Decoys |
|---|---|---|---|---|---|
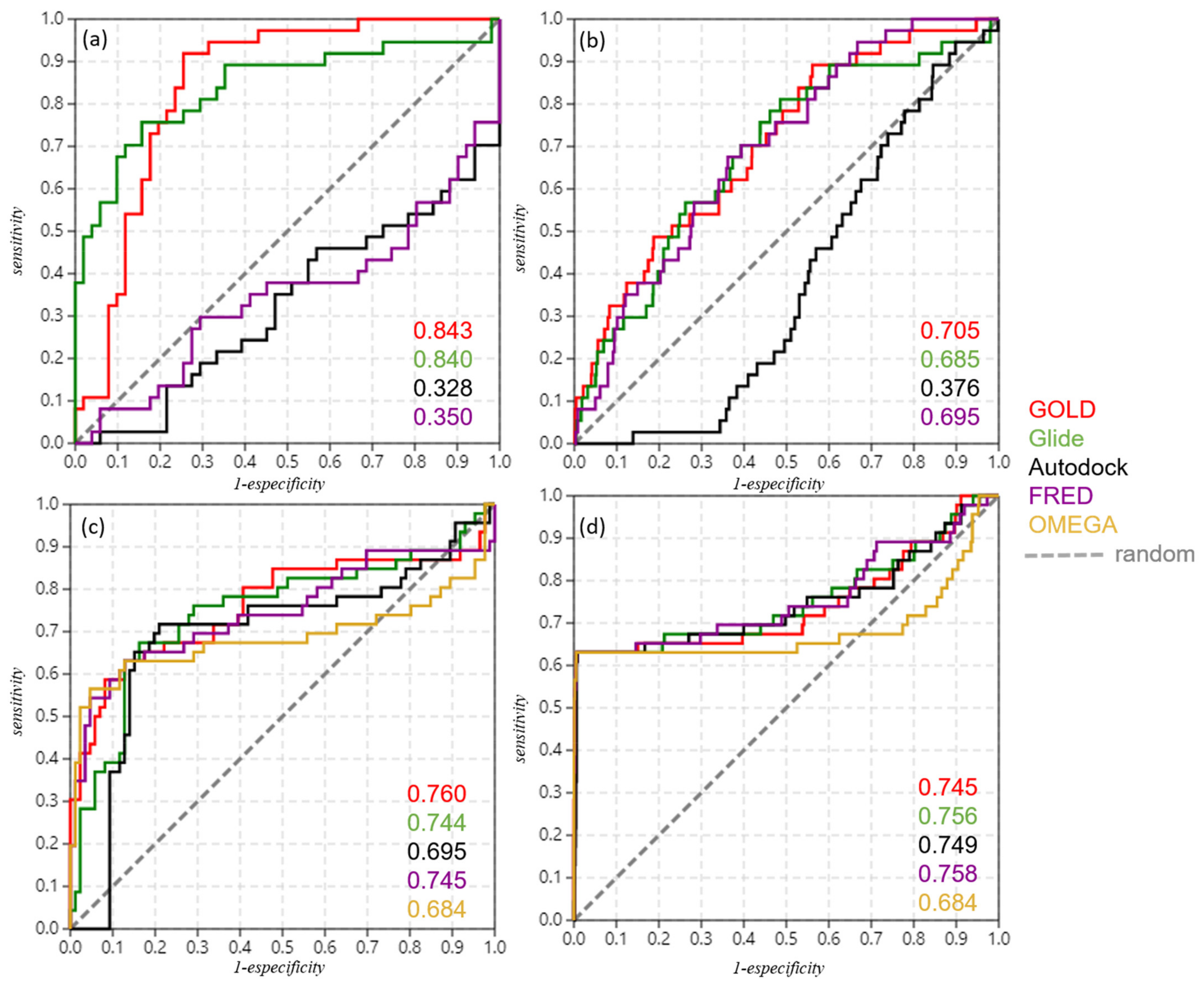
| GOLD | 0.843 | 0.705 | GOLD query | 0.760 | 0.745 |
| Glide | 0.840 | 0.685 | Glide query | 0.744 | 0.756 |
| Autodock | 0.328 | 0.376 | Autodock query | 0.695 | 0.749 |
| FRED | 0.350 | 0.695 | FRED query | 0.745 | 0.758 |
| OMEGA query | 0.684 | 0.684 |
| Compound | ROCS TC | EON TC | Glide XPscore | MW | PSA | (QP)logPo/w | (QP)logBB | HOA% | (QP)PCaco | (QP)PMDCK |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.000 | 2.000 | −6.13 | 429.55 | 118.58 | 5.84 | −2.04 | 94.2 | 375.19 | 171.46 |
| LCQFGS01 | 0.884 | 0.720 | −5.919 | 312.81 | 48.79 | 5.13 | −0.42 | 100 | 2782.34 | 4074.68 |
| LCQFGS02 | 0.917 | 0.672 | −5.622 | 269.34 | 57.19 | 3.01 | −0.35 | 100 | 1421.35 | 1121.06 |
| Compound | HOMO (eV) | LUMO (eV) | GAP * | IP (Kcal.mol−1) |
|---|---|---|---|---|
| 1 | −6.42 | −1.94 | 4.48 | 181.06 |
| 18 | −6.19 | −1.79 | 4.40 | 174.75 |
| 24 | −6.46 | −1.86 | 4.60 | 182.23 |
| LCQFGS01 | −6.59 | −1.77 | 4.82 | 192.97 |
| LCQFGS02 | −6.10 | −1.03 | 5.07 | 177.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, G.M.; Borges, R.S.; Santos, K.L.B.; Federico, L.B.; Francischini, I.A.G.; Gomes, S.Q.; Barcelos, M.P.; Silva, R.C.; Santos, C.B.R.; Silva, C.H.T.P. Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8252. https://doi.org/10.3390/ijms22158252
Silva GM, Borges RS, Santos KLB, Federico LB, Francischini IAG, Gomes SQ, Barcelos MP, Silva RC, Santos CBR, Silva CHTP. Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(15):8252. https://doi.org/10.3390/ijms22158252
Chicago/Turabian StyleSilva, Guilherme M., Rosivaldo S. Borges, Kelton L. B. Santos, Leonardo B. Federico, Isaque A. G. Francischini, Suzane Q. Gomes, Mariana P. Barcelos, Rai C. Silva, Cleydson B. R. Santos, and Carlos H. T. P. Silva. 2021. "Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 15: 8252. https://doi.org/10.3390/ijms22158252
APA StyleSilva, G. M., Borges, R. S., Santos, K. L. B., Federico, L. B., Francischini, I. A. G., Gomes, S. Q., Barcelos, M. P., Silva, R. C., Santos, C. B. R., & Silva, C. H. T. P. (2021). Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(15), 8252. https://doi.org/10.3390/ijms22158252