
An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches


Abstract
:1. Introduction
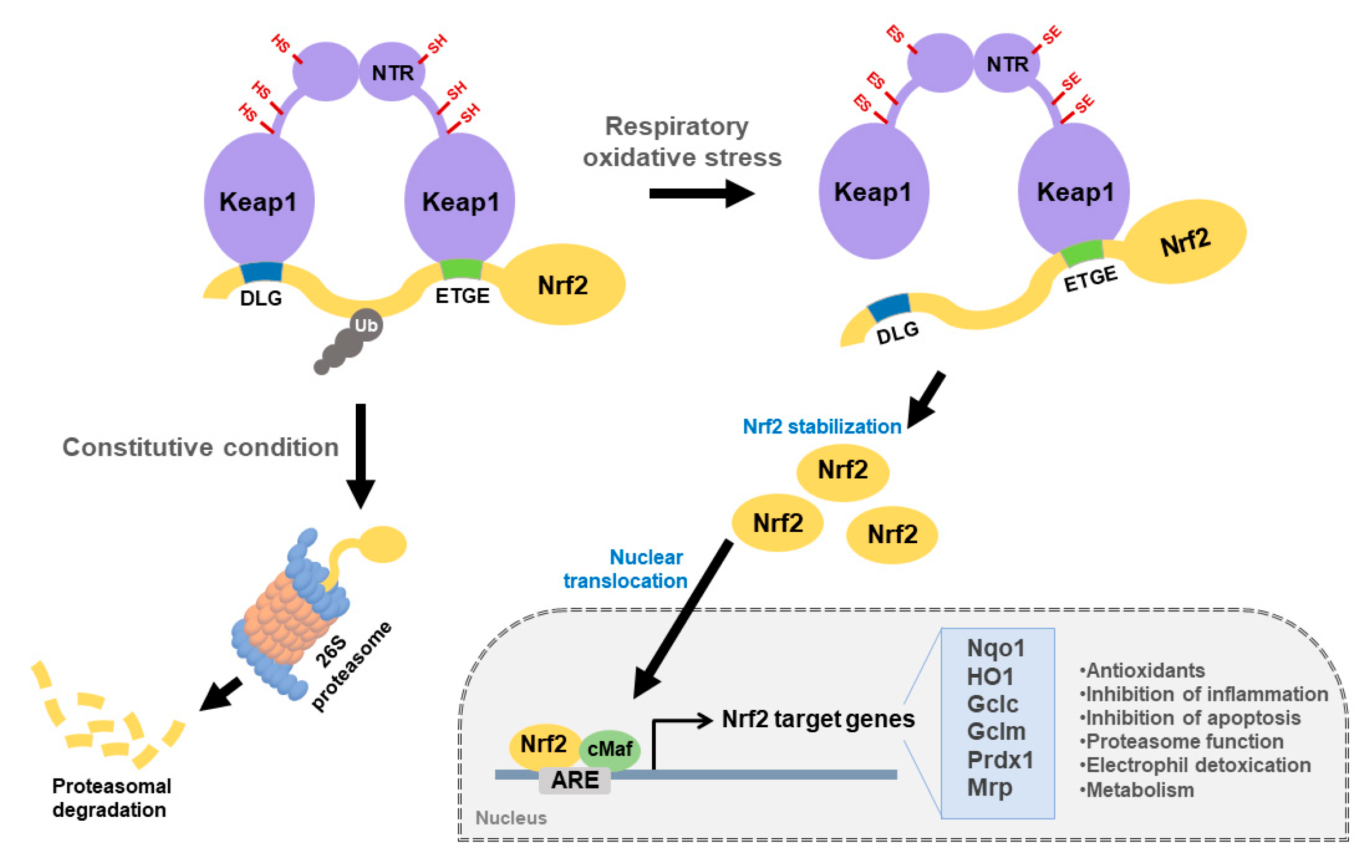
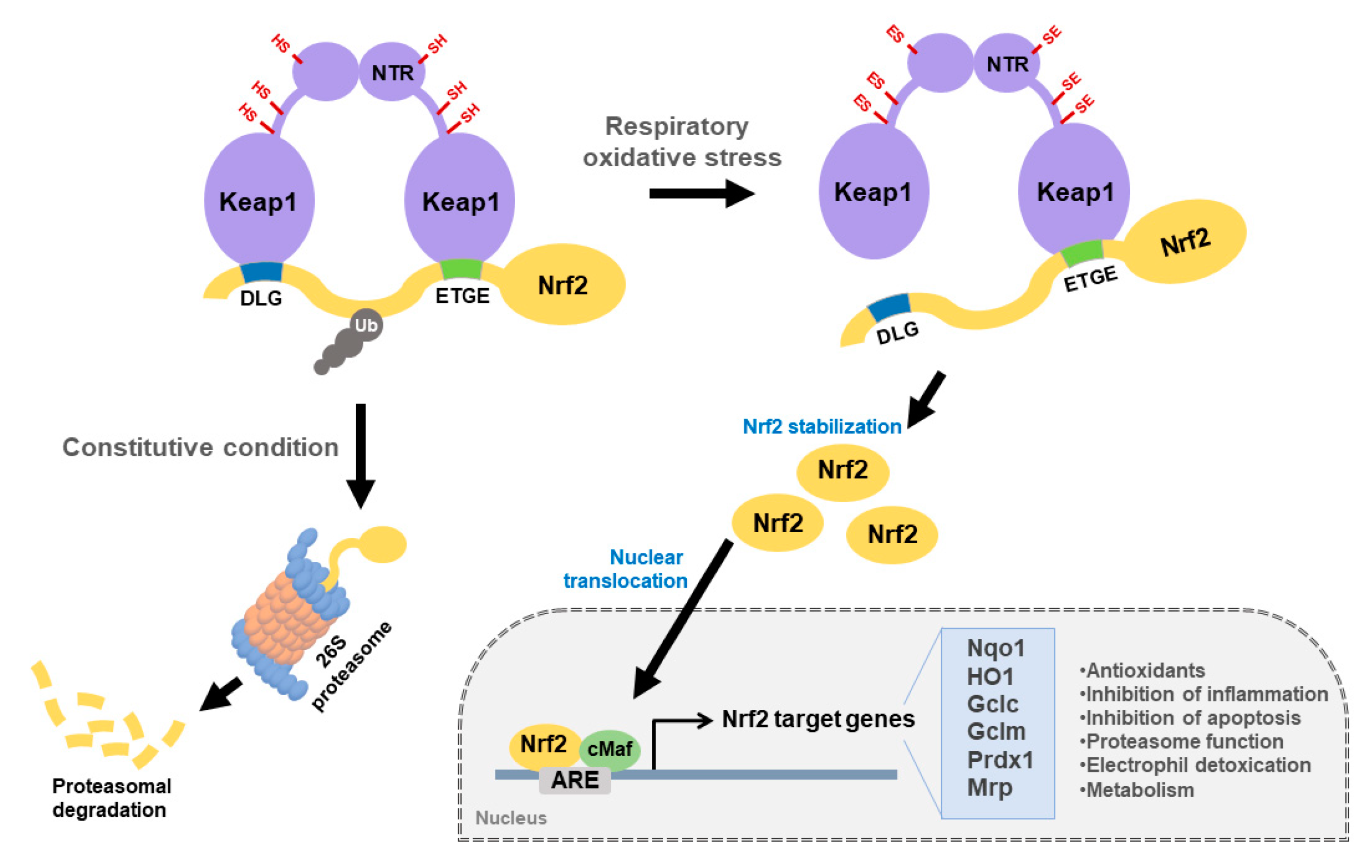
2. Keap1-Nrf2 Signaling Pathway
3. Keap1-Nrf2 in Respiratory Diseases
3.1. ARDS/ALI
3.2. COPD
3.3. Idiopathic Pulmonary Fibrosis (IPF)
3.4. Asthma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Rosanna, D.P.; Salvatore, C. Reactive oxygen species, inflammation, and lung diseases. Curr. Pharm. Des. 2012, 18, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Kisaoglu, A.; Borekci, B.; Yapca, O.E.; Bilen, H.; Suleyman, H. Tissue damage and oxidant/antioxidant balance. Eurasian J. Med. 2013, 45, 47–49. [Google Scholar] [CrossRef]
- Victoni, T.; Barreto, E.; Lagente, V.; Carvalho, V.F. Oxidative Imbalance as a Crucial Factor in Inflammatory Lung Diseases: Could Antioxidant Treatment Constitute a New Therapeutic Strategy? Oxidative Med. Cell. Longev. 2021, 2021, 6646923. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc Czech Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, C.B.; Je, J.Y.; Kim, Y.S.; Park, S.J.; Kim, B.I. Induction of Nrf2-mediated phase II detoxifying/antioxidant enzymes in vitro by chitosan-caffeic acid against hydrogen peroxide-induced hepatotoxicity through JNK/ERK pathway. Mol. Cell. Biochem. 2017, 424, 79–86. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Song, M.Y.; Kim, E.H. Role of Oxidative Stress and Nrf2/KEAP1 Signaling in Colorectal Cancer: Mechanisms and Therapeutic Perspectives with Phytochemicals. Antioxidants 2021, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Brandt, J.P.; Mandiga, P. Histology, Alveolar Cells; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 2015, 16, 36–44. [Google Scholar] [CrossRef]
- Franks, T.J.; Colby, T.V.; Travis, W.D.; Tuder, R.M.; Reynolds, H.Y.; Brody, A.R.; Cardoso, W.V.; Crystal, R.G.; Drake, C.J.; Engelhardt, J.; et al. Resident cellular components of the human lung: Current knowledge and goals for research on cell phenotyping and function. Proc. Am. Thorac. Soc. 2008, 5, 763–766. [Google Scholar] [CrossRef]
- Liao, D.; Li, H. Dissecting the Niche for Alveolar Type II Cells With Alveolar Organoids. Front. Cell Dev. Biol. 2020, 8, 419. [Google Scholar] [CrossRef]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044.e18. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Hu, G.; Christman, J.W. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front. Immunol. 2019, 10, 2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Sun, N.N.; Gao, H.N.; Chen, Z.Y.; Yang, Y.; Ju, B.; Tang, L.L. Risk factors analysis of COVID-19 patients with ARDS and prediction based on machine learning. Sci. Rep. 2021, 11, 2933. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 2015, 194, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Englert, J.A.; Bobba, C.; Baron, R.M. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Schweikl, H.; Gallorini, M.; Poschl, G.; Urmann, V.; Petzel, C.; Bolay, C.; Hiller, K.A.; Cataldi, A.; Buchalla, W. Functions of transcription factors NF-kappaB and Nrf2 in the inhibition of LPS-stimulated cytokine release by the resin monomer HEMA. Dent. Mater. 2018, 34, 1661–1678. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Gladwell, W.; Marzec, J.; McCaw, Z.R.; Bienstock, R.J.; Kleeberger, S.R. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid. Redox Signal. 2015, 22, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Marzec, J.; Kleeberger, S.R. Functional polymorphisms in Nrf2: Implications for human disease. Free Radic. Biol. Med. 2015, 88, 362–372. [Google Scholar] [CrossRef]
- Wei, J.; Chen, G.; Shi, X.; Zhou, H.; Liu, M.; Chen, Y.; Feng, D.; Zhang, P.; Wu, L.; Lv, X. Nrf2 activation protects against intratracheal LPS induced mouse/murine acute respiratory distress syndrome by regulating macrophage polarization. Biochem. Biophys. Res. Commun. 2018, 500, 790–796. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Grebenchikov, O.A. Transcription Factor Nrf2 as a Potential Therapeutic Target for Prevention of Cytokine Storm in COVID-19 Patients. Biochemistry 2020, 85, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Kozumbo, W.J.; Kapoor, R.; Dhawan, G.; Lara, P.C.; Giordano, J. Nrf2 activation putatively mediates clinical benefits of low-dose radiotherapy in COVID-19 pneumonia and acute respiratory distress syndrome (ARDS): Novel mechanistic considerations. Radiother. Oncol. 2021, 160, 125–131. [Google Scholar] [CrossRef]
- Augusti, P.R.; Conterato, G.M.M.; Denardin, C.C.; Prazeres, I.D.; Serra, A.T.; Bronze, M.R.; Emanuelli, T. Bioactivity, Bioavailability, and Gut Microbiota Transformations of Dietary Phenolic Compounds: Implications for Covid-19. J. Nutr. Biochem. 2021, 108787. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Geraci, K.P.; Gao, B. Nrf2 Activator PB125((R)) as a Potential Therapeutic Agent against COVID-19. Antioxidants 2020, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Kozumbo, W.J. Radiotherapy treatment of human inflammatory diseases and conditions: Optimal dose. Hum. Exp. Toxicol. 2019, 38, 888–898. [Google Scholar] [CrossRef]
- Hu, L.Y.; Cui, J.B.; Xu, X.M.; Huang, Z.H.; Jiao, H.T. Expression of Nrf2-Keap1-ARE signal pathway in traumatic lung injury and functional study. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1402–1408. [Google Scholar] [CrossRef]
- Yan, X.; Fu, X.; Jia, Y.; Ma, X.; Tao, J.; Yang, T.; Ma, H.; Liang, X.; Liu, X.; Yang, J.; et al. Nrf2/Keap1/ARE Signaling Mediated an Antioxidative Protection of Human Placental Mesenchymal Stem Cells of Fetal Origin in Alveolar Epithelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2654910. [Google Scholar] [CrossRef]
- Lei, L.; Guo, Y.; Lin, J.; Lin, X.; He, S.; Qin, Z.; Lin, Q. Inhibition of endotoxin-induced acute lung injury in rats by bone marrow-derived mesenchymal stem cells: Role of Nrf2/HO-1 signal axis in inhibition of NLRP3 activation. Biochem. Biophys. Res. Commun. 2021, 551, 7–13. [Google Scholar] [CrossRef]
- Reddy, N.M.; Suryanaraya, V.; Yates, M.S.; Kleeberger, S.R.; Hassoun, P.M.; Yamamoto, M.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; Reddy, S.P. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Kensler, T.W.; Yamamoto, M.; Hassoun, P.M.; Reddy, S.P. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J. Immunol. 2009, 182, 7264–7271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, L.; Chen, H.; Zhang, W.; Xing, C.; Qu, Z.; Yu, J.; Zhuang, C. Structure-based molecular hybridization design of Keap1-Nrf2 inhibitors as novel protective agents of acute lung injury. Eur. J. Med. Chem. 2021, 222, 113599. [Google Scholar] [CrossRef]
- Cho, H.Y.; Miller-DeGraff, L.; Blankenship-Paris, T.; Wang, X.; Bell, D.A.; Lih, F.; Deterding, L.; Panduri, V.; Morgan, D.L.; Yamamoto, M.; et al. Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice. Toxicol. Appl. Pharmacol. 2019, 364, 29–44. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.; Zhang, X.; Liu, F.; Yang, K.; Du, G.; Rui, X. Dasatinib protects against acute respiratory distress syndrome via Nrf2-regulated M2 macrophages polarization. Drug Dev. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, H.; Li, H.; Ci, X.; Peng, L. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-kappaB pathways. Cell Commun. Signal. 2019, 17, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, P.; Zhang, Z.; Cao, J. Isorhapontigenin alleviates lipopolysaccharide-induced acute lung injury via modulating Nrf2 signaling. Respir. Physiol. Neurobiol. 2021, 289, 103667. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, C.H.; Lee, J.; Jeong, Y.; Park, J.H.; Nam, I.J.; Lee, D.S.; Lee, H.M.; Lee, J.; Yun, N.; et al. Botanical formulation, TADIOS, alleviates lipopolysaccharide (LPS)-Induced acute lung injury in mice via modulation of the Nrf2-HO-1 signaling pathway. J. Ethnopharmacol. 2021, 270, 113795. [Google Scholar] [CrossRef]
- Qing, R.; Huang, Z.; Tang, Y.; Xiang, Q.; Yang, F. Cordycepin alleviates lipopolysaccharide-induced acute lung injury via Nrf2/HO-1 pathway. Int. Immunopharmacol. 2018, 60, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Wei, Y.; Song, P.; Li, Y.; Zhang, T.; Feng, Q.; Xu, G. Cordycepin inhibits LPS-induced acute lung injury by inhibiting inflammation and oxidative stress. Eur. J. Pharmacol. 2018, 818, 110–114. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, Z.; Sheng, L.; Wu, H. Protective effects of syringin against lipopolysaccharide-induced acute lung injury in mice. J. Surg. Res. 2017, 209, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Yao, H.; Meister, M.; Gardenhire, D.S.; Mo, H. Tocotrienols: Dietary Supplements for Chronic Obstructive Pulmonary Disease. Antioxidants 2021, 10, 883. [Google Scholar] [CrossRef] [PubMed]
- Lugg, S.T.; Scott, A.; Parekh, D.; Naidu, B.; Thickett, D.R. Cigarette smoke exposure and alveolar macrophages: Mechanisms for lung disease. Thorax 2021. [Google Scholar] [CrossRef]
- Fratta Pasini, A.M.; Stranieri, C.; Ferrari, M.; Garbin, U.; Cazzoletti, L.; Mozzini, C.; Spelta, F.; Peserico, D.; Cominacini, L. Oxidative stress and Nrf2 expression in peripheral blood mononuclear cells derived from COPD patients: An observational longitudinal study. Respir. Res. 2020, 21, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhaye, V.K.; Holbrook, J.T.; Burke, A.; Sudini, K.R.; Sethi, S.; Criner, G.J.; Fahey, J.W.; Berenson, C.S.; Jacobs, M.R.; Thimmulappa, R.; et al. Compartmentalization of anti-oxidant and anti-inflammatory gene expression in current and former smokers with COPD. Respir. Res. 2019, 20, 190. [Google Scholar] [CrossRef]
- Lee, H.; Park, J.R.; Kim, W.J.; Sundar, I.K.; Rahman, I.; Park, S.M.; Yang, S.R. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J. 2017, 31, 2076–2089. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Iizuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.; Hong, S.H.; Rahman, I.; Yang, S.R. Inhibition of RAGE Attenuates Cigarette Smoke-Induced Lung Epithelial Cell Damage via RAGE-Mediated Nrf2/DAMP Signaling. Front. Pharmacol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Qu, J.; Yue, L.; Gao, J.; Yao, H. Perspectives on Wnt Signal Pathway in the Pathogenesis and Therapeutics of Chronic Obstructive Pulmonary Disease. J. Pharmacol. Exp. Ther. 2019, 369, 473–480. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Z.; Zhang, P.; Qu, J.; Zheng, C.; Mo, X.; Zhou, W.; Xu, L.; Yao, H.; Gao, J. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/beta-catenin and AMPK pathways. J. Cell. Mol. Med. 2018, 22, 3514–3525. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef]
- Russo, M.; Spagnuolo, C.; Russo, G.L.; Skalicka-Wozniak, K.; Daglia, M.; Sobarzo-Sanchez, E.; Nabavi, S.F.; Nabavi, S.M. Nrf2 targeting by sulforaphane: A potential therapy for cancer treatment. Crit. Rev. Food Sci. Nutr. 2018, 58, 1391–1405. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of Effect of Oral Sulforaphane Administration on Nrf2 Expression in COPD: A Randomized, Double-Blind, Placebo Controlled Trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, T.; Ding, X.; Li, J.; Wu, Y.; Ren, B.; Li, J.; Lv, H.; Chen, J.; Li, W. Triterpene Acids of Loquat Leaf Improve Inflammation in Cigarette Smoking Induced COPD by Regulating AMPK/Nrf2 and NFkappaB Pathways. Nutrients 2020, 12, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Liu, X.; Zhang, G.; Ming, Z.; Wang, T. Isoliquiritigenin Inhibits Cigarette Smoke-Induced COPD by Attenuating Inflammation and Oxidative Stress via the Regulation of the Nrf2 and NF-kappaB Signaling Pathways. Front. Pharmacol. 2018, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Guo, J.; Niu, G.; Chen, M. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-kappaB pathways. Respir. Res. 2020, 21, 95. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Crestani, B.; Hernandez, P.; Inoue, Y.; Wachtlin, D.; Loaiza, L.; Quaresma, M.; Stowasser, S.; Richeldi, L. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: Pooled data from six clinical trials. BMJ Open Respir. Res. 2019, 6, e000397. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Selman, M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am. J. Respir. Crit. Care Med. 2015, 191, 252–254. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e117. [Google Scholar] [CrossRef]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, G.; Klotsche, J.; Kreuter, M.; Prasse, A.; Wirtz, H.; Pittrow, D.; Frankenberger, M.; Behr, J.; Kneidinger, N.; Group, I.-I.R. Idiopathic Pulmonary Fibrosis in Elderly Patients: Analysis of the INSIGHTS-IPF Observational Study. Front. Med. 2020, 7, 601279. [Google Scholar] [CrossRef]
- Kurundkar, A.; Thannickal, V.J. Redox mechanisms in age-related lung fibrosis. Redox Biol. 2016, 9, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Warraich, U.E.; Hussain, F.; Kayani, H.U.R. Aging-Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Inghilleri, S.; Morbini, P.; Campo, I.; Zorzetto, M.; Oggionni, T.; Pozzi, E.; Luisetti, M. Factors influencing oxidative imbalance in pulmonary fibrosis: An immunohistochemical study. Pulm. Med. 2011, 2011, 421409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliogiannis, P.; Fois, A.G.; Collu, C.; Bandinu, A.; Zinellu, E.; Carru, C.; Pirina, P.; Mangoni, A.A.; Zinellu, A. Oxidative stress-linked biomarkers in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Biomark. Med. 2018, 12, 1175–1184. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef]
- Ghatak, S.; Hascall, V.C.; Markwald, R.R.; Feghali-Bostwick, C.; Artlett, C.M.; Gooz, M.; Bogatkevich, G.S.; Atanelishvili, I.; Silver, R.M.; Wood, J.; et al. Transforming growth factor beta1 (TGFbeta1)-induced CD44V6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J. Biol. Chem. 2017, 292, 10490–10519. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Alkharfy, K.M.; Jan, B.L.; Ahad, A.; Ansari, M.A.; Al-Jenoobi, F.I.; Raish, M. Thymoquinone treatment modulates the Nrf2/HO-1 signaling pathway and abrogates the inflammatory response in an animal model of lung fibrosis. Exp. Lung Res. 2020, 46, 53–63. [Google Scholar] [CrossRef]
- Zhou, W.; Mo, X.; Cui, W.; Zhang, Z.; Li, D.; Li, L.; Xu, L.; Yao, H.; Gao, J. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci. Rep. 2016, 6, 38646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxidative Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [Green Version]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Ma, Z.; Shi, S.; Hu, Y.; Ma, T.; Rong, G.; Yang, J. Sulforaphane prevents bleomycininduced pulmonary fibrosis in mice by inhibiting oxidative stress via nuclear factor erythroid 2related factor2 activation. Mol. Med. Rep. 2017, 15, 4005–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Zuo, R.; Wang, Y.; Li, J.; Wu, J.; Wang, W.; Li, B.; Sun, C.; Wang, Z.; Shi, C.; Zhou, Y.; et al. Rapamycin Induced Autophagy Inhibits Inflammation-Mediated Endplate Degeneration by Enhancing Nrf2/Keap1 Signaling of Cartilage Endplate Stem Cells. Stem Cells 2019, 37, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Deng, S.; Wu, W.; Li, Z.; Lei, W.; Wang, Y.; Vongphouttha, C.; Zhang, T.; Dong, Z. Rapamycin attenuates the paraquat-induced pulmonary fibrosis through activating Nrf2 pathway. J. Cell. Physiol. 2020, 235, 1759–1768. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Plate, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, T.F.; Bleecker, E. Asthma heterogeneity and severity. World Allergy Organ. J. 2016, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, J.R.; Lloyd, C.M. Chronic inflammation and asthma. Mutat. Res. 2010, 690, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.S.; Mistry, K.J.; Kakade, A.M.; Niphadkar, P.V. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India 2010, 27, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, L.H.C.; Ferreira, S.R.D.; Silva, M.; Ferreira, P.B.; de Souza, I.L.L.; Cavalcante, F.A.; da Silva, B.A. Uncovering the Role of Oxidative Imbalance in the Development and Progression of Bronchial Asthma. Oxidative Med. Cell. Longev. 2021, 2021, 6692110. [Google Scholar] [CrossRef]
- Yousefi, S.; Simon, D.; Simon, H.U. Eosinophil extracellular DNA traps: Molecular mechanisms and potential roles in disease. Curr. Opin. Immunol. 2012, 24, 736–739. [Google Scholar] [CrossRef]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L27–L36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Macias, H.; Romieu, I. Effects of antioxidant supplements and nutrients on patients with asthma and allergies. J. Allergy Clin. Immunol. 2014, 133, 1237–1244, quiz 1245. [Google Scholar] [CrossRef]
- Hubbard, R.; Fogarty, A. The developing story of antioxidants and asthma. Thorax 2004, 59, 3–4. [Google Scholar] [PubMed]
- Pan, Y.; Li, W.; Feng, Y.; Xu, J.; Cao, H. Edaravone attenuates experimental asthma in mice through induction of HO-1 and the Keap1/Nrf2 pathway. Exp. Ther. Med. 2020, 19, 1407–1416. [Google Scholar] [CrossRef]
- Zhang, J.H.; Yang, X.; Chen, Y.P.; Zhang, J.F.; Li, C.Q. Nrf2 Activator RTA-408 Protects Against Ozone-Induced Acute Asthma Exacerbation by Suppressing ROS and gammadeltaT17 Cells. Inflammation 2019, 42, 1843–1856. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, H.; Morishima, Y.; Ishii, Y.; Yoshida, K.; Nakajima, M.; Tsunoda, Y.; Hayashi, S.Y.; Kiwamoto, T.; Matsuno, Y.; Kawaguchi, M.; et al. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free Radic. Biol. Med. 2018, 129, 473–485. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Respiratory Disease | Keap1-Nrf2 Regulator | Therapeutic Mechanism | Reference |
|---|---|---|---|
| ARDS | SCN-16 | Inflammation reduction in LPS-induced ARDS model. | [44] |
| ARDS | Dasatinib | Inhibition of recruitment neutrophils, lymphocytes, and macrophages in LPS-administrated mouse model. | [46] |
| ARDS | Oridonin | Nrf2, HO-1, GCLM upregulation and NF-kB pathway inhibition in RAW 264.7. | [47] |
| ARDS | CDDO-Im | Nrf2, Gclc, NQO1, and GPX2 upregulation in hyperoxia-induced lung injury mouse model. | [42] |
| ARDS | Sulphoraphane | Reduction in hypoxic lung injury via enhanced energy metabolism in mitochondria and cardiovascular function. | [45] |
| COPD | Triterpene Acids | Reduction in pulmonary edema, inflammatory cytokines. Activation of AMPK phosphorylation and Nrf2 expression in CS-induced COPD mouse model. | [69] |
| COPD | Isoliquiritigenin | Reduced inflammatory cytokines, decreased NF-κB expression, increased Nrf2 and HO-1 expression in CS-induced COPD model. | [70] |
| COPD | Alantolactone | Inflammatory cytokine and MDA reduction, activation of Nrf2/HO-1 pathway in CSE-induced NHBE and Beas-2B. | [71] |
| IPF | Sulphoraphane | Regulation of TGF- β-mediated EMT signaling in A549 cells, and pro-fibrotic proteins (e.g., TGF-β1, vimentin), and inflammation in mice. | [74,75] |
| IPF | Rapamycin | Increase in Nrf2 signaling pathway in paraquat-induced IPF rat model. | [78] |
| Asthma | Trifluoromethyl-methoxychalone | Decrease in the number of eosinophils and allergic-related cytokines such as IL-4 and IL13 in OVA-induced asthma mice. | [86] |
| Asthma | Edaravone | Decrease in ROS levels, and restore antioxidants (e.g., SOD, CAT, and GSH-Px). Activation of Keap1-Nrf2 pathway and HO-1 expression in the OVA-induced asthma mice. | [89] |
| Asthma | RTA-408 | Decrease in pro-inflammatory cytokine production and suppression of the percentage of IL-17+ γδT cells in OVA/ozone-induced acute exacerbation asthma model. | [90] |
| Asthma | Surphoraphane | Activation of Nrf2 and HDAC2 in cigarette smoking-related asthma mice. | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Jang, J.; Park, S.-M.; Yang, S.-R. An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 8406. https://doi.org/10.3390/ijms22168406
Lee J, Jang J, Park S-M, Yang S-R. An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches. International Journal of Molecular Sciences. 2021; 22(16):8406. https://doi.org/10.3390/ijms22168406
Chicago/Turabian StyleLee, Jooyeon, Jimin Jang, Sung-Min Park, and Se-Ran Yang. 2021. "An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches" International Journal of Molecular Sciences 22, no. 16: 8406. https://doi.org/10.3390/ijms22168406