Abstract
Tauopathy refers to a group of progressive neurodegenerative diseases, including frontotemporal lobar degeneration and Alzheimer’s disease, which correlate with the malfunction of microtubule-associated protein Tau (MAPT) due to abnormal hyperphosphorylation, leading to the formation of intracellular aggregates in the brain. Despite extensive efforts to understand tauopathy and develop an efficient therapy, our knowledge is still far from complete. To find a solution for this group of devastating diseases, several animal models that mimic diverse disease phenotypes of tauopathy have been developed. Rodents are the dominating tauopathy models because of their similarity to humans and established disease lines, as well as experimental approaches. However, powerful genetic animal models using Drosophila, zebrafish, and C. elegans have also been developed for modeling tauopathy and have contributed to understanding the pathophysiology of tauopathy. The success of these models stems from the short lifespans, versatile genetic tools, real-time in-vivo imaging, low maintenance costs, and the capability for high-throughput screening. In this review, we summarize the main findings on mechanisms of tauopathy and discuss the current tauopathy models of these non-rodent genetic animals, highlighting their key advantages and limitations in tauopathy research.
1. Introduction
Tauopathy refers to a group of neurodegenerative disorders that share a common feature of neurofibrillary tangles (NFT) originating from the aggregation of abnormal hyperphosphorylated and paired helical filament (PHF) Tau proteins [1]. The term tauopathy was first coined in 1997 to describe a familial dementia characterized by abundant and widespread intracellular NFT with the absence of amyloid β (Aβ) plaque deposits [2]. Since then, other neurodegenerative diseases have also been classified as tauopathy, including Alzheimer’s Disease (AD), frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), cortical basal degeneration, amyotrophic lateral sclerosis, dementia pugilistica, tangle-only dementia, progressive supranuclear palsy, and Pick’s disease [1,3].
Although the Tau protein was first identified almost a half century ago [4], scientists did not pay attention to this protein until the discovery of Tau as a major component of the abnormal aggregated tangles in AD patient brains a decade later [5,6,7,8,9]. Tau is one of the first characterized and well-known factors to maintain the stability of microtubules (MTs) in the central nervous system (CNS) [10,11]. Tau protein is subject to diverse post-translational modifications (PTMs), including glycosylation, glycation, nitration, polyamination, phosphorylation, ubiquitination, oxidation, cleavage or truncation, small ubiquitin-like modifier (SUMO) modification, and aggregation [12].
Thus far, 19 Tau-targeting therapeutic compounds have been proposed and tested based on diverse mechanisms by which Tau protein aggregation causes neurodegeneration. Unfortunately, all compounds have failed to pass phase 3 of clinical trials [13]. Therefore, tauopathies and AD, in particular, remain incurable diseases, and with the number of tauopathy-related dementias increasing every year, it is urgent to discover an effective therapeutic for tauopathy. In this review, we first summarize underlying mechanisms of tauopathy and then discuss research on tauopathy using non-rodent genetic animal models. We focus on Drosophila, zebrafish, and C. elegans, highlighting their advantages and limitations as tauopathy models in order to unveil mechanistic aspects of tauopathy and to develop novel therapeutic strategies. The most popular tauopathy model, the rodent model, has been extensively reviewed elsewhere [14] and is not covered here.
2. Underlying Mechanisms of Tauopathy
2.1. Normal Function of Tau
In humans, the Tau protein consists of six isoforms translated from alternatively spliced mRNAs of a single microtubule-associated protein Tau gene (MAPT) containing 16 exons (Figure 1A). These isoforms are classified by the combination of numbers of N (N-terminal projection) and R (microtubule-binding repeats) domains (i.e., 2N4R, 1N4R, 0N4R, 2N3R, 1N3R, and 0N3R). In particular, the number of N is determined by alternative splicing of exon 2 and 3 at the N terminal, whereas the number of R is defined by whether they contain repeat 2 encoded by exon 10 in mature mRNA [15,16]. In humans, mRNAs encoding 3R isoforms are found in both fetal and adult brains, whereas only mRNAs of 4R isoforms are found exclusively in the adult brain [15]. Moreover, the level of 3R and 4R isoforms in the adult brain is about equal to that which any disturbances are associated with neurodegeneration [17].
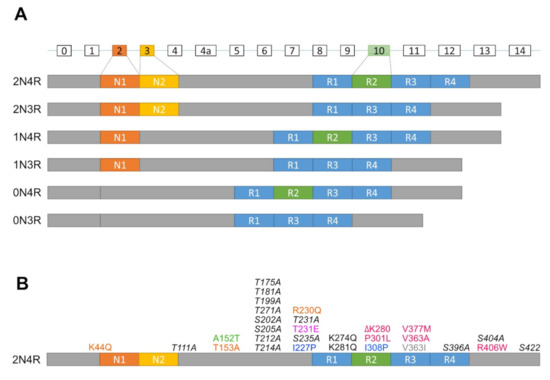
Figure 1.
Diagrams of isoforms and mutations of the MAPT gene. (A) The MAPT gene contains 16 exons (rectangles with exon numbers on top), where exons with white rectangles are constitutive whereas exon 2 (dark orange box), exon 3 (orange box) and exon 10 (green box) are subject to alternative splicing. 0N, 1N, or 2N isoform depends on the presence of exon 2 and 3, while 3R or 4R isoform is determined by the presence of exon 10. (B) Mutations of the MAPT gene used to generate tauopathy models in Drosophila, zebrafish and C. elegans. dark orange, Calpain-resistant mutations; red, reduced microtubule-binding and increased aggregation mutations; green, a reduced microtubule-binding and increased oligomerization, but not fibrillization mutation; pink, an increased phosphorylation mutation; blue, anti-aggregation mutations; grey, promote microtubule assembly and increased oligomerization, but not fibrillization mutation; black, increased disease-associated lysine acetylation mutations; italic black, phosphorylation-incompetent mutations.
In general, Tau protein is ubiquitously expressed from the cell body to neurites in immature neurons but mainly found in the axon, interacting with MTs in mature neurons, where Tau regulates their stability [17]. However, Tau is also present in other cellular compartments, such as in both pre- and post-synaptic compartments [18,19]; at the post-synapse, Tau plays a role in establishing the proper localization of Fyn and is involved in regulating N-methyl-D-aspartate receptors (NMDARs) and post-synaptic density protein 95 (PSD95) complex [20,21]. In addition, Tau is found in the nucleus, where it binds to the heterochromatin and protects the integrity of DNA and RNA [22,23], and is localized in the neural plasma membrane in a MT-independent manner [24,25].
2.2. Phosphorylation of Tau
In AD and other tauopathies, phosphorylation of Tau is one of the main events among several PTMs that the Tau protein undergoes [26]. A 2N4R Tau usually carries approximately two phosphates per molecule [27]. Tau phosphorylation at Thr205, Thr231, and Ser396 is highly correlative with adult hippocampal neurogenesis as a compensatory mechanism to neuronal loss in AD [28]. In fact, similar Tau phosphorylation could reduce the endoplasmic reticulum (ER) stress and kinase-induced apoptosis [29,30]. However, in pathological conditions, the phosphorylation of Tau is approximately eight phosphates per molecule, an increase of three to four times more compared to normal individuals [6,26,31]. When hyperphosphorylated, Tau partially loses its binding affinity to MTs affecting their dynamics [32]. Hyperphosphorylated Tau also causes defects in axonal neurotransmitter transport, release, and uptake, thereby resulting in axon degeneration and finally neurodegeneration [33]. Therefore, Tau hyperphosphorylation is considered as an early-onset pathological process [34]. Known protein kinases and phosphatases that determine the degree of phosphorylation of Tau include glycogen synthase kinase 3β (GSK3β), mitogen-activated protein kinase (MAPK), cyclin AMP (cAMP)-dependent protein kinase A (PKA), calmodulin kinase II (CaMK II), protein kinase (PKC), microtubule affinity regulating kinase (MARK), and protein phosphatase family, especially type 2A (PP2A) [26,35].
In the longest isoform of Tau, 2N4R, there are approximately 85 potential phosphorylation sites (80 Serine or Threonine, and 5 Tyrosine residues). Among them, there are 17 Thr-Pro or Ser-Pro motifs that are abnormally hyperphosphorylated in AD and other Tauopathies [36], and the phosphorylation of Tau at Tyr394 and Tyr18 is detected in paired helix filaments (PHFs) in the brains of AD patients [37]. A study using the mouse tauopathy model also suggests that the hyperphosphorylation of Tau at tyrosine residues correlates with Tau aggregation [38]. Although all members of the protein phosphatase family are involved in dephosphorylation of Tau, PP2A is the major phosphatase responsible for almost 70% of the Tau phosphatase activity in human brains, and a loss of function of this protein phosphatase may be a cause of disease onset [36]. Consistently, PP2A activity is decreased by 20% to 40% in AD brains [39,40,41].
2.3. Aggregation of Tau
Both in vitro and in vivo studies have shown that Tau aggregation occurs in a “nucleation-elongation” mechanism [42]. The initiation of Tau aggregation can be affected by motif sequences, cofactors, phosphorylation status, and cleavage. In Tau protein, there are two small fractions at the beginning of R2 and R3 domains, with VQIINK and VQIVYK motifs, respectively. These hexapeptides are essential for forming β-sheet structures that tightly interlink the β-sheets together in a steric “zippers” manner, thereby leading to Tau aggregation [42,43]. Accordingly, the disruption or reinforcement of these motifs abolish or enhance the tendency of β-sheet structure and Tau aggregation, respectively [44]. Tau aggregation is also modulated by Tau cofactors. Negative-charged polyanions, such as glycosaminoglycans, and their sulfated forms (e.g., heparin) or RNA (e.g., polyU RNA) can promote Tau aggregation in vitro [45,46,47,48]. Another cofactor, FKBP52 (the 52 kD FK506-binding protein), also induces Tau aggregation in vitro and accelerates memory impairments in vivo [49,50].
In addition, among many post-translational modifications affecting Tau aggregation, Tau hyperphosphorylation was shown to precede aggregation significantly in patient samples and transgenic mice [51]. Such abnormal hyperphosphorylated Tau from AD brains can self-assemble into PHFs form in vitro [52]. However, it has been also shown that not all phosphorylation of Tau contributes to aggregation but rather the spatial domain of phosphorylation sites on Tau have an impact on the aggregation. For instance, the phosphorylation at Ser241 and Ser262 on the repeat domain of Tau prevents the aggregation [53], and in Sf9 cell culture, Tau is not aggregated despite the high concentration and extensive hyperphosphorylation of the protein, presumably due to the additional contribution of other factors that promote Tau aggregation [54]. Furthermore, regardless of the phosphorylation status, truncated Tau fragments containing the repeat domain are highly prone to aggregation. Human AD brains and rat transgenic models burdened with Tau fragment 151–391 develop Tau aggregation and neurofibrillary pathology [55]. Another Tau fragment, 255–368, formed by cleavage of an asparagine endopeptidase or long fragments (e.g., Tau 1–368 or 1–421), also causes neurofibrillary pathology in vitro [56,57]. In a cell model of tauopathy that expresses the ΔK280 (a repeat-domain-mutant), Tau accelerates robust aggregation and neurodegeneration [58,59].
Of note, pre-formed PHFs can act as external seeds to accelerate the aggregation in cultured cells without an endogenous nucleation step [42]. Similarly, when preformed PHFs are injected into the mouse brain, Tau seeds initiate widespread tauopathy and cause neuronal loss in a time-dependent manner [60,61].
2.4. Toxicity of Tau
Since there is a strong correlation between the presence of NFTs in the AD brain and the harshness of the memory impairment, NFTs have been suspected to be the direct culprit for neurodegeneration. However, the formation of NFTs turned out to be neither necessary nor sufficient to provoke neurodegeneration. For example, neurons with NFTs may live more than a decade in AD patients [62], which is in line with the observations from transgenic mouse models [63]. Neurons died before the formation of NFTs, and NFT-bearing neurons can survive without cellular activity defect [63]. Remarkably, in two reversible Tau expression transgenic mouse models, rTg4510 and ΔK280, which express human TauP301L mutation and pro-aggregation mutant, respectively, the decline in cognitive function was halted or even reversed after switching off Tau expression, regardless of the presence of NFTs. This finding strongly suggests that NFTs are not sufficient for the neurodegeneration [64,65,66]. Therefore, the toxicity that leads to neurodegeneration of Tau species is still under debate. Nonetheless, recent studies have indicated that Tau oligomers are the bona fide toxic species for neurodegeneration in tauopathy. Levels of soluble Tau oligomers in AD and PSP brains are significantly elevated [67,68], and a host of in vitro data has shown that Tau oligomers are neurotoxic [69,70,71]. It is also feasible that instead of being directly harmful, NFTs may alleviate the toxicity of Tau oligomers and thus protect neurons from degeneration [72]. However, it is still plausible that NFTs may be toxic by sequestering and occupying the cytosol space and thus inducing other cellular defects in the long term [73]. The normal neuron and pathological neuron in tauopathy are briefly depicted in Figure 2.
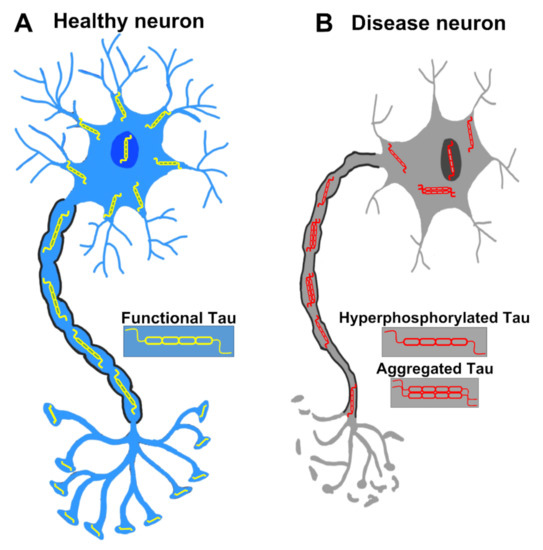
Figure 2.
A healthy neuron and a pathological neuron in tauopathy. (A) In a healthy neuron, Tau is usually found in axons but also located in dendrites, the nucleus, the plasma membrane and synapses. (B) In the tauopathy condition, Tau undergoes PTMs such as hyperphosphorylation, resulting in the detachment from microtubules and the formation of aggregates. Tau dysfunction causes many neurodegenerative phenotypes such as pre- and post-synaptic decays, axonal degeneration, and eventually neuronal death.
2.5. Clearance of Tau
Clearance of excessive and pathological Tau is a critical step for neurons to maintain the homeostasis of Tau protein and prevent Tau toxicity. Tau protein is cleared mainly via proteasome- and autophagy-dependent pathways. The general structures and function of the proteasome and autophagy are extensively reviewed elsewhere [74], and only Tau-related issues will be briefly covered in this review.
2.5.1. Tau Is Degraded by the Proteasome
Tau is subject to degradation by the proteasome in an ubiquitylation-dependent or -independent manner. Tau can be ubiquitylated in vitro [75,76], and the modification at K6 inhibits ubiquitin-dependent degradation by the 26S proteasome contributes to the formation of NFTs [77]. Moreover, the impaired function of the 26S proteasome has been reported in the rTg4510 mouse model where accumulated TauP301L binds to the 26S proteasome and inhibits its hydrolyzing activity against ubiquitinated proteins [78]. Nonetheless, through different approaches, such as ATP depletion, ubiquitylation-deficient cells, knockdown of a 19S proteasomal regulator subunit, and in-vitro ubiquitylation studies, it was shown in several cell models that ubiquitylation was not required for Tau degradation by the proteasome 20S [79].
2.5.2. Tau Is Degraded by Autophagy
Autophagy is a process known as “self-eating” that degrades substrates by the lysosome. There are three forms of autophagy: macroautophagy, chaperone-mediated autophagy, and microautophagy. The most common and well understood is macroautophagy [80] (hereafter, “autophagy” in short). An electron microscopic analysis of brain tissues from confirmed AD patients revealed that autophagic vacuole accumulates in dystrophic neurites and is correlated with the presence of NFTs [81]. In line with this clinical observation, a large number of studies have provided evidence that Tau is significantly degraded by autophagy. Cathepsin D is a protease that degrades Tau in the lysosome. When cathepsin D was added to the rat brain homogenates, Tau degradation was observed [82], and depletion of cathepsin D advanced Tau-induced neurotoxicity in a Drosophila tauopathy model [83]. In addition, when hippocampal slices were treated with chloroquine, a chemical well-known to raise lysosomal pH and inhibit autophagic flux, Tau protein levels (especially the PHF1 form) were significantly increased [84]. When the autophagy gene Atg7 was deleted in forebrain neurons in a mouse knockout model, accumulation of phosphorylated Tau, together with NFT formation, was observed with age-dependent neurodegeneration [85]. All of these studies collectively indicate that autophagy plays a key role in mediating Tau clearance.
Mutations of Tau that are covered in this review are summarized in Figure 1B.
3. Drosophila Tauopathy Model
3.1. Advantages
Drosophila melanogaster has been extensively used as an excellent model system due to their short lifespan, small size, and increased propagation. In addition, Drosophila melanogaster allows in-depth reverse genetic studies and large-scale forward genetic screening [86]. Approximately 70% of the genes related to disease conditions in mammals are also present in Drosophila [87]. Generation of loss-of-function mutants or transgenic lines for the desired gene of interest is relatively easier than other model systems [88]. Based on these advantages, Drosophila has been an attractive model system for studying human neurodegenerative disorders, including tauopathies [89,90,91,92], helping us to understand the molecular mechanisms underlying neurodegeneration phenotypes.
3.2. Current Drosophila Tauopathy Models
Drosophila has a single homologous gene to human Tau in the genome. Its protein contains five microtubule-binding domains, which are 46% and 66% similar to 3R and 4R of human Tau, respectively [93]. As a well-established genetic model, nearly 100 Drosophila Tau models, using at least 37 human Tau constructs, have been generated [90] (Table 1). Different human Tau isoforms, as well as numerous Tau mutants and truncated variants, were overexpressed under pan-neuronal- or eye-specific promoters using a GAL4/UAS system, a well-established binary gene expression system in Drosophila [94]. Although Drosophila tauopathy models mostly do not show tau aggregation, Tau is found phosphorylated [14]. In addition, numerous tauopathy-related phenotypes can be observed in these flies, such as rough eye phenotypes (REP), neurodegeneration (ND) in the brain and eyes, learning and memory defects (LMD), reduced lifespan (RL), neuromuscular junction (NMJ), and locomotion defects. These phenotypes can be scored simply and quickly and have been successfully used in large-scale screens for searching modifiers of Tau pathology [92]. Thus, these fly models provide a versatile genetic tool to explore the vast diversity of tauopathies. The representative Drosophila tauopathy models and their phenotypes are listed and summarized in Table 1. (More information about Drosophila models can be found at https://flybase.org (accessed on 4 August 2021)).

Table 1.
Drosophila tauopathy models. GMR-GAL4: eye-specific GAL4 driver; Elav-GAL4: pan-neural GAL4 driver; OK-GAL4: motor neuron-specific GAL4 driver; MB-GAL4: Mushroom Body-specific GAL4 driver; C161-GAL4: sensory neuron-specific GAL4 driver; D42-GAL4: motor neuron-specific GAL4 driver. hTauWT: human Tau wildtype; LMD: learning and memory defects; MT: microtubule; ND: neurodegeneration; NMJ: neuromuscular junction; pTL: pTau levels; REP: rough eye phenotype; RL: reduced lifespan.
3.3. Representative Assays
3.3.1. REP
REP is the most commonly used phenotype for screening of molecular players in Drosophila because it provides a visible phenotype that can be easily quantifiable [110], hence making Drosophila useful for enhancer or suppressor screens of Tau toxicity [96,110,111]. Expression of the Tau gene in the eyes using eye-specific GMR-GAL4 generates roughening of eyes with reduced eye size due to retinal degeneration as a readout for Tau toxicity [95,112]. Drosophila REP screening was applied to identify orthologs of candidate risk genes using data from a genome-wide association study-AD (GWAS-AD) data involved in Tau neurotoxicity. The screening identified Amph, p130CAS, Eph, Fak, and Rab3-GEF as modifier genes [111]. Apart from genetic screening analysis, REP was also useful in analyzing the extent of neurotoxicity of different phosphorylation forms of Tau protein [113] and assessing electrophysiological properties of Tau using electroretinogram recordings [114].
3.3.2. Neuronal Cell Death and Neurodegeneration
A shrunken brain, which is the consequence of cell loss in the brain, is the common feature of many neurodegenerative diseases, including tauopathy [115]. Therefore, neuronal cell death is examined regularly as one of the most representative phenotypes when a tauopathy animal model is generated. Tauopathy flies show increased vacuolar phenotype either in the brain (Elav-GAL4) or in the eye (GMR-GAL4) for neuronal degeneration due to neuronal cell loss. This phenotype can be visualized by immunohistochemical and histological methods using hematoxylin staining for vacuoles or TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assays for apoptotic cells in the tauopathy models [89,97,99,101,103].
3.3.3. Learning and Memory Assays
The Drosophila mushroom body (MB) is the functionally homologous brain structure to the hippocampus of mammals, which is the epicenter of regulating learning and memory [116,117,118,119,120]. Expression of human Tau using a pan-neuronal driver (Elav-GAL4) affects olfactory learning and memory functions in Drosophila [105]. Restricted Tau expression to MBs also results in defects in learning and memory that were determined by testing responses to attractive and repulsive odors in Tau-overexpressing flies [121]. Similarly, an aversive phototaxis suppression assay was used in Drosophila tauopathy models to measure learning and memory function [122].
3.3.4. Lifespan
The ultimate consequence of Tau toxicity is the reduction of lifespan in tauopathy flies, which can be analyzed by counting the number of surviving flies. Under normal conditions, wildtype flies live approximately 60 to 90 days, and neuronal expression of Tau isoforms and mutants reduce lifespan according to their toxicity [89,123]. Apart from the lifespan assay, pan-neuronal expression of Tau also leads to pupal lethality, which can be analyzed for Tau toxicity [102]. Overall, these phenotypes can be used to identify genetic modifiers of Tau toxicity.
3.3.5. NMJ and Locomotion
The larval NMJ is a widely analyzed structure used to understand the mechanisms of synapse formation, growth, and maintenance [124,125]. The most commonly used approach for NMJ analysis is to drive expression of the gene of interest (e.g., Tau) in larval motor neurons, using drivers such as D42-GAL4 and OK6-GAL4, and then visualize its activity within readily accessible larval motor neuron axons and NMJ using assays for axonal transport and synaptic function, respectively [91]. Expression of 0N3R Tau in NMJ using D42-GAL4 affects morphological and functional disruption of NMJ with reduction in size and numbers of boutons, as well as reduction in mitochondria at the synapse [126]. In addition, hyper-phosphorylated Tau strongly affected vesicle motion in axons and synaptic function [102]. Synaptic abnormalities led to defects in larval locomotion, and Drosophila AD larvae expressing Aβ42, or Tau exhibited reduction of NMJ bouton number and larval locomotion [92,127,128]. Hence, the locomotion behavior in conjunction with the NMJ phenotype is quite useful in addressing neuronal morphological defects followed by associated behavioral changes upon expression of diverse Tau species and manipulations of Tau modifiers.
3.3.6. Transposon Mobility
Transposable elements (TEs) are known as ‘jumping genes’ and constitutes approximately 45% of the human genome. Transposable elements are the mobile genetic sequences seen in all the eukaryotic genomes [129]. Somatic transposition of TEs, retrotransposons, which mobilize through an RNA intermediate—are observed in human brains [130,131,132], mouse [133], and fly models [134]. In addition, TEs have been also shown to carry transcriptional control elements that regulate the transcription of their own genes as well as neighboring host genes, which was intensively reviewed in a recent review [135]. Recent studies of TEs in Drosophila have shown TE activation to be in association with Tau pathology in AD [136]. They also showed that Tau alters TE activity in the Drosophila brain expressing human wild-type or mutant Tau, suggesting that genomic instability in Tau-mediated AD mechanisms occurs due to TE activation. Similarly, transposable element dysregulation was identified as a key mediator of neuronal death in tauopathies, where heterochromatin decondensation and reduction of piwi and piwi-interacting RNAs (piRNAs) drive transposable element dysregulation in tauopathy [137].
3.4. Limitations
Despite many advantages of Drosophila as a tauopathy model, a few limitations need to be considered. First, compared to other vertebrate species, such as mice, the anatomy of the Drosophila brain is quite different from that of the human, in that brain connectivity is relatively simple and asymmetric [138,139]. Second, established behavioral assays are relatively simple and limited to directly addressing in-depth cognitive function [140]. Lastly, the lack of some critical organs/tissues homologous to those of vertebrates that may contribute to tauopathy, such as the adaptive immune system, closed blood circulatory system, and blood-brain barrier, might cause inconsistent and unpredictable results when applied to humans [141,142].
3.5. The Utility of the Drosophila Tauopathy Models and Their Translational Applications
A study from the Drosophila tauopathy model showed that the PAR-1 kinase can trigger tau toxicity by directly phosphorylating Tau at Ser262 and Ser356 [99]. This phosphorylation is an important step for the activity of downstream kinases, such as GSK-3β/sgg and Cdk5, to phosphorylate other sites in Tau and generate phosphoepitopes in diseased conditions [99]. Such phosphorylation of Tau protein may cause dysfunction of microtubule stabilization and neurodegeneration, demonstrating that phosphorylation of Tau can occur in a sequential manner dictated by previous phosphorylation events [97]. Pharmacological inhibition of GSK-3β by treating with SB415286 and lithium [143,144,145] promotes synapse formation and lifespan, respectively. Using Elav-GAL4/UAS-WT or hTauR406W Drosophila tauopathy models, nicotinamide mononucleotide (NAD) adenylyl transferase (NMNAT) were shown to be involved in a neuroprotective role of rescuing TauR406W mutant phenotypes by clearing toxic phosphorylated Tau proteins and facilitating proper neuronal function [122]. Apart from GSK3β signaling regulated by Insulin signaling pathway, other pathways JAK/STAT signaling pathway [146], and Nuak1, an AMP-activated protein kinase (AMPK)-related kinase [147] were identified in causing hyperphosphorylation and Tau toxicity. In addition, genes involved in regulators of actin network, viz, cheerio (a fly ortholog of filamin), chd64 (a fly ortholog of transgelin-3), jaguar (a fly ortholog of myosinVI), paxillin, were identified as modifiers of the TauV337M [148].
Ambegaokar and Jackson (2011) carried out one of the most exhaustive genetic screens using P-element insertion collections and identified a loss-of function collection consisting of 920 genomically mapped lethal P-elements [149]. In total, 1905 lines were screened and 37 modifiers of tau toxicity regulating autophagy, the cell cycle, and gene expression as well as phosphorylation were identified. Recent studies have also investigated the ability of small therapeutic compounds using a custom chemical library to improve tau-induced rough-eye phenotype in a Drosophila melanogaster model of frontotemporal dementia (FTD) and showed that Ro 31-8220, a potent inhibitor of PKCα, improved the rough-eye phenotype, reduced phosphorylated Tau species in vitro and in vivo, reversed Tau-induced memory impairment, and improved the fly motor functions [150].
As invaluable resources, the UAS-dsRNAi line libraries that cover more than 90% of the entire fly genome can be utilized for Tau regulator screening (Vienna Drosophila Research Centre, The Harvard Drosophila RNAi Resource Project, NIG-FLY Stock Centre and Bloomington Stock Centre), where various UAS-dsRNAi flies can be screened to identify the candidates involved in regulating Tau toxicity by crossing with UAS-Tau-expressing transgenic flies. Tissue specific knock down of UAS-dsRNAi in flies expressing UAS-Tau will give a clear picture in mediating Tau pathogenesis and its interaction with Tau proteins. For example, RNAi functional screening in Drosophila models of human Tau-mediated neurodegeneration found that the downregulation of the Drosophila REEP1 homolog enhanced Tau toxicity with increased formation of Tau insoluble aggregates [151]. They further showed that the overexpression of either the Drosophila or the human REEP1 protein was able to revert these phenotypes and promote neuronal resistance to ER stress. Another genetic screening using GMR-GAL4/UAS-hTauV377M transgene have identified 16 enhancers and 8 suppressors of Tau toxicity which encode kinases and phosphatases as the major determinants of neurotoxicity in vivo [110]. In addition, a genetic screen for modifiers of 2N4R hTau toxicity has identified components of the ERK/MAPK and p38/MAPK pathways [149]. More specifically, a genetic modifier RNAi screening designed to validate AD susceptibility genes have identified cindr, fit1/2, and aret, the fly orthologs of the human CD2AP, FERMT2, and CELF1, respectively, as Tau modifiers implicated mainly in cell adhesion, together with identification of ITGAM and ITGA9, a fly homolog of the human integrin adhesion receptors [152]. This genome-wide genetic screening strategy can be modified by beginning the screening with a Drosophila genome-wide UAS-miRNA library followed by another RNAi screening for a selected set of identified miRNA-targets, yielding an E3/E4 ubiquitin ligase UBE4B as a novel tauopathy modifier to clear accumulated wild type Tau protein through autophagy [92].
4. Zebrafish Tauopathy Model
4.1. Advantages
The zebrafish is a small vertebrate animal model with high generation capacity [153]. Similar to other vertebrates, the anatomical structure of the zebrafish central nervous system is conserved, divided into the forebrain, midbrain, hindbrain, and the spinal cord, together with optical transparency at the larvae stage for superb observation capability [154,155]. Zebrafish and humans share approximately 70% similarity at the genome level, with 84% of counterpart genes related to human genetic diseases, including neurodegenerative diseases found in the zebrafish genome [156]. In addition, transgenic zebrafish can be efficiently generated with the Tol2 transposase system using specific tissue promoters by conventional expression [157] or conditionally using GAL4-UAS system [158] or Cre/loxP system [159]. Furthermore, targeted gene knockouts, such as Zinc Finger Nucleases (ZFNs), Transcription Activator-like Effector Nucleases (TALEN), and recently Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 [160], aided by a number of fluorescence-based transgenic lines, allow for the creation of disease models with high productivity [161].
Drug screening for neurodegenerative diseases including tauopathy requires a high-throughput in-vivo screening platform with large-scale screening capability because of the intricate etiologies of neurodegenerative diseases [162]. Zebrafish can provide a unique opportunity for the phenotype-based large-scale high-throughput screening in larval, as well as adult stages as a vertebrate animal model [163,164,165]. Indeed, through zebrafish chemical screening, several drugs have been discovered and translated into clinical trial [166]. For example, Lorcaserin, an FDA-approved serotonin agonist, was identified from a drug screening using scn1 mutants to reduce the seizure severity and/or frequency in five Dravet syndrome patients [167].
4.2. Current Zebrafish Tauopathy Models
In zebrafish, one MAPT in humans is duplicated as paralogous genes mapta and maptb: mapta can be spliced into isoforms with four and six tubulin-binding repeats (4R, 6R), while maptb is mostly spliced into 3R isoforms [168]. Both paralogous genes are predominantly expressed in the CNS, with maptb largely expressed in the trigeminal ganglion and dorsal sensory neurons during early embryogenesis [168].
Since 2002, several zebrafish genetic tauopathy models have been reported using different strategies (Table 2). Among them, the most acknowledged model is the one from Haass group, in which the human TauP301L mutant form was overexpressed under pan-neuron-specific HuC promoter by utilizing a Gal4/UAS-based bidirectional expression system [169]. This transgenic zebrafish model exhibited pathological tauopathy characteristics like multiple pathological Tau-specific phosphorylation, defective motor axons/synapses, neuronal cell death in the spinal cord, the escape response defect at larval stages, and NFT formation at young adult stages [169]. Another zebrafish tauopathy model from the Rubinsztein group successfully confirmed the pathogenicity of Tau A152T variant (TauA125T) as a risk factor for FTD and AD by generating a novel transgenic line overexpressing this human Tau variant using the Gal4/UAS system [170]. This TauA125T transgenic model also exhibits tauopathy phenotypes, including pathological Tau phosphorylation of AT270, PHF, and AT8, the presence of NFT, increased neuronal cell death, abnormal axonal pathfinding and branching, and the motility defect against touch response. In particular, the proteasomal activity was shown to be compromised in this model delaying Tau clearance, and induction of autophagy activity was suggested as a potential therapy [170]. Other zebrafish tauopathy models in which human Tau was directly expressed under a pan-neuronal promoter also showed frank phosphorylation of Tau, but other classical tauopathy phenotypes were either vague or undetectable in these models [171,172,173,174,175]. The reason for this inconsistency is not clear, but the expression level of Tau protein may be a key difference. Nonetheless, due to encouraging results of zebrafish as a tauopathy model, more transgenic lines are expected to follow, especially to take full advantage of zebrafish as a genetic tauopathy model. The current zebrafish tauopathy models and their phenotypes are listed and summarized in Table 2 (More information about zebrafish can be found at https://zfin.org (accessed on 4 August 2021)).
Table 2.
Zebrafish tauopathy models. hTau: human Tau; N/A: not applicable; TBI: traumatic brain injury; WT: wildtype; +: positive.
4.3. Representative Assays
4.3.1. Neuronal Cell Death
In the zebrafish, assays for apoptosis detection were well developed and described either in whole-mount or in section by TUNEL staining or by acridine orange staining that detects dead cells in live zebrafish embryos without fixation [174,176,177]. Several antibodies for detecting apoptotic pathways can also be applied to zebrafish tauopathy models, such as anti-cleaved caspase-3 antibody [176].
4.3.2. Axonopathies
Axonopathies, such as dystrophic axons, axonal swelling, and demyelination, are some of the distinctive neuropathological clinical features of tauopathy [178,179]. In developing zebrafish, primary motor axons with a large diameter appear at ~11 h postfertilization from the spinal segments and protrudes ventrally [180]. During developmental stages, these axons can be visualized live with high resolution using motor neuron-specific transgenic lines such as Tg(mnx1:GFP)ml2 and Tg(insm1a: EGFP)ntu804 [181] or using motor axon-specific antibody, Znp1 [182]. The continuous development of motor neurons has been examined without missing crucial events in live embryo with time lapse imaging [183] and can be applied to examine Tau- or Tau modifier-induced axonal defects with great detail in a zebrafish tauopathy model.
4.3.3. Locomotion
Neuron-specific expression of Tau in tauopathy models affects the nervous system, resulting in morphological and functional defects at multiple levels, and the locomotion phenotype is an essential assay that allows evaluation of those defects collectively. Zebrafish locomotion has been intensively utilized due to its small size and high-throughput capacity at both larvae and adult stages [184,185]. For convenience of researchers, various kinds of the commercially available apparatus and software, plus detailed protocols have been well developed specifically for analyzing the locomotion in zebrafish, making such an assay feasible in zebrafish laboratories on a regular basis.
4.3.4. Ubiquitin-Proteasome System (UPS) and Autophagy-Lysosomal Pathway (ALP)
In general, monomeric Tau protein is degraded by both 20S and 26S UPS [186,187,188,189], but such UPS is likely unable to clear aggregated Tau but is instead impaired by Tau toxicity [78,190]. In the zebrafish tauopathy model, apart from conventional methods such as western blotting for UPS markers, the UPS activity can be easily monitored in vivo by injecting Ub-R-YFP (substrate tagged with YFP, known to be degraded by the UPS) construct into embryos [191]. Thus, the examination of UPS malfunction is worth considering when characterizing zebrafish tauopathy phenotypes. Similarly, the ALP, another degradative pathway for Tau protein, can be visualized in vivo by expressing GFP-Lc3-RFP-Lc3∆G construct, as well as western blots of autophagy markers and specific inhibitor treatment in zebrafish models [192].
4.4. Limitations
Despite several advantages, the number of zebrafish tauopathy models is still limited compared to those of rodents, Drosophila, or C. elegans tauopathy models. More zebrafish tauopathy models can be developed and further characterized with a battery of tauopathy phenotypes in great detail in vivo. In particular, a few neurobehavioral assays in zebrafish during both larva and adult stages that may be directly relevant to tauopathy have yet to be applied to current and future zebrafish tauopathy models. For example, the anxiety level and a memory-based adaptive behavior, possibly disrupted in tauopathy, can be represented by simple neurobehavioral tests such as thigmotaxis/open field test and habituation of both larva and adult zebrafish, which can be devised for high-throughput screening.
4.5. The Utility of the Zebrafish Tauopathy Models and Their Translational Applications
Although zebrafish tauopathy models are limited, several new findings on pathological features and signaling factors for tauopathy have been discovered, and novel GSK3β inhibitors and Tau assembly modulators were validated as potential therapeutic targets for tauopathy using these zebrafish Tau models.
By combining human TauP301L-overexrpessing zebrafish model with the “mitofish”, a transgenic zebrafish that was developed to study the dynamic and life cycle of mitochondria in real time in vivo, a profound mitochondrial transport deficit was visualized under a tauopathy condition [193]. This phenotype was rescued by overexpressing MARK2, the kinase that regulates the binding affinity of Tau to microtubules [194], thus directly demonstrating the impact of MARK2 on axonal transport in a Tau-dependent manner in vivo [193]. Similarly, the behavior of microglia in a tauopathy disease context in vivo was investigated by combining human TauP301L-overexpressing zebrafish model with the Tg(apoe:EGFP) that marks the microglia [195]. Compared to the non-disease condition, the microglia in the zebrafish tauopathy model became highly mobile and dynamically changed their morphology with a fewer and shorter branching: more interestingly, the phagocytosis process by the microglia engulfing apoptotic neurons was also observed in real time, and furthermore, the genetic ablation of microglia increased the Tau hyperphosphorylation level, suggesting that microglia possesses a neuroprotective role in the tauopathy condition [195]. Also, as traumatic brain injury (TBI) is a conspicuous risk factor for dementias including Tauopathies, subjecting a novel TBI paradigm to the human TauWT zebrafish larval model increased neuronal death and Tau inclusion of which phenotypes were rescued by dynamin inhibitors or anticonvulsant drugs [175], suggesting that the seizure activity has the strong impact on prion-like seeding and spreading of Tau following TBI.
Potential candidate factors that may modulate tauopathy were functionally validated using zebrafish tauopathy models. For example, dysregulated signaling of brain-derived neurotrophic factor (BDNF) is strongly associated with neurodegenerative diseases [196], but how BDNF signaling affects tauopathy or vice versa remains unclear. By exploiting the human TauP301L zebrafish model, it was shown that the BDNF level was reduced and associated with axonal developmental defects, but not neuronal death, in the tauopathy condition, since exogenous BDNF supplementation was able to rescue the primary axonal growth but had no effect on Tau-induced apoptotic cells [197]. In addition, HS3ST2 (heparan sulphate glucosamine 3-O-sulphotransferase 2), also known as 3OST2, and REG-1α (regenerating islet-derived 1α) are two new factors that were shown to be critical for the abnormal phosphorylation of Tau in the hTauP301L zebrafish model [198]. A loss of function of HS3ST2 result in an apparent inhibition of abnormal Tau phosphorylation in the brain and the spinal cord, leading to a complete rescue of axonal length and touch response phenotypes [198]. Reversely, overexpression of REG-1α increased tau hyperphosphorylation through the AKT/GSK3β pathway in the same zebrafish tauopathy model [199]. In terms of testing a Tau aggregation modulator, FKBP52 (FK506-binding protein with a molecular mass of ~52 kDa) was validated as a new player that is potentially applicable for therapeutics [49]. This immunophilin protein directly interacted with Tau protein and induced the formation of Tau oligomers in vitro, with EM analysis showing that FKBP52-induced oligomers assembled into filaments. In the human TauP301L zebrafish model, knockdown of FKBP52 debilitated the pathological Tau activity and recovered axonal outgrowth and branching in defective motoneurons, confirming the functional implication of FKBP52 in modulating Tau conformational change and assembly in vivo [49].
To take advantage of zebrafish as an in vivo animal model for chemical genetics, zebrafish tauopathy models were also utilized for candidate small molecule validation. GSK3β, one of the main kinases that phosphorylates Tau, is therefore considered as a key therapeutic target for tauopathy. The human TauP301L transgenic zebrafish was adopted to validate the efficacy of a newly developed GSK3β inhibitor, AR-534, confirming its stronger effect on diminishing pathologic Tau phosphorylation with a high selectivity and good bioavailability in vivo than SB-216763 and SB415286, two well-known GSK3β inhibitors [169,200]. In addition, a few FDA-approved clinical drugs were tested in the human TauP301L zebrafish model. Given that HS3ST2 induced abnormal Tau phosphorylation as aforementioned, surfen and oxalyl surfen are small molecules harboring heparan sulfate antagonist properties and well tolerated in clinical setting in cancer treatment [201,202]. These two small molecules were capable of mitigating Tau hyperphosphorylation and rescuing spinal motoneuron defects, leading to recover the touch escape response in the zebrafish TauP301L tauopathy model [203]. Sildenafil or tadalafil, an inhibitor of phosphodiesterase 5, widely used for erectile dysfunction and pulmonary hypertension treatment [130], was also shown to be a promising strategy to prevent Tau toxicity: by activating cGMP (cyclic guanosine monophosphate)-dependent protein kinase, the stimulated proteasome activity reduced the level of Tau protein and decreased the associated morphological abnormalities in the zebrafish human TauA152T model [204].
Finally, zebrafish tauopathy models can be also applied for mid- to high-throughput in vivo chemical library screening. An embryonic zebrafish tauopathy model that transiently overexpresses Tau-GFP fusion was used to screen 400 herbal extracts using neuronal death as an in vivo readout [205]. 45 out of 400 extracts were confirmed to be effective in reducing Tau-induced neuronal death, and among them, the extract from Tripterygium wilfordii stem showed the highest effect. HPLC analysis showed that its major compound was epicatechin that activated Nrf2-dependent antioxidant responses and thus abated Tau-induced neuronal death [205]. Of note, overexpression of multiple signaling factors such as Bcl2-L1 or GDNF was also capable of effectively protecting neurons from Tau-induced apoptosis in the same zebrafish tauopathy model [173,205].
5. C. elegans Tauopathy Models
5.1. Advantages
Caenorhabditis elegans (C. elegans) is a small invertebrate genetic animal model that possesses a high reproduction ability (once every 3–4 days) and a short lifespan (approximately 2–3 weeks) [206,207]. A major advantage of C. elegans is that desired phenotypes can be assessed faster than any other model organisms [208,209]. Regardless of its simplicity, C. elegans is a multicellular organism composed of the brain, pharynx, intestine, gonads, muscle, and rectum. Furthermore, the transparency of its body during its lifetime facilitates live imaging with the aid of fluorescent protein reporters under tissue-specific promoters, including neuronal promoters. C. elegans has 302 lineage-identifiable neurons, thus increasing the accuracy of neuronal analyses while reducing the complexity [210]. In the context of neurodegenerative disorders research, neuronal cell death or protein aggregation can be easily observed and quantified, allowing single cell-lineage analysis for a phenotype at an organism level [211]. Practically, the maintenance of C. elegans is very convenient due to its small size even in the adult stage (~1 mm) and the ability to be stored in liquid nitrogen and recovered years later [212].
C. elegans have 60–80% orthologous genes with humans, with more than half of them associated with human diseases [213]. Along with the completed sequenced genome [214], the compendium of neuronal connectomes and the whole neuronal expression map by the CeNGEN project of C. elegans using state-of-the-art technologies are available. These tools help to understand neuronal function with single cell resolution at the organism level [210,215]. Transgenic C. elegans models for neurodegenerative diseases can be generated easily by traditional microinjecting transgene DNA [216] or biolistic bombardment using DNA-coated gold particles to integrate exogenous DNA with high efficiency [217]. Altogether, C. elegans is a still convenient and promising in vivo system for modeling human mutation-associated diseases while maintaining favorable features of in vitro systems.
5.2. Current C. elegans Tauopathy Models
C. elegans has two PTL-1 isoforms, PTL1a and PTL1b, homologous to human Tau. The two isoforms are differentiated by the number of tandem repeats (5 repeats in PTL-1a and 4 repeats in PTL-1b) and share a homologous microtubule-binding region with mammalian Tau [218]. To date, nearly 20 lines of C. elegans tauopathy models from seven groups have been established by taking advantage of the convenient genetic manipulation. Various isoforms (0N4R, 1N4R, 2N4R), as well as wild type (WT) and mutation forms (P301L, V337M, R406W, A152T, V363I, V363A, T231E, K274/281Q, ∆K280, pseudo-hyperphosphorylation form) of human Tau were expressed under pan-neuronal or touch-neuronal promoters in the transgenic worms (Table 3). In these tauopathy models, most of the pathological features of human tauopathy, such as Tau phosphorylation, axonal degeneration, neuronal loss, synapse dysfunction, and mitochondria defect, were in an age-dependent manner. Typical C. elegans behavioral phenotypes, including uncoordinated moving, touch response, or thrash rates, were affected to different degrees of severity depending on the expressed forms of human Tau species [59,219,220,221,222,223,224]. The representative C. elegans tauopathy models and their phenotypes are listed and summarized in Table 3 (More information about C. elegans can be found at https://wormbase.org (accessed on 4 August 2021)).
Table 3.
hTau: human Tau; N/A: not applicable; PHP: pseudohyperphosphorylated; Unc: uncoordinated; TAI: Tau aggregation inhibitors; ThS: thioflavin S; WT: wildtype; +: positive.
5.3. Representative Assays
5.3.1. Lifespan
Due to the short life cycle of C. elegans (reproduction, every 3~4 days; lifespan, 2~3 weeks), different stimuli that influence survival are widely used [225]. For example, genetic manipulations or temperature effects on the lifespan of C. elegans can be tested on the solid bacteria agar plate, whereas the treatment of diet compounds or chemicals can be more feasible in liquid media [226,227].
5.3.2. Neuronal Death
Similar to Drosophila or zebrafish tauopathy models, neuronal cell death is one of the indispensable pathological phenotypes in C. elegans tauopathy models. Various staining techniques, such as TUNEL, acridine orange, or immunostaining, can be used to visualize this phenotype in worm models [228].
5.3.3. Axonal Defects
In C. elegans tauopathy models, the simple morphology of cells and tissues labeled with fluorescence markers have made it possible to use live imaging to examine pathophenotypes of motor and mechanosensory axons in great detail and to assess axonal discontinuities, morphological abnormality, synapse defects, and faulty commissure [229,230,231,232].
5.3.4. Behavior Phenotypes
Generally, behavioral phenotype assays to test Tau toxicity in C. elegans are mobility-based [233]. In the thrashing assay, a well-established motility-based assay, the frequency and lateral swimming movements of worms placed in liquid media are measured [234]. Other mobility-dependent indexes, such as uncoordinated movement, distorted wave motion, or locomotion speed, are also commonly used as behavioral assays in C. elegans models to test effects of drugs, chemicals, or genetic mutations [235,236].
5.4. Limitations
Ironically, the aforementioned advantages of C. elegans can be potentially disadvantageous in tauopathy research under certain conditions. For instance, the small size and limited number of neurons of C. elegans can be a drawback in biochemical approaches because the small number of touch neurons overexpressing human Tau (a total of only six neurons) causes difficulty in detecting Tau protein abnormalities such as thioflavin staining [224]. In addition, although C. elegans carries 60–80% orthologous genes with human and mutant line generation in C. elegans is straightforward, only 30% of genes of C. elegans can be mutated because of high lethality [237]. Although gene-silencing techniques such as RNAi can be complemented to knock down the gene of interest, such knockdowns in the C. elegans nervous system are often refractory [238,239,240], thereby hampering the tauopathy modifier studies. Furthermore, lack of features of vertebrate neurons, such as DNA methylation or myelination of axons, in addition to absence of several defined vertebrate organs, such as brain and circulatory system, may lead to an inconsistency in both physiological and pathological outcomes of tauopathy models of C. elegans and human cases and in screening results for tauopathy drugs or compounds [241]. Finally, despite recent studies showing the ability of this model to learn and remember experiences [242,243], it may be too simple to deduce the cognitive behavioral phenotypes observed in mammalian neurodegenerative disease.
5.5. The Utility of the C. elegans Tauopathy Models and Their Translational Applications
Even though C. elegans is a very simple model and appears to be far from being an alternative for vertebrate models as a clinical testing system for neurodegenerative diseases, the C. elegans model research for tauopathy, covering the discovery of its characteristics and the identification of the drugs or therapeutic targets, has been quite informative and worth being validated in higher model systems before transferring to clinical trials.
For instance, to assess the significance of the Tau aggregation process in tauopathy, a pro-aggregation C. elegans model overexpressing human TauΔK280 was examined in parallel with another transgenic line co-expressing anti-aggregation substitutions I277P and I308P of Tau that prevent β-sheet formation and subsequent aggregation (human TauΔK280, plus I277P and I308P) [59]. As a result, the pro-aggregation strain showed severe motility impairment, neuronal dysfunction as well as disturbed axonal transport of mitochondria which was not observed in the strain that co-expresses the anti-aggregation substitutions. Furthermore, several Tau aggregation inhibitors showed the ability to prevent Tau toxicity in the pro-aggregation strain, highlighting that inhibiting the Tau aggregate formation may be a potential target and Tau aggregation inhibitors could be encouraged to test in clinical trials [59]. Another example is the function validation of dihydrolipoamide dehydrogenase, one of the major metabolic enzymes of mitochondria [244]. In the human TauWT C. elegans model, suppression of this gene either by RNAi or inhibitor resulted in increased whole-body glucose levels and Tau phosphorylation, consistent with the observation in AD patients [245], suggesting that the disturbance in energy metabolism can significantly induce the neurotoxicity of pathological Tau [246]. Also, co-expression of wild type LRRK2, known as a genetic cause of Parkinson’s disease and some cases of Tauopathy [247], led to increased expression of several 60S ribosomal, mitochondrial, and V-type proton ATPase proteins without altering the redox status in the human TauV337M C. elegans model. Moreover, co-expression with mutant LRRK2 (G2019S or R1441C) showed similar effects additionally with increased protein oxidation and lipid peroxidation, revealing roles of the LRRK2 activity in a Tau-dependent context [248]. In addition, a selective loss of glutamatergic neurons was observed in a human TauA152T overexpressing transgenic C. elegans [249]. In this strain, the glutamatergic nervous system was shown to be particularly vulnerable, largely due to necrotic cell death pathway. Genetic analysis also revealed this phenomenon occurred through several mechanisms, including type 9 adenylyl cyclase signaling, signaling through glutamate receptor complexes, aging-related signaling, and Ca2+ dyshomeostasis with increased Ca2+ released from the ER [249].
In a recent study, a “PTM-mimetics” C. elegans strain was generated to clarify the precise impact of PTMs on neural toxicity of Tau. This strain contained mutations of T231E that mimics phosphorylation of pathological epitope or K274Q and K281Q that mimic disease-associated lysine acetylation as a single-copy gene insertion by CRISPR-Cas9 genome editing [224]. This “PTM-mimetics” strain showed reduced touch response and exhibited neuronal morphological abnormalities in an age-dependent manner, as well as mitophagy defect in response to mitochondrial stress. By limiting Tau expression as a single-copy, not as overexpression, the decisive role of PTMs in the pathogenesis of tauopathy has been emphasized, thus providing a new perspective for the therapeutic approach [224].
Curcumin, one of the common used spices and ancient medicines, has been validated epidemiologically and currently tested for clinical trials of various diseases [250]. Interestingly, epidemiological studies showed that the prevalence of AD in India is less than in US, which may be explained by the curcumin-rich diet [251]. Based on this observation, Yasuo Ihara group have tested the effects of curcumin on C. elegans human TauWT and TauR406W models [252]. As a result, curcumin rescued not only the uncoordinated (Unc) phenotype but also the neuritic abnormalities of both human TauWT and TauR406W expressing worms. However, curcumin did not prevent Tau hyperphosphorylation nor the aggregation, but instead increased the level of acetylated α-tubulin, suggesting tubulin may play a critical role in controlling Tau toxicity [252]. The following study has disclosed the important of balanced expression of Tau and tubulin in tauopathy by examining several different strains of C. elegans expressing various levels of WT Tau and assessing the effect of the tubulin [253]. The knockdown of tbce-1, a human tubulin-folding cofactor E homologue, or α-tubulin led to enhanced tau toxicity in a Tau level-dependent manner. Thus, the neurotoxicity of Tau can be triggered by reducing tubulin levels, even in a very low Tau-expressing worm that showed no abnormal behaviors per se. More strikingly, co-incubation of purified tubulin was able to inhibit Tau aggregation in vitro. Therefore, the ratio of expression between Tau and tubulin may be a determinative character of the tauopathy cascade [253].
One of the obvious advantages of C. elegans tauopathy model is a large-scale genetic screening capability. Indeed, a RNAi library screening was conducted using human TauV337M C. elegans model: out of 16757 genes screened, 60 genes showed increased Unc phenotype in the Tau transgenic worm. Confirmed by loss-of-function studies, aex-1, acr-14, lin-44, sir-2.3, pxn-1, and vap-1 were finally identified as enhancer factors of Tau toxicity, being novel candidate genes associated with tauopathy since human homologs were found [254]. Similarly, from a forward genetic screening using human TauV337M C. elegans model, recessive mutations of sut-1 and sut-2 partially suppressed the Unc phenotype, Tau aggregation and pathological features of tauopathy. SUT-1 protein was shown to interact with UNC-34 protein, while SUT-2 physically bound to ZYG-12, required for tau neurotoxicity. Moreover, mammalian SUT-2, which is MSUT2, was found markedly decreased in the post-mortem temporal lobe brain region of AD patients. In line with this, in vitro data also showed that high levels Tau led to increased expression of MSUT2 protein [255,256,257]. Taken together, screening of tauopathy C. elegans models allowed the identification of several conserved molecular pathways participating in Tau neurotoxicity, which are potentially utilized to be novel strategies for treating tauopathy.
6. Conclusions
The etiology of tauopathy is very complex and multi-faceted, and finding an efficient cure is imperative. Appropriate utilization of animal models that mimic essential pathophenotypes of tauopathy will reveal underlying mechanisms and identify key modifiers for disease prevention or novel drug target discovery. Despite their imperfections, the small genetic animal models discussed in this review (Drosophila, zebrafish, and C. elegans) have greatly helped scientists to appreciate diverse aspects of Tau biology as well as to understand detailed molecular pathways of Tauopathy (Figure 3, Table 4). Through their major advantages, including (1) low cost in infrastructure and maintenance, (2) short lifespan, (3) strong flexibilities of genetic manipulation, and (4) real-time, live imaging, and/or (5) high-throughput screen capability, tauopathy models of Drosophila, zebrafish, and C. elegans will continue to contribute to the discovery of a novel therapy for Tauopathy.
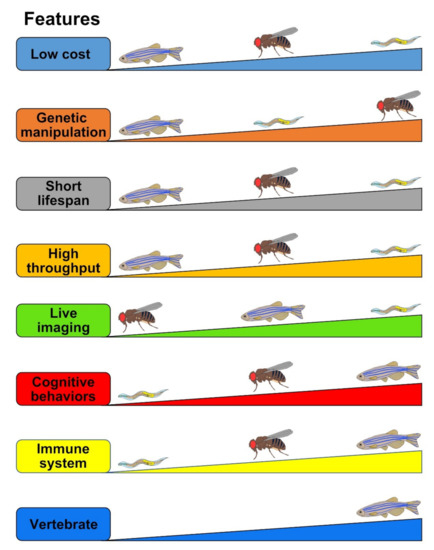
Figure 3.
Comparisons of the strength and limitations of Drosophila, zebrafish, and C. elegans as tauopathy models. The evaluated criteria (“Features”) are represented as the thicker means the better. For example, in the Low cost feature, C. elegans stands in the first place which is the cheapest, followed by Drosophila in the second and zebrafish in the last.
Table 4.
Summary of tauopathy models of Drosophila, zebrafish, and C. elegans.
Author Contributions
H.-K.G., K.Y. and J.-S.L. designed the format of the manuscript, and all authors contributed to writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Research Foundation (NRF) and National Research Council of Science & Technology (NST) of Ministry of Science and ICT of Korea (NRF-2019R1A2C1010661, 2019R1A2C2004149, CRC-15-04-KIST), and KRIBB Research Initiative Program (KGM5352113, KGM2112133).
Acknowledgments
Authors thank all lab members for thoughtful comments and discussion on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chi, H.; Sang, T.-K.; Chang, H.-Y. Tauopathy. In Cognitive Disorders; IntechOpen: London, UK, 2018. [Google Scholar]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu Rev. Pathol 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Brion, J.P.; Couck, A.; Passareiro, E.; Flament-Durand, J. Neurofibrillary tangles of Alzheimer’s disease: AN immunohistochemical study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Wischik, C.; Crowther, R.; Walker, J.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Novak, M.; Edwards, P.; Klug, A.; Tichelaar, W.; Crowther, R. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Novak, M.; Thøgersen, H.; Edwards, P.; Runswick, M.; Jakes, R.; Walker, J.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Colin, M.; Buée, L. Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol. Appl. Neurobiol. 2015, 41, 59–80. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.; Potier, M.; Ulrich, J.; Crowther, R. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- D’Souza, I.; Schellenberg, G.D. Regulation of tau isoform expression and dementia. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Goda, Y. The actin cytoskeleton: Integrating form and function at the synapse. Annu. Rev. Neurosci. 2005, 28, 25–55. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.-C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431.e27–431.e38. [Google Scholar] [CrossRef]
- Oliveira, J.; Costa, M.; de Almeida, M.S.C.; da Cruz e Silva, O.A.; Henriques, A.G. Protein phosphorylation is a key mechanism in Alzheimer’s disease. J. Alzheimer Dis. 2017, 58, 953–978. [Google Scholar] [CrossRef]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the τ in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.P.; Peng, C.X.; Wei, W.; Tian, Q.; Liu, Y.H.; Yao, X.Q.; Zhang, Y.; Cao, F.Y.; Wang, Q.; Wang, J.Z. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 2010, 20, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-H.; Chang, J.-R.; Zhang, J.; Zhang, B.-H.; Li, Y.-L.; Teng, X.; Zhu, Y.; Du, J.; Tang, C.-S.; Qi, Y.-F. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis 2013, 18, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.d.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Götz, J. Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.-H.; Zhang, Y.; Hong, X.-Y.; Zhang, J.-F.; Wang, J.-Z.; Liu, G.-P. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J. Huazhong Univ. Sci. Technol. [Med. Sci.] 2017, 37, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rosseels, J.; Van den Brande, J.; Violet, M.; Jacobs, D.; Grognet, P.; Lopez, J.; Huvent, I.; Caldara, M.; Swinnen, E.; Papegaey, A. Tau monoclonal antibody generation based on humanized yeast models: Impact on Tau oligomerization and diagnostics. J. Biol. Chem. 2015, 290, 4059–4074. [Google Scholar] [CrossRef]
- Vega, I.E.; Cui, L.; Propst, J.A.; Hutton, M.L.; Lee, G.; Yen, S.-H. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Mol. Brain Res. 2005, 138, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef]
- Sontag, E.; Hladik, C.; Montgomery, L.; Luangpirom, A.; Mudrak, I.; Ogris, E.; White, C.L., III. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. I PP2A 1 affects Tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J. Biol. Chem. 2008, 283, 10513–10521. [Google Scholar] [CrossRef]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.-M.; Mandelkow, E. Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Khlistunova, I.; Biernat, J.; Wang, Y.; Pickhardt, M.; von Bergen, M.; Gazova, Z.; Mandelkow, E.; Mandelkow, E.-M. Inducible expression of Tau repeat domain in cell models of tauopathy: Aggregation is toxic to cells but can be reversed by inhibitor drugs. J. Biol. Chem. 2006, 281, 1205–1214. [Google Scholar] [CrossRef]
- Wille, H.; Drewes, G.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992, 118, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.; Hasegawa, M.; Smith, M.; Crowther, R. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Fichou, Y.; Oberholtzer, Z.R.; Ngo, H.; Cheng, C.-Y.; Keller, T.J.; Eschmann, N.A.; Han, S. Tau-cofactor complexes as building blocks of tau fibrils. Front. Neurosci. 2019, 13, 1339. [Google Scholar] [CrossRef]
- Giustiniani, J.; Chambraud, B.; Sardin, E.; Dounane, O.; Guillemeau, K.; Nakatani, H.; Paquet, D.; Kamah, A.; Landrieu, I.; Lippens, G. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Criado-Marrero, M.; Gebru, N.T.; Gould, L.A.; Blazier, D.M.; Vidal-Aguiar, Y.; Smith, T.M.; Abdelmaboud, S.S.; Shelton, L.B.; Wang, X.; Dahrendorff, J. FKBP52 overexpression accelerates hippocampal-dependent memory impairments in a tau transgenic mouse model. NPJ Aging Mech. Dis. 2021, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Braak, F.; Braak, H.; Mandelkow, E.-M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.d.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Biernat, J.; Von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.-M.; Müller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef]
- de Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.-S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.-Z. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [PubMed]
- Fatouros, C.; Pir, G.J.; Biernat, J.; Koushika, S.P.; Mandelkow, E.; Mandelkow, E.-M.; Schmidt, E.; Baumeister, R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 2012, 21, 3587–3603. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Morsch, R.; Simon, W.; Coleman, P.D. Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; De Calignon, A.; Matsui, T.; Zehr, C.; Pitstick, R.; Wu, H.-Y.; Osetek, J.D.; Jones, P.B.; Bacskai, B.J.; Feany, M.B. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J. Neurosci. 2008, 28, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Hochgräfe, K.; Ahmed, T.; Decker, J.M.; Sydow, A.; Hofmann, A.; Wu, D.; Messing, L.; Balschun, D.; D’Hooge, R. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef]
- Flach, K.; Hilbrich, I.; Schiffmann, A.; Gärtner, U.; Krüger, M.; Leonhardt, M.; Waschipky, H.; Wick, L.; Arendt, T.; Holzer, M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012, 287, 43223–43233. [Google Scholar] [CrossRef]
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013, 2013, 260787. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Murillo, M.C.; Sengupta, U.; Barrett, A.; Kayed, R. Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. USA 2006, 103, 8864–8869. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Petrucelli, L.; Dawson, T. Mechanism of neurodegenerative disease: Role of the ubiquitin proteasome system. Ann. Med. 2004, 36, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef] [PubMed]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Botzen, D.; Engels, M.; Voss, P.; Kaiser, B.; Jung, T.; Grimm, S.; Ermak, G.; Davies, K.J. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 2010, 500, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Bednarski, E.; Lynch, G. Cytosolic proteolysis of τ by cathepsin D in hippocampus following suppression of cathepsins B and L. J. Neurochem. 1996, 67, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Elson-Schwab, I.; Fulga, T.A.; Sharp, K.A.; Loewen, C.A.; Mulkearns, E.; Tyynela, J.; Scherzer, C.R.; Feany, M.B. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010, 6, e1001026. [Google Scholar] [CrossRef] [PubMed]
- Bendiske, J.; Bahr, B.A. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis—an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 2003, 62, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- St Johnston, D. The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 2002, 3, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Tickoo, S.; Russell, S. Drosophila melanogaster as a model system for drug discovery and pathway screening. Curr. Opin. Pharmacol. 2002, 2, 555–560. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef]
- Gistelinck, M.; Lambert, J.C.; Callaerts, P.; Dermaut, B.; Dourlen, P. Drosophila models of tauopathies: What have we learned? Int. J. Alzheimer Dis. 2012, 2012, 970980. [Google Scholar] [CrossRef]
- Sivanantharajah, L.; Mudher, A.; Shepherd, D. An evaluation of Drosophila as a model system for studying tauopathies such as Alzheimer’s disease. J. Neurosci. Methods 2019, 319, 77–88. [Google Scholar] [CrossRef]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 3291. [Google Scholar] [CrossRef]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Traven, A.; Jelicic, B.; Sopta, M. Yeast Gal4: A transcriptional paradigm revisited. EMBO Rep. 2006, 7, 496–499. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Karsten, S.L.; Sang, T.K.; Gehman, L.T.; Chatterjee, S.; Liu, J.; Lawless, G.M.; Sengupta, S.; Berry, R.W.; Pomakian, J.; Oh, H.S.; et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006, 51, 549–560. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sang, T.K.; Lawless, G.M.; Jackson, G.R. Dissociation of tau toxicity and phosphorylation: Role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 2009, 18, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, S.; Grammenoudi, S.; Papanikolopoulou, K.; Skoulakis, E.M. Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 2010, 30, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, I.; Yang, Y.; Lu, B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 2004, 116, 671–682. [Google Scholar] [CrossRef]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 2007, 18, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Iijima-Ando, K.; Zhao, L.; Gatt, A.; Shenton, C.; Iijima, K. A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Hum. Mol. Genet. 2010, 19, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Talmat-Amar, Y.; Arribat, Y.; Redt-Clouet, C.; Feuillette, S.; Bouge, A.L.; Lecourtois, M.; Parmentier, M.L. Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum. Mol. Genet. 2011, 20, 3738–3745. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Lu, Y.; Steinhilb, M.L.; Oldham, S.; Shulman, J.M.; Feany, M.B. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 2006, 16, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, J.B.; DeVos, S.L.; McGrath, J.P.; Shepard, A.M.; Goncharoff, D.K.; Tait, D.N.; Fleming, S.R.; Vincent, M.P.; Steinhilb, M.L. Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE 2011, 6, e23865. [Google Scholar] [CrossRef] [PubMed]
- Beharry, C.; Alaniz, M.E.; Alonso Adel, C. Expression of Alzheimer-like pathological human tau induces a behavioral motor and olfactory learning deficit in Drosophila melanogaster. J. Alzheimer Dis. 2013, 37, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Tyrer, M.; Shepherd, D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J. Comp. Neurol 2000, 428, 630–640. [Google Scholar] [CrossRef]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, H.; Benton, R.; Shulman, J.M.; St Johnston, D. The role of PAR-1 in regulating the polarised microtubule cytoskeleton in the Drosophila follicular epithelium. Development 2003, 130, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [CrossRef]
- Dourlen, P. Identification of Tau Toxicity Modifiers in the Drosophila Eye. Methods Mol. Biol. 2017, 1523, 375–389. [Google Scholar] [CrossRef]
- Bolkan, B.J.; Kretzschmar, D. Loss of Tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 2014, 74, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Abeta42-Induced Tau Toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef]
- Dermaut, B.; Norga, K.K.; Kania, A.; Verstreken, P.; Pan, H.; Zhou, Y.; Callaerts, P.; Bellen, H.J. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 2005, 170, 127–139. [Google Scholar] [CrossRef]
- Quintas-Neves, M.; Teylan, M.A.; Besser, L.; Soares-Fernandes, J.; Mock, C.N.; Kukull, W.A.; Crary, J.F.; Oliveira, T.G. Magnetic resonance imaging brain atrophy assessment in primary age-related tauopathy (PART). Acta Neuropathol. Commun. 2019, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, M.; Borst, A.; Wagner, S.; Byers, D. Drosophila mushroom body mutants are deficient in olfactory learning. J. Neurogenet. 1985, 2, 1–30. [Google Scholar] [CrossRef] [PubMed]
- de Belle, J.S.; Heisenberg, M. Associative odor learning in Drosophila abolished by chemical ablation of mushroom bodies. Science 1994, 263, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, J.; Grady, L.; Kitamoto, T.; Tully, T. Disruption of neurotransmission in Drosophila mushroom body blocks retrieval but not acquisition of memory. Nature 2001, 411, 476–480. [Google Scholar] [CrossRef]
- McGuire, S.E.; Le, P.T.; Davis, R.L. The role of Drosophila mushroom body signaling in olfactory memory. Science 2001, 293, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Davis, R.L. Molecular biology and anatomy of Drosophila olfactory associative learning. Bioessays 2001, 23, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Ruan, K.; Zhai, R.G. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum. Mol. Genet. 2012, 21, 237–250. [Google Scholar] [CrossRef]
- Kim, M.; Subramanian, M.; Cho, Y.H.; Kim, G.H.; Lee, E.; Park, J.J. Short-term exposure to dim light at night disrupts rhythmic behaviors and causes neurodegeneration in fly models of tauopathy and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 495, 1722–1729. [Google Scholar] [CrossRef]
- Keshishian, H.; Broadie, K.; Chiba, A.; Bate, M. The drosophila neuromuscular junction: A model system for studying synaptic development and function. Annu. Rev. Neurosci. 1996, 19, 545–575. [Google Scholar] [CrossRef]
- Menon, K.P.; Carrillo, R.A.; Zinn, K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 647–670. [Google Scholar] [CrossRef]
- Chee, F.C.; Mudher, A.; Cuttle, M.F.; Newman, T.A.; MacKay, D.; Lovestone, S.; Shepherd, D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005, 20, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.D.; Satyasi, V.; Killen, M.; Paddock, B.E.; Moir, R.D.; Saunders, A.J.; Marenda, D.R. Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis. Models Mech. 2014, 7, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.I.; Suh, Y.S.; Chung, Y.J.; Yu, K.; Park, C.B. Shedding Light on Alzheimer’s beta-Amyloidosis: Photosensitized Methylene Blue Inhibits Self-Assembly of beta-Amyloid Peptides and Disintegrates Their Aggregates. Sci. Rep. 2017, 7, 7523. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sanchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Perrat, P.N.; DasGupta, S.; Wang, J.; Theurkauf, W.; Weng, Z.; Rosbash, M.; Waddell, S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science 2013, 340, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, R.; Palazzo, A.; Lorusso, P.; Viggiano, L.; Marsano, R.M. “What You Need, Baby, I Got It”: Transposable Elements as Suppliers of Cis-Operating Sequences in Drosophila. Biology 2020, 9, 25. [Google Scholar] [CrossRef]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Peng, H.; Chung, P.; Long, F.; Qu, L.; Jenett, A.; Seeds, A.M.; Myers, E.W.; Simpson, J.H. BrainAligner: 3D registration atlases of Drosophila brains. Nat. Methods 2011, 8, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M. Neuroanatomy: Connectome connects fly and mammalian brain networks. Curr. Biol. 2015, 25, R416–R418. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Li, H.; Li, Y.; Liu, L.; Liu, Q.; Lu, H.; Pan, Y.; Wu, Z.; Zhang, K.; Zhu, Y. Vision, memory, and cognition in Drosophila. Learn. Theory Behav. 2017, 483–503. [Google Scholar]
- Hindle, S.J.; Bainton, R.J. Barrier mechanisms in the Drosophila blood-brain barrier. Front. Neurosci. 2014, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Salminen, T.S.; Vale, P.F. Drosophila as a model system to investigate the effects of mitochondrial variation on innate immunity. Front. Immunol. 2020, 11, 521. [Google Scholar] [CrossRef]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, C.; Zheng, M.; Liu, S.; Shi, R. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci. 2019, 239, 117043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, C.; Du, Y.; Gaur, U.; Yang, M. Pharmacological Treatment of Alzheimer’s Disease: Insights from Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 4621. [Google Scholar] [CrossRef]
- Colodner, K.J.; Feany, M.B. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J. Neurosci. 2010, 30, 16102–16113. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; de Haro, M.; Hao, S.; Park, J.; Rousseaux, M.W.; Al-Ramahi, I.; Jafar-Nejad, P.; Vilanova-Velez, L.; See, L.; De Maio, A.; et al. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 2016, 92, 407–418. [Google Scholar] [CrossRef]
- Blard, O.; Feuillette, S.; Bou, J.; Chaumette, B.; Frebourg, T.; Campion, D.; Lecourtois, M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum. Mol. Genet. 2007, 16, 555–566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef]
- Shim, K.H.; Kim, S.H.; Hur, J.; Kim, D.H.; Demirev, A.V.; Yoon, S.Y. Small-molecule drug screening identifies drug Ro 31-8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol. Dis. 2019, 130, 104519. [Google Scholar] [CrossRef]
- Appocher, C.; Klima, R.; Feiguin, F. Functional screening in Drosophila reveals the conserved role of REEP1 in promoting stress resistance and preventing the formation of Tau aggregates. Hum. Mol. Genet. 2014, 23, 6762–6772. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Imboywa, S.; Giagtzoglou, N.; Powers, M.P.; Hu, Y.; Devenport, D.; Chipendo, P.; Chibnik, L.B.; Diamond, A.; Perrimon, N.; et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet. 2014, 23, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B. Patterning the brain of the zebrafish embryo. Annu. Rev. Neurosci. 1993, 16, 707–732. [Google Scholar] [CrossRef] [PubMed]
- Rupp, B.; Reichert, H. Neuroanatomy of the Zebrafish Brain: A Topological Atlas; Birkhauser: Basel, Switzerland, 1996. [Google Scholar]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Transposon tools and methods in zebrafish. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 234, 244–254. [Google Scholar] [CrossRef]
- Halpern, M.E.; Rhee, J.; Goll, M.G.; Akitake, C.M.; Parsons, M.; Leach, S.D. Gal4/UAS transgenic tools and their application to zebrafish. Zebrafish 2008, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Hans, S.; Kaslin, J.; Freudenreich, D.; Brand, M. Temporally-controlled site-specific recombination in zebrafish. PLoS ONE 2009, 4, e4640. [Google Scholar] [CrossRef]
- Schmid, B.; Haass, C. Genomic editing opens new avenues for zebrafish as a model for neurodegeneration. J. Neurochem. 2013, 127, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Poskanzer, K.E.; Freeman, M.R.; Monk, K.R. Live-imaging of astrocyte morphogenesis and function in zebrafish neural circuits. Nat. Neurosci. 2020, 23, 1297–1306. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Overall, C.M.; Power, C. Deciphering complex mechanisms in neurodegenerative diseases: The advent of systems biology. Trends Neurosci. 2009, 32, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Sabaliauskas, N.A.; Foutz, C.A.; Mest, J.R.; Budgeon, L.R.; Sidor, A.T.; Gershenson, J.A.; Joshi, S.B.; Cheng, K.C. High-throughput zebrafish histology. Methods 2006, 39, 246–254. [Google Scholar] [CrossRef]
- Green, J.; Collins, C.; Kyzar, E.J.; Pham, M.; Roth, A.; Gaikwad, S.; Cachat, J.; Stewart, A.M.; Landsman, S.; Grieco, F. Automated high-throughput neurophenotyping of zebrafish social behavior. J. Neurosci. Methods 2012, 210, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lee, J.G.; Kim, J.H.; Kim, S.Y.; Huh, Y.H.; Kim, H.J.; Lee, K.S.; Yu, K.; Lee, J.S. Vascular defects of DYRK1A knockouts are ameliorated by modulating calcium signaling in zebrafish. Dis. Model. Mech. 2019, 12, dmm037044. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Zon, L.I.; Langenau, D.M. Zebrafish disease models in drug discovery: From preclinical modelling to clinical trials. Nat. Rev. Drug Discov. 2021, 20, 611–628. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Martins, R.N.; Lardelli, M. Complex splicing and neural expression of duplicated tau genes in zebrafish embryos. J. Alzheimer Dis. 2009, 18, 305–317. [Google Scholar] [CrossRef]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.-M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Lee, S.E.; Wojta, K.; Ramos, E.M.; Klein, E.; Chen, J.; Boxer, A.L.; Gorno-Tempini, M.L.; Geschwind, D.H.; Schlotawa, L. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 2017, 140, 1128–1146. [Google Scholar] [CrossRef]
- Tomasiewicz, H.G.; Flaherty, D.B.; Soria, J.; Wood, J.G. Transgenic zebrafish model of neurodegeneration. J. Neurosci. Res. 2002, 70, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Garver, J.A.; Hukriede, N.A.; Burton, E.A. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007, 35, 6501–6516. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.-K.; Yuan, R.-Y.; Lien, H.-W.; Hung, C.-C.; Hwang, P.-P.; Chen, R.P.-Y.; Chang, C.-C.; Liao, Y.-F.; Huang, C.-J. Multiple signaling factors and drugs alleviate neuronal death induced by expression of human and zebrafish tau proteins in vivo. J. Biomed. Sci. 2016, 23, 25. [Google Scholar] [CrossRef]
- Cosacak, M.I.; Bhattarai, P.; Bocova, L.; Dzewas, T.; Mashkaryan, V.; Papadimitriou, C.; Brandt, K.; Hollak, H.; Antos, C.L.; Kizil, C. Human TAU P301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 2017, 7, 12959. [Google Scholar] [CrossRef]
- Alyenbaawi, H.; Kanyo, R.; Locskai, L.F.; Kamali-Jamil, R.; DuVal, M.G.; Bai, Q.; Wille, H.; Burton, E.A.; Allison, W.T. Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. eLife 2021, 10, e58744. [Google Scholar] [CrossRef] [PubMed]
- Curado, S.; Anderson, R.M.; Jungblut, B.; Mumm, J.; Schroeter, E.; Stainier, D.Y. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1025–1035. [Google Scholar] [CrossRef]
- van Ham, T.J.; Mapes, J.; Kokel, D.; Peterson, R.T. Live imaging of apoptotic cells in zebrafish. FASEB J. 2010, 24, 4336–4342. [Google Scholar] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Zelenchuk, T.A.; Brusés, J.L. In vivo labeling of zebrafish motor neurons using an mnx1 enhancer and Gal4/UAS. Genesis 2011, 49, 546–554. [Google Scholar] [CrossRef]
- Trevarrow, B.; Marks, D.L.; Kimmel, C.B. Organization of hindbrain segments in the zebrafish embryo. Neuron 1990, 4, 669–679. [Google Scholar] [CrossRef]
- Kamei, M.; Weinstein, B.M. Long-term time-lapse fluorescence imaging of developing zebrafish. Zebrafish 2005, 2, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Colwill, R.M.; Creton, R. Locomotor behaviors in zebrafish (Danio rerio) larvae. Behav. Process. 2011, 86, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Kyriakatos, A.; Mahmood, R.; Ausborn, J.; Porres, C.P.; Büschges, A.; El Manira, A. Initiation of locomotion in adult zebrafish. J. Neurosci. 2011, 31, 8422–8431. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the α-ring channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.-C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.-N. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef] [PubMed]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small molecule modulation of proteasome assembly. Biochemistry 2018, 57, 4214–4224. [Google Scholar] [CrossRef]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef]
- Imamura, S.; Yabu, T.; Yamashita, M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J. Biol. Chem. 2012, 287, 23047–23056. [Google Scholar] [CrossRef]
- Mathai, B.J.; Meijer, A.H.; Simonsen, A. Studying Autophagy in Zebrafish. Cells 2017, 6, 21. [Google Scholar] [CrossRef]
- Plucinska, G.; Paquet, D.; Hruscha, A.; Godinho, L.; Haass, C.; Schmid, B.; Misgeld, T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 2012, 32, 16203–16212. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.-M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Hassan-Abdi, R.; Brenet, A.; Bennis, M.; Yanicostas, C.; Soussi-Yanicostas, N. Neurons Expressing Pathological Tau Protein Trigger Dramatic Changes in Microglial Morphology and Dynamics. Front. Neurosci. 2019, 13, 1199. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharm. Res. 2020, 158, 104865. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Diaz, J.E.; Alavi Naini, S.M.; Huynh, M.B.; Ouidja, M.O.; Yanicostas, C.; Chantepie, S.; Villares, J.; Lamari, F.; Jospin, E.; van Kuppevelt, T.H.; et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 2015, 138, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- Moussaed, M.; Huc-Brandt, S.; Cubedo, N.; Silhol, M.; Murat, S.; Lebart, M.C.; Kovacs, G.; Verdier, J.M.; Trousse, F.; Rossel, M.; et al. Regenerating islet-derived 1alpha (REG-1alpha) protein increases tau phosphorylation in cell and animal models of tauopathies. Neurobiol. Dis. 2018, 119, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Buffet, M.; Fenwick, A.E.; Haigh, D.; Ife, R.J.; Saunders, M.; Slingsby, B.P.; Stacey, R.; Ward, R.W. 3-Anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorganic Med. Chem. Lett. 2001, 11, 635–639. [Google Scholar] [CrossRef]
- Faria-Ramos, I.; Poças, J.; Marques, C.; Santos-Antunes, J.; Macedo, G.; Reis, C.A.; Magalhães, A. Heparan Sulfate Glycosaminoglycans:(Un) Expected Allies in Cancer Clinical Management. Biomolecules 2021, 11, 136. [Google Scholar] [CrossRef]
- Lanzi, C.; Cassinelli, G. Heparan sulfate mimetics in cancer therapy: The challenge to define structural determinants and the relevance of targets for optimal activity. Molecules 2018, 23, 2915. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Yanicostas, C.; Hassan-Abdi, R.; Blondeel, S.; Bennis, M.; Weiss, R.J.; Tor, Y.; Esko, J.D.; Soussi-Yanicostas, N. Surfen and oxalyl surfen decrease tau hyperphosphorylation and mitigate neuron deficits in vivo in a zebrafish model of tauopathy. Transl. Neurodegener. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220–14230. [Google Scholar] [CrossRef]
- Wu, B.K.; Yuan, R.Y.; Chang, Y.P.; Lien, H.W.; Chen, T.S.; Chien, H.C.; Tong, T.S.; Tsai, H.P.; Fang, C.L.; Liao, Y.F.; et al. Epicatechin isolated from Tripterygium wilfordii extract reduces tau-GFP-induced neurotoxicity in zebrafish embryo through the activation of Nrf2. Biochem. Biophys. Res. Commun. 2016, 477, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.; Carrel, J.S. Fertilization and sperm competition in the nematodeCaenorhabditis elegans. Dev. Biol. 1979, 73, 304–321. [Google Scholar] [CrossRef]
- Kimble, J.; Crittenden, S.L. Germline Proliferation and Its Control. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2005. [Google Scholar] [CrossRef]
- Kobet, R.A.; Pan, X.; Zhang, B.; Pak, S.C.; Asch, A.S.; Lee, M.-H. Caenorhabditis elegans: A model system for anti-cancer drug discovery and therapeutic target identification. Biomol. Ther. 2014, 22, 371. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Hendricks, G.L.; Lee, K.; Mylonakis, E. An update on the use of C. elegans for preclinical drug discovery: Screening and identifying anti-infective drugs. Expert Opin. Drug Discov. 2017, 12, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Jarrell, T.A.; Brittin, C.A.; Wang, Y.; Bloniarz, A.E.; Yakovlev, M.A.; Nguyen, K.C.; Tang, L.T.-H.; Bayer, E.A.; Duerr, J.S. Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 2019, 571, 63–71. [Google Scholar] [CrossRef]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 1–20. [Google Scholar] [CrossRef]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. OrthoList 2: A new comparative genomic analysis of human and Caenorhabditis elegans genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef]
- The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, M.; Hobert, O.; Miller, D.M., 3rd; Sestan, N. The CeNGEN project: The complete gene expression map of an entire nervous system. Neuron 2018, 99, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.A.; Knight, A.L.; Caldwell, G.A.; Caldwell, K.A. Generation of stable transgenic C. elegans using microinjection. J. Vis. Exp. 2008. [Google Scholar] [CrossRef] [PubMed]
- Schweinsberg, P.J.; Grant, B.D. C. elegans gene transformation by microparticle bombardment. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2018. [Google Scholar] [CrossRef]
- McDermott, J.B.; Aamodt, S.; Aamodt, E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the τ microtubule-associated proteins. Biochemistry 1996, 35, 9415–9423. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.-M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 1–21. [Google Scholar] [CrossRef]
- Morelli, F.; Romeo, M.; Barzago, M.M.; Bolis, M.; Mattioni, D.; Rossi, G.; Tagliavini, F.; Bastone, A.; Salmona, M.; Diomede, L. V363I and V363A mutated tau affect aggregation and neuronal dysfunction differently in C. elegans. Neurobiol. Dis. 2018, 117, 226–234. [Google Scholar] [CrossRef]
- Guha, S.; Fischer, S.; Johnson, G.V.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Park, H.-E.H.; Jung, Y.; Lee, S.-J.V. Survival assays using Caenorhabditis elegans. Mol. Cells 2017, 40, 90. [Google Scholar] [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. C. Elegans 1999, 2, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Amrit, F.R.G.; Ratnappan, R.; Keith, S.A.; Ghazi, A. The C. elegans lifespan assay toolkit. Methods 2014, 68, 465–475. [Google Scholar] [CrossRef]
- Lettre, G.; Hengartner, M.O. Developmental apoptosis in C. elegans: A complex CEDnario. Nat. Rev. Mol. Cell Biol. 2006, 7, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Bargmann, C.I. Chemosensation in C. elegans. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2006. [Google Scholar] [CrossRef]
- Laughlin, S.T.; Bertozzi, C.R. In vivo imaging of Caenorhabditis elegans glycans. ACS Chem. Biol. 2009, 4, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Kerk, S.Y.; Kratsios, P.; Hart, M.; Mourao, R.; Hobert, O. Diversification of C. elegans motor neuron identity via selective effector gene repression. Neuron 2017, 93, 80–98. [Google Scholar] [CrossRef]
- Van Krugten, J.; TARIS, K.K.; Peterman, E.J. Imaging adult C. elegans live using light-sheet microscopy. J. Microsc. 2021, 281, 214–223. [Google Scholar] [CrossRef]
- Atakan, H.B.; Alkanat, T.; Cornaglia, M.; Trouillon, R.; Gijs, M.A. Automated phenotyping of Caenorhabditis elegans embryos with a high-throughput-screening microfluidic platform. Microsyst. Nanoeng. 2020, 6, 24. [Google Scholar] [CrossRef]
- Nawa, M.; Matsuoka, M. The method of the body bending assay using Caenorhabditis elegans. Bio-Protocol 2012, 2, e253. [Google Scholar] [CrossRef]
- Buckingham, S.D.; Sattelle, D.B. Strategies for automated analysis of C. elegans locomotion. Invertebr. Neurosci. 2008, 8, 121. [Google Scholar] [CrossRef]
- Zhen, M.; Samuel, A.D. C. elegans locomotion: Small circuits, complex functions. Curr. Opin. Neurobiol. 2015, 33, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Celniker, S.E.; Dillon, L.A.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M. Unlocking the secrets of the genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Timmons, L.; Tabara, H.; Mello, C.C.; Fire, A.Z. Inducible systemic RNA silencing in Caenorhabditis elegans. Mol. Biol. Cell 2003, 14, 2972–2983. [Google Scholar] [CrossRef]
- Asikainen, S.; Vartiainen, S.; Lakso, M.; Nass, R.; Wong, G. Selective sensitivity of Caenorhabditis elegans neurons to RNA interference. Neuroreport 2005, 16, 1995–1999. [Google Scholar] [CrossRef]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A transparent window into biology: A primer on Caenorhabditis elegans. Genetics 2015, 200, 387–407. [Google Scholar] [CrossRef]
- Katz, M.; Shaham, S. Learning and Memory: Mind over Matter in C. elegans. Curr. Biol. 2019, 29, R365–R367. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.H. But can they learn? My accidental discovery of learning and memory in C. elegans. J. Neurogenet. 2020, 34, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Schlipalius, D.I.; Valmas, N.; Tuck, A.G.; Jagadeesan, R.; Ma, L.; Kaur, R.; Goldinger, A.; Anderson, C.; Kuang, J.; Zuryn, S. A core metabolic enzyme mediates resistance to phosphine gas. Science 2012, 338, 807–810. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 1–10. [Google Scholar] [CrossRef]
- Ahmad, W. Dihydrolipoamide dehydrogenase suppression induces human tau phosphorylation by increasing whole body glucose levels in a C. elegans model of Alzheimer’s Disease. Exp. Brain Res. 2018, 236, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Dickson, D.; Robinson, C.; Ross, O.; Dächsel, J.; Lincoln, S.; Cobb, S.; Rajput, M.; Farrer, M. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Sultana, R.; Ferree, A.; Smith, K.; Barone, E.; Perluigi, M.; Coccia, R.; Pierce, W.; Cai, J.; Mancuso, C.; et al. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: Relevance to Parkinson disease. Antioxid Redox Signal. 2012, 17, 1490–1506. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Mandelkow, E.; Mandelkow, E.M.; Pir, G.J. Glutamatergic nervous system degeneration in a C. elegans Tau(A152T) tauopathy model involves pathways of excitotoxicity and Ca(2+) dysregulation. Neurobiol. Dis. 2018, 117, 189–202. [Google Scholar] [CrossRef]
- Salehi, B.; Stojanović-Radić, Z.; Matejić, J.; Sharifi-Rad, M.; Kumar, N.V.A.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Pandav, R.; Dodge, H.; Johnston, J.; Belle, S.; DeKosky, S.; Ganguli, M. Incidence of Alzheimer’s disease in a rural community in India: The Indo–US study. Neurology 2001, 57, 985–989. [Google Scholar] [CrossRef]
- Miyasaka, T.; Xie, C.; Yoshimura, S.; Shinzaki, Y.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Curcumin improves tau-induced neuronal dysfunction of nematodes. Neurobiol. Aging 2016, 39, 69–81. [Google Scholar] [CrossRef]
- Miyasaka, T.; Shinzaki, Y.; Yoshimura, S.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Imbalanced Expression of Tau and Tubulin Induces Neuronal Dysfunction in C. elegans Models of Tauopathy. Front. Neurosci. 2018, 12, 415. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C.R.; Greenup, L.; Leverenz, J.B.; Kraemer, B.C. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011, 20, 1989–1999. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).