
NGS in Hereditary Ataxia: When Rare Becomes Frequent


 , , ,
, , ,  ,
,  , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment
4.2. DNA Extraction and Preliminary Analyses of Repeated Nucleotide Expansions
4.3. Massive Parallel Sequencing and Data Analysis
4.4. Sanger Sequencing
4.5. Multiplex Ligation-Dependent Probe Amplification (MLPA) Analysis
4.6. Computational Analysis of Protein Stability
4.7. Literature Revision
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synofzik, M.; Schüle, R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef]
- Ruano, L.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Galatolo, D.; Tessa, A.; Filla, A.; Santorelli, F.M. Clinical application of next generation sequencing in hereditary spinocerebellar ataxia: Increasing the diagnostic yield and broadening the ataxia-spasticity spectrum. A retrospective analysis. Neurogenetics 2018, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.H.; Kwasniewska, A.C.; Lise, S.; Parolin Schnekenberg, R.; Becker, E.B.E.; Bera, K.D.; Shanks, M.E.; Gregory, L.; Buck, D.; Zameel Cader, M.; et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013, 136, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- De Michele, G.; Galatolo, D.; Barghigiani, M.; Dello Iacovo, D.; Trovato, R.; Tessa, A.; Salvatore, E.; Filla, A.; De Michele, G.; Santorelli, F.M. Spinocerebellar ataxia type 48: Last but not least. Neurol. Sci. 2020, 41, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- De Michele, G.; Galatolo, D.; Lieto, M.; Maione, L.; Cocozza, S.; Santorelli, F.M.; Filla, A. New AARS2 mutations in two siblings with tremor, downbeat nystagmus, and primary amenorrhea: A benign phenotype without leukoencephalopathy. Mov. Disord. Clin. Pract. 2020, 7, 684–687. [Google Scholar] [CrossRef]
- Korenke, G.C.; Roth, C.; Krasemann, E.; Hüfner, M.; Hunneman, D.H.; Hanefeld, F. Variability of endocrinological dysfunction in 55 patients with X- linked adrenoleucodystrophy: Clinical, laboratory and genetic findings. Eur. J. Endocrinol. 1997, 137, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Tunc, S.; Dulovic-Mahlow, M.; Baumann, H.; Baaske, M.K.; Jahn, M.; Junker, J.; Münchau, A.; Brüggemann, N.; Lohmann, K. Spinocerebellar ataxia type 28—Phenotypic and molecular characterization of a family with heterozygous and compound-heterozygous mutations in AFG3L2. Cerebellum 2019, 18, 817–822. [Google Scholar] [CrossRef]
- Dosi, C.; Galatolo, D.; Rubegni, A.; Doccini, S.; Pasquariello, R.; Nesti, C.; Sicca, F.; Barghigiani, M.; Battini, R.; Tessa, A.; et al. Expanding the clinical and genetic heterogeneity of SPAX5. Ann. Clin. Transl. Neurol. 2020, 7, 595–601. [Google Scholar] [CrossRef]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Brugman, F.; Scheffer, H.; Wokke, J.H.J.; Nillesen, W.M.; De Visser, M.; Aronica, E.; Veldink, J.H.; Van Den Berg, L.H. Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology 2008, 71, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Renaud, M.; Anheim, M.; Kamsteeg, E.J.; Mallaret, M.; Mochel, F.; Vermeer, S.; Drouot, N.; Pouget, J.; Redin, C.; Salort-Campana, E.; et al. Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: Delineation and genotype-phenotype correlation study. JAMA Neurol. 2014, 71, 1305–1310. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Mancini, P.; Menchise, V.; Saccà, F.; De Michele, G.; Banfi, S.; Filla, A. Very late onset in ataxia oculomotor apraxia type I. Ann. Neurol. 2005, 57, 777. [Google Scholar] [CrossRef]
- McConville, C.M.; Stankovic, T.; Byrd, P.J.; McGuire, G.M.; Yao, Q.Y.; Lennox, G.G.; Taylor, A.M.R. Mutations associated with variant phenotypes in ataxia-telangiectasia. Am. J. Hum. Genet. 1996, 59, 320–330. [Google Scholar]
- Teraoka, S.N.; Telatar, M.; Becker-catania, S.; Liang, T.; Suna, O.; Tolun, A.; Chessa, L.; Sanal, O.; Bernatowska, E.; Gatti, R.A.; et al. Splicing defects in the ataxia-telangiectasia gene, ATM: Underlying mutations and consequences. Am. J. Hum. Genet. 1999, 64, 1617–1631. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.P.; Bendix, R.; Chen, P.; Clark, R.; Dörk, T.; Lavin, M.F. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 925–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Michele, G.; Galatolo, D.; Lieto, M.; Fico, T.; Saccà, F.; Santorelli, F.M.; Filla, A. Ataxia-myoclonus syndrome due to a novel homozygous ATP13A2 mutation. Park. Relat. Disord. 2020, 76, 42–43. [Google Scholar] [CrossRef]
- Coutelier, M.; Blesneac, I.; Monteil, A.; Monin, M.; Ando, K.; Mundwiller, E.; Brusco, A.; Le Ber, I.; Anheim, M.; Castrioto, A.; et al. A recurrent mutation in CACNA1G alters Cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar ataxia. Am. J. Hum. Genet. 2015, 97, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Mero, S.; Salviati, L.; Leuzzi, V.; Rubegni, A.; Calderan, C.; Nardecchia, F.; Galatolo, D.; Desbats, M.A.; Naef, V.; Gemignani, F.; et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J. Neurol. 2021, 1–9. [Google Scholar] [CrossRef]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Burgi, S.; et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Mignot, C.; Apartis, E.; Durr, A.; Marques Lourenco, C.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerards, M.; van den Bosch, B.; Calis, C.; Schoonderwoerd, K.; van Engelen, K.; Tijssen, M.; de Coo, R.; van der Kooi, A.; Smeets, H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Coarelli, G.; Monin, M.; Konop, J.; Davoine, C.; Tesson, C.; Valter, R.; Anheim, M.; Behin, A.; Castelnovo, G.; et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 2017, 140, 1579–1594. [Google Scholar] [CrossRef]
- Winkelmann, J.; Lin, L.; Schormair, B.; Kornum, B.R.; Faraco, J.; Plazzi, G.; Melberg, A.; Cornelio, F.; Urban, A.E.; Pizza, F.; et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 2012, 21, 2205–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Yourshaw, M.; Mamsa, H.; Rudnik-schöneborn, S.; Menezes, M.P.; Hong, J.E.; Leong, D.W.; Senderek, J.; Salman, M.S.; Chitayat, D.; et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 2012, 44, 704–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatolo, D.; Kuo, M.E.; Mullen, P.; Meyer-Schuman, R.; Doccini, S.; Battini, R.; Lieto, M.; Tessa, A.; Filla, A.; Francklyn, C.; et al. Bi-allelic mutations in HARS1 severely impair histidyl-tRNA synthetase expression and enzymatic activity causing a novel multisystem ataxic syndrome. Hum. Mutat. 2020, 41, 1232–1237. [Google Scholar] [CrossRef]
- Möller, G.; Van Grunsven, E.G.; Wanders, R.J.A.; Adamski, J. Molecular basis of D-bifunctional protein deficiency. Mol. Cell. Endocrinol. 2001, 171, 61–70. [Google Scholar] [CrossRef]
- Barresi, S.; Niceta, M.; Alfieri, P.; Brankovich, V.; Piccini, G.; Bruselles, A.; Barone, M.; Cusmai, R.; Tartaglia, M.; Bertini, E.; et al. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin. Genet. 2017, 91, 86–91. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.; Coimbra, R. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: Proposal for a new channelopathy. Clin. Genet. 2015, 87, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.L.; Hedrich, U.B.S.; Shinde, D.N.; Krey, I.; Teichmann, A.; Hentschel, J.; Schubert, J.; Chamberlin, A.C.; Huether, R.; Lu, H.; et al. A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann. Neurol. 2016, 80, 638–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, K.P.; Minassian, N.A.; Stevanin, G.; Waters, M.; Garibyan, V.; Forlani, S.; Strzelczyk, A.; Bürk, K.; Brice, A.; Dürr, A.; et al. KCNC3: Phenotype, mutations, channel biophysics—A study of 260 familial ataxia patients. Hum. Mutat. 2010, 31, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasdemir, C.; Juang, J.; Kose, S.; Kocabas, U.; Orman, M.; Payzin, S.; Sahin, H.; Celen, C.; Ozcan, E.; Chen, C.Y.J.C.; et al. Co-existence of Atrioventricular Accessory Pathways and Drug-Induced Type 1 Brugada Pattern. Physiol. Behav. 2018, 41, 1078–1092. [Google Scholar] [CrossRef]
- Lee, Y.; Durr, A.; Majczenko, K.; Huang, Y.; Liu, Y.; Lien, C.; Tsai, P.; Ichikawa, Y.; Goto, J.; Monin, M.-L.; et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Nemani, T.; Steel, D.; Kaliakatsos, M.; DeVile, C.; Ververi, A.; Scott, R.; Getov, S.; Sudhakar, S.; Male, A.; Mankad, K.; et al. KIF1A-related disorders in children: A wide spectrum of central and peripheral nervous system involvement. J. Peripher. Nerv. Syst. 2020, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, A.; Poretti, A.; Romani, M.; Ginevrino, M.; Mazza, T.; Aiello, C.; Zanni, G.; Baumgartner, B.; Borgatti, R.; Brockmann, K.; et al. Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti-Boltshauser syndrome). Eur. J. Hum. Genet. 2016, 24, 1262–1267. [Google Scholar] [CrossRef]
- Di Meglio, C.; Bonello-Palot, N.; Boulay, C.; Milh, M.; Ovaert, C.; Levy, N.; Chabrol, B. Clinical and allelic heterogeneity in a pediatric cohort of 11 patients carrying MFN2 mutation. Brain Dev. 2016, 38, 498–506. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Doré, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef]
- Nair, P.; Sabbagh, S.; Mansour, H.; Fawaz, A.; Hmaimess, G.; Noun, P.; Dagher, R.; Megarbane, H.; Hana, S.; Alame, S.; et al. Contribution of next generation sequencing in pediatric practice in Lebanon. A Study on 213 cases. Mol. Genet. Genomic Med. 2018, 6, 1041–1052. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jiang, Y.; Gao, Z.; Wang, J.; Yuan, Y.; Xiong, H.; Chang, X.; Bao, X.; Zhang, Y.; Xiao, J.; et al. Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur. J. Neurol. 2009, 16, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Schollen, E.; Pardon, E.; Veiga-Da-Cunha, M.; Jaeken, J.; Cassiman, J.; Van Schaftingen, E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat. Genet. 1997, 16, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Lomniczi, A.; Kretzschmar, D.; Dissen, G.A.; Kotan, L.D.; McArdle, C.A.; Koc, A.F.; Hamel, B.C.; Guclu, M.; Papatya, E.D.; et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J. Clin. Endocrinol. Metab. 2014, 99, e2067–e2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufnagel, R.B.; Arno, G.; Hein, N.D.; Hersheson, J.; Prasad, M.; Anderson, Y.; Krueger, L.A.; Gregory, L.C.; Stoetzel, C.; Jaworek, T.J.; et al. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J. Med. Genet. 2015, 52, 85–94. [Google Scholar] [CrossRef]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Lima-Martinez, M.M.; et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014, 137, 69–77. [Google Scholar] [CrossRef]
- Teive, H.A.G.; Camargo, C.H.F.; Sato, M.T.; Shiokawa, N.; Boguszewski, C.L.; Raskin, S.; Buck, C.; Seminara, S.B.; Munhoz, R.P. Different cerebellar ataxia phenotypes associated with mutations of the PNPLA6 gene in Brazilian patients with recessive ataxias. Cerebellum 2018, 17, 380–385. [Google Scholar] [CrossRef]
- Lamantea, E.; Tiranti, V.; Bordoni, A.; Toscano, A.; Bono, F.; Servidei, S.; Papadimitriou, A.; Spelbrink, H.; Silvestri, L.; Casari, G.; et al. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann. Neurol. 2002, 52, 211–219. [Google Scholar] [CrossRef]
- Van Goethem, G.; Schwartz, M.; Löfgren, A.; Dermaut, B.; Van Broeckhoven, C.; Vissing, J. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur. J. Hum. Genet. 2003, 11, 547–549. [Google Scholar] [CrossRef]
- Van Goethem, G.; Luoma, P.; Rantamäki, M.; Al Memar, A.; Kaakkola, S.; Hackman, P.; Krahe, R.; Löfgren, A.; Martin, J.J.; De Jonghe, P.; et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004, 63, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Minnerop, M.; Kurzwelly, D.; Wagner, H.; Soehn, A.S.; Reichbauer, J.; Tao, F.; Rattay, T.W.; Peitz, M.; Rehbach, K.; Giorgetti, A.; et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 2017, 140, 1561–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Michele, G.; Galatolo, D.; Galosi, S.; Mignarri, A.; Silvestri, G.; Casali, C.; Leuzzi, V.; Ricca, I.; Barghigiani, M.; Tessa, A.; et al. Episodic ataxia and severe infantile phenotype in spinocerebellar ataxia type 14: Expansion of the phenotype and novel mutations. J. Neurol. 2021, 1–9. [Google Scholar] [CrossRef]
- Vlak, M.H.M.; Sinke, R.J.; Rabelink, G.M.; Kremer, B.P.H.; van de Warrenburg, B.P.C. Novel PRKCG/SCA14 mutation in a Dutch spinocerebellar ataxia family: Expanding the phenotype. Mov. Disord. 2006, 21, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Riso, V.; Rossi, S.; Perna, A.; Nicoletti, T.; Bosco, L.; Zanni, G.; Silvestri, G. NGS-based detection of a novel mutation in PRKCG (SCA14) in sporadic adult-onset ataxia plus dystonic tremor. Neurol. Sci. 2020, 41, 2989–2991. [Google Scholar] [CrossRef]
- Lieto, M.; Galatolo, D.; Roca, A.; Cocozza, S.; Pontillo, G.; Fico, T.; Pane, C.; Saccà, F.; De Michele, G.; Santorelli, F.M.; et al. Overt hypogonadism may not be a sentinel sign of RING finger protein 216: Two novel mutations associated with ataxia, chorea, and fertility. Mov. Disord. Clin. Pract. 2019, 6, 724–726. [Google Scholar] [CrossRef]
- Leen, W.G.; Mewasingh, L.; Verbeek, M.M.; Kamsteeg, E.J.; van de Warrenburg, B.P.; Willemsen, M.A. Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov. Disord. 2013, 28, 1439–1442. [Google Scholar] [CrossRef]
- Martinuzzi, A.; Montanaro, D.; Vavla, M.; Paparella, G.; Bonanni, P.; Musumeci, O.; Brighina, E.; Hlavata, H.; Rossi, G.; Aghakhanyan, G.; et al. Clinical and paraclinical indicators of motor system impairment in hereditary spastic paraplegia: A pilot study. PLoS ONE 2016, 11, e0153283. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.L.; Davoine, C.S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Jacob, F.; Ho, E.S.; Martinez-ojeda, M.; Darras, B.T.; Khwaja, O.S. Case of infantile onset spinocerebellar ataxia type 5. J. Child Neurol. 2013, 28, 1292–1295. [Google Scholar] [CrossRef]
- De Michele, G.; Lieto, M.; Galatolo, D.; Salvatore, E.; Cocozza, S.; Barghigiani, M.; Tessa, A.; Baldacci, J.; Pappatà, S.; Filla, A.; et al. Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: A report of two Italian families. Park. Relat. Disord. 2019, 65, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Depondt, C.; Donatello, S.; Simonis, N.; Rai, M.; van Heurck, R.; Abramowicz, M.; D’Hooghe, M.; Pandolfo, M. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology 2014, 82, 1749–1750. [Google Scholar] [CrossRef]
- Lieto, M.; Riso, V.; Galatolo, D.; De Michele, G.; Rossi, S.; Barghigiani, M.; Cocozza, S.; Pontillo, G.; Trovato, R.; Saccà, F.; et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur. J. Neurol. 2020, 27, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Genis, D.; Ortega-Cubero, S.; Nicolas, H.S.; Corral, J.; Gardenyes, J.; De Jorge, L.; Lopez, E.; Campos, B.; Lorenzo, E.; Tonda, R.; et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 2018, 91, e1988–e1998. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef]
- Delplanque, J.; Devos, D.; Huin, V.; Genet, A.; Sand, O.; Moreau, C.; Goizet, C.; Charles, P.; Anheim, M.; Monin, M.L.; et al. TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain 2014, 137, 2657–2663. [Google Scholar] [CrossRef] [Green Version]
- Riso, V.; Galatolo, D.; Barghigiani, M.; Galosi, S.; Tessa, A.; Ricca, I.; Rossi, S.; Caputi, C.; Cioffi, E.; Leuzzi, V.; et al. A next generation sequencing-based analysis of a large cohort of ataxic patients refines the clinical spectrum associated with spinocerebellar ataxia 21. Eur. J. Neurol. 2021, 28, 2784–2788. [Google Scholar] [CrossRef]
- Sleat, D.E.; Gin, R.M.; Sohar, I.; Wisniewski, K.; Sklower-Brooks, S.; Pullarkat, R.K.; Palmer, D.N.; Lerner, T.J.; Boustany, R.M.; Uldall, P.; et al. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am. J. Hum. Genet. 1999, 64, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Rigoli, L.; Aloi, C.; Salina, A.; Di Bella, C.; Salzano, G.; Caruso, R.; Mazzon, E.; Maghnie, M.; Patti, G.; D’Annunzio, G.; et al. Wolfram syndrome 1 in the Italian population: Genotype–Phenotype correlations. Pediatr. Res. 2020, 87, 456–462. [Google Scholar] [CrossRef]
- Ohba, C.; Osaka, H.; Iai, M.; Yamashita, S.; Suzuki, Y.; Aida, N.; Shimozawa, N.; Takamura, A.; Doi, H.; Tomita-Katsumoto, A.; et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013, 14, 225–232. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Schwartzentruber, J.; Beaulieu, C.L.; Dyment, D.; Smith, A.; Chardon, J.W.; Yoon, G.; Rouleau, G.A.; Suchowersky, O.; Siu, V.; et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum. Mutat. 2014, 35, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.; Lee, H.; Deignan, J.; Strom, S.; Kantarci, S.; Wang, X.; Quintero-Rivera, F.; Vilain, E.; Grody, W.; Perlman, S.; et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014, 71, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Smertenko, T.; Bargiela, D.; Griffin, H.; Duff, J.; Appleton, M.; Douroudis, K.; Pfeffer, G.; Santibanez-Koref, M.; Eglon, G.; et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015, 138, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Steele, H.; Douroudis, K.; Pyle, A.; Duff, J.; Hussain, R.; Smertenko, T.; Griffin, H.; Santibanez-Koref, M.; Horvath, R.; et al. Frequency of rare recessive mutations in unexplained late onset cerebellar ataxia. J. Neurol. 2015, 262, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Mallaret, M.; Renaud, M.; Redin, C.; Drouot, N.; Muller, J.; Severac, F.; Mandel, J.L.; Hamza, W.; Benhassine, T.; Ali-Pacha, L.; et al. Validation of a clinical practice-based algorithm for the diagnosis of autosomal recessive cerebellar ataxias based on NGS identified cases. J. Neurol. 2016, 263, 1314–1322. [Google Scholar] [CrossRef]
- van de Warrenburg, B.P.; Schouten, M.I.; de Bot, S.T.; Vermeer, S.; Meijer, R.; Pennings, M.; Gilissen, C.; Willemsen, A.A.P.; Scheffer, H.; Kamsteeg, E.-J. Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur. J. Hum. Genet. 2016, 24, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Marelli, C.; Guissart, C.; Hubsch, C.; Renaud, M.; Villemin, J.P.; Larrieu, L.; Charles, P.; Ayrignac, X.; Sacconi, S.; Collignon, P.; et al. Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Hum. Mutat. 2016, 37, 1340–1353. [Google Scholar] [CrossRef]
- Kuperberg, M.; Lev, D.; Blumkin, L.; Zerem, A.; Ginsberg, M.; Linder, I.; Carmi, N.; Kivity, S.; Lerman-Sagie, T.; Leshinsky-Silver, E. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J. Child Neurol. 2016, 31, 1534–1539. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Rydning, S.L.; Wedding, I.M.; Koht, J.; Pihlstrøm, L.; Rengmark, A.H.; Henriksen, S.P.; Tallaksen, C.M.E.; Toft, M. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLoS ONE 2017, 12, e0174667. [Google Scholar] [CrossRef] [Green Version]
- Nibbeling, E.A.R.; Duarri, A.; Verschuuren-Bemelmans, C.C.; Fokkens, M.R.; Karjalainen, J.M.; Smeets, C.J.L.M.; De Boer-Bergsma, J.J.; Van Der Vries, G.; Dooijes, D.; Bampi, G.B.; et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 2017, 140, 2860–2878. [Google Scholar] [CrossRef]
- Montaut, S.; Tranchant, C.; Drouot, N.; Rudolf, G.; Guissart, C.; Tarabeux, J.; Stemmelen, T.; Velt, A.; Fourrage, C.; Nitschké, P.; et al. Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurol. 2018, 75, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.L.; Ma, Y.; Li, Q.F.; Du, Y.C.; Yang, L.; Chen, S.; Wu, Z.Y. Genetic and clinical features of Chinese patients with mitochondrial ataxia identified by targeted next-generation sequencing. CNS Neurosci. Ther. 2019, 25, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Liang, C.; Ahmad, K.E.; Gu, Y.; Siow, S.F.; Colebatch, J.G.; Whyte, S.; Ng, K.; Cremer, P.D.; Corbett, A.J.; et al. High degree of genetic heterogeneity for hereditary cerebellar ataxias in Australia. Cerebellum 2019, 18, 137–146. [Google Scholar] [CrossRef]
- Shakya, S.; Kumari, R.; Suroliya, V.; Tyagi, N.; Joshi, A.; Garg, A.; Singh, I.; Kalikavil Puthanveedu, D.; Cherian, A.; Mukerji, M.; et al. Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. Clin. Genet. 2019, 96, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2019, 21, 195–206. [Google Scholar] [CrossRef]
- Arslan, E.A.; Öncel, İ.; Ceylan, A.C.; Topçu, M.; Topaloğlu, H. Genetic and phenotypic features of patients with childhood ataxias diagnosed by next-generation sequencing gene panel. Brain Dev. 2020, 42, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, K.J.; Rexach, J.E.; Lee, H.; Petty, L.E.; Perlman, S.; Valera, J.M.; Deignan, J.L.; Mao, Y.; Aker, M.; Posey, J.E.; et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 2020, 41, 487–501. [Google Scholar] [CrossRef]
- Mutlu-Albayrak, H.; Kırat, E.; Gürbüz, G. Childhood-onset autosomal recessive ataxias: A cross-sectional study from Turkey. Neurogenetics 2020, 21, 59–66. [Google Scholar] [CrossRef]
- Gauquelin, L.; Hartley, T.; Tarnopolsky, M.; Dyment, D.A.; Brais, B.; Geraghty, M.T.; Tétreault, M.; Ahmed, S.; Rojas, S.; Choquet, K.; et al. Channelopathies are a frequent cause of genetic ataxias associated with cerebellar atrophy. Mov. Disord. Clin. Pract. 2020, 7, 940–949. [Google Scholar] [CrossRef]
- Ignatius, E.; Isohanni, P.; Pohjanpelto, M.; Lahermo, P.; Ojanen, S.; Brilhante, V.; Palin, E.; Suomalainen, A.; Lönnqvist, T.; Carroll, C.J. Genetic background of ataxia in children younger than 5 years in Finland. Neurol. Genet. 2020, 6, e444. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, A.R.; Kim, J.S.; Park, J.; Youn, J.; Ahn, J.H.; Mun, J.K.; Lee, C.; Kim, N.S.; Kim, N.K.D.; et al. Clarification of undiagnosed ataxia using whole-exome sequencing with clinical implications. Park. Relat. Disord. 2020, 80, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova-Mihaylova, P.; Hebert, J.; Moran, S.; Murphy, M.; Ward, D.; Walsh, R.A.; Murphy, S.M. Inherited cerebellar ataxias: 5-year experience of the Irish National Ataxia Clinic. Cerebellum 2021, 20, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Németh, A. Recessive ataxias. Handb. Clin. Neurol. 2018, 155, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Roux, T.; Barbier, M.; Papin, M.; Davoine, C.S.; Sayah, S.; Coarelli, G.; Charles, P.; Marelli, C.; Parodi, L.; Tranchant, C.; et al. Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: A frequent cause of predominant cognitive impairment. Genet. Med. 2020, 22, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.M.; Benkirane, M.; Calmels, N.; Marelli, C.; Ory-Magne, F.; Ewenczyk, C.; Halleb, Y.; Tison, F.; Lecocq, C.; Pische, G.; et al. Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J. Neurol. 2021, 268, 1927–1937. [Google Scholar] [CrossRef]
- Coarelli, G.; Schule, R.; van de Warrenburg, B.; De Jonghe, P.; Ewenczyk, C.; Martinuzzi, A.; Synofzik, M.; Hamer, E.; Baets, J.; Anheim, M.; et al. Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7. Neurology 2019, 92, e2679–e2690. [Google Scholar] [CrossRef]
- Mancini, C.; Giorgio, E.; Rubegni, A.; Pradotto, L.; Bagnoli, S.; Rubino, E.; Prontera, P.; Cavalieri, S.; Di Gregorio, E.; Ferrero, M.; et al. Prevalence and phenotype of the c.1529C>T SPG7 variant in adult-onset cerebellar ataxia in Italy. Eur. J. Neurol. 2019, 26, 80–86. [Google Scholar] [CrossRef]
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Traschütz, A.; van Gaalen, J.; Oosterloo, M.; Vreeburg, M.; Kamsteeg, E.J.; Deininger, N.; Rieß, O.; Reimold, M.; Haack, T.; Schöls, L.; et al. The movement disorder spectrum of SCA21 (ATX-TMEM240): 3 novel families and systematic review of the literature. Park. Relat. Disord. 2019, 62, 215–220. [Google Scholar] [CrossRef]
- Synofzik, M.; Puccio, H.M.; Mochel, F.; Schols, L. Autosomal recessive cerebellar ataxias: Paving the way toward targeted molecular therapies. Neuron 2019, 101, 560–583. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, T.; Barth, P.; Reneman, L.; Appelhof, B.; Baas, F.; Poll-The, B.T. A de novo missense mutation in the inositol 1,4,5-triphosphate receptor type 1 gene causing severe pontine and cerebellar hypoplasia: Expanding the phenotype of ITPR1-related spinocerebellar ataxias. Am. J. Med. Genet. Part A 2017, 173, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Falk, M.J.; Gai, X.; Postrel, R.; Schule, R.; Zuchner, S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum. Mutat. 2015, 36, 950–956. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Kiezun, A.; Sunyaev, S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011, 12, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riso, V.; Rossi, S.; Nicoletti, T.F.; Tessa, A.; Travaglini, L.; Zanni, G.; Aiello, C.; Perna, A.; Barghigiani, M.; Pomponi, M.G.; et al. Application of a clinical workflow may lead to increased diagnostic precision in hereditary spastic paraplegias and cerebellar ataxias: A single center experience. Brain Sci. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Baroni, M.G.; Oelbaum, R.S.; Pozzilli, P.; Stocks, J.; Li, S.-R.; Fiore, V.; Galton, D.J. Polymorphisms at the GLUT1 (HepG2) and GLUT4 (muscle/adipocyte) glucose transporter genes and non-insulin-dependent diabetes mellitus (NIDDM). Hum. Genet. 1992, 88, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Pontillo, G.; De Michele, G.; Perillo, T.; Guerriero, E.; Ugga, L.; Salvatore, E.; Galatolo, D.; Riso, V.; Saccà, F.; et al. The “crab sign”: An imaging feature of spinocerebellar ataxia type 48. Neuroradiology 2020, 62, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Marques Matos, C.; Alonso, I.; Leão, M. Diagnostic yield of next-generation sequencing applied to neurological disorders. J. Clin. Neurosci. 2019, 67, 14–18. [Google Scholar] [CrossRef]
- Verdura, E.; Schlüter, A.; Fernández-Eulate, G.; Ramos-Martín, R.; Zulaica, M.; Planas-Serra, L.; Ruiz, M.; Fourcade, S.; Casasnovas, C.; López de Munain, A.; et al. A deep intronic splice variant advises reexamination of presumably dominant SPG7 cases. Ann. Clin. Transl. Neurol. 2020, 7, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Magri, S.; Fracasso, V.; Plumari, M.; Alfei, E.; Ghezzi, D.; Gellera, C.; Rusmini, P.; Poletti, A.; Di Bella, D.; Elia, A.; et al. Concurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation. Hum. Mutat. 2018, 39, 2060–2071. [Google Scholar] [CrossRef]
- Bis-Brewer, D.M.; Gan-Or, Z.; Sleiman, P.; Rodriguez, A.; Bacha, A.; Kosikowski, A.; Wood, B.; McCray, B.; Blume, B.; Siskind, C.; et al. Assessing non-Mendelian inheritance in inherited axonopathies. Genet. Med. 2020, 22, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.P.; Barron, L.H.; Goudie, D.; Kelly, K.; Dow, D.; Fitzpatrick, D.R.; Brock, D.J. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J. Med. Genet. Genet. 1996, 33, 1022–1026. [Google Scholar] [CrossRef] [Green Version]
- Cagnoli, C.; Stevanin, G.; Michielotto, C.; Promis, G.G.; Brussino, A.; Pappi, P.; Durr, A.; Dragone, E.; Viemont, M.; Gellera, C.; et al. Large pathogenic expansions in the SCA2 and SCA7 genes can be detected by fluorescent repeat-primed polymerase chain reaction assay. J. Mol. Diagn. 2006, 8, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Duclos, F.; Monticelli, A.; Zara, F.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Frappier, V.; Najmanovich, R.J. A Coarse-Grained Elastic Network Atom Contact Model and Its Use in the Simulation of Protein Dynamics and the Prediction of the Effect of Mutations. PLoS Comput. Biol. 2014, 10, e1003569. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. MCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, 314–319. [Google Scholar] [CrossRef] [PubMed]
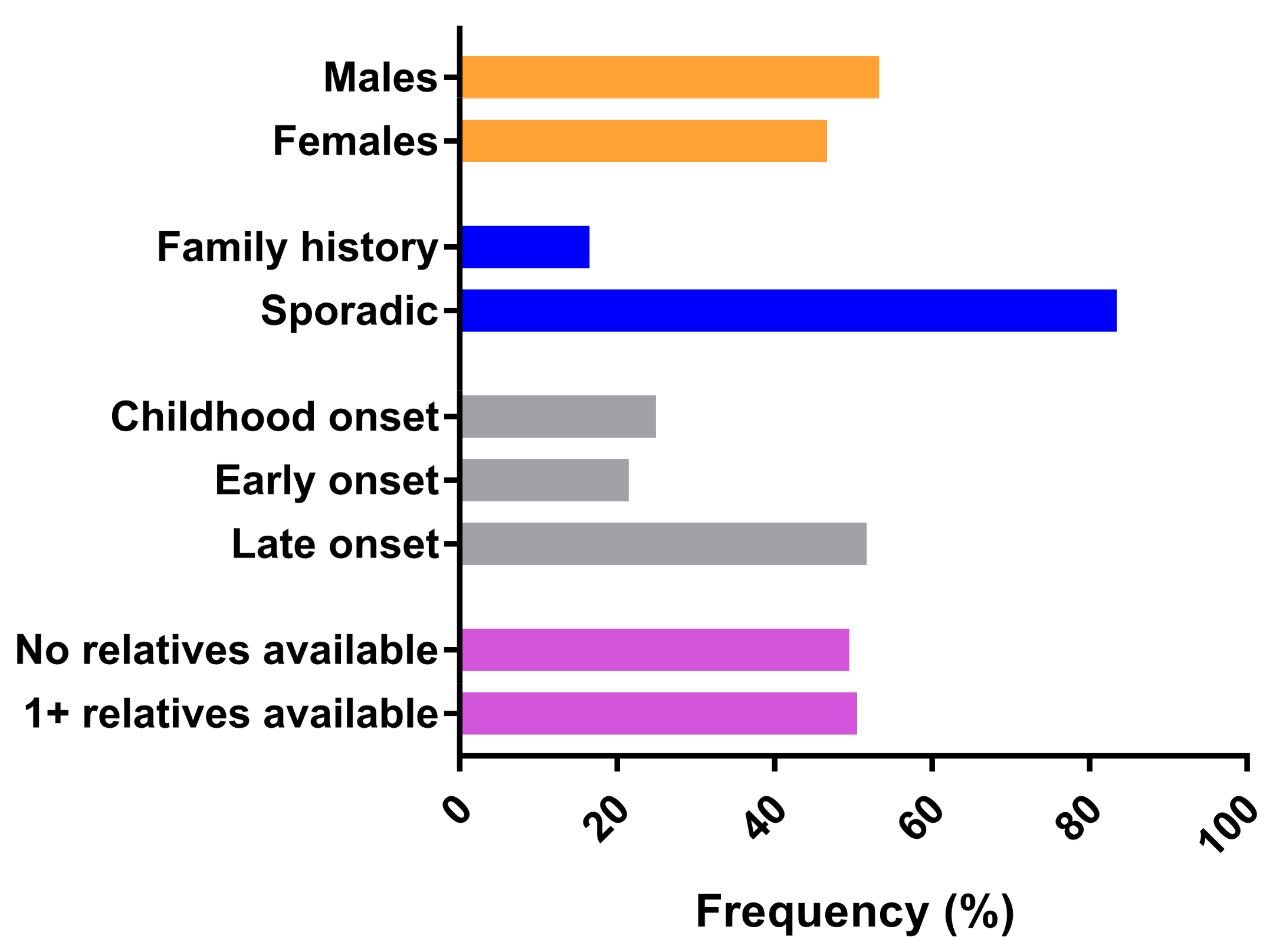
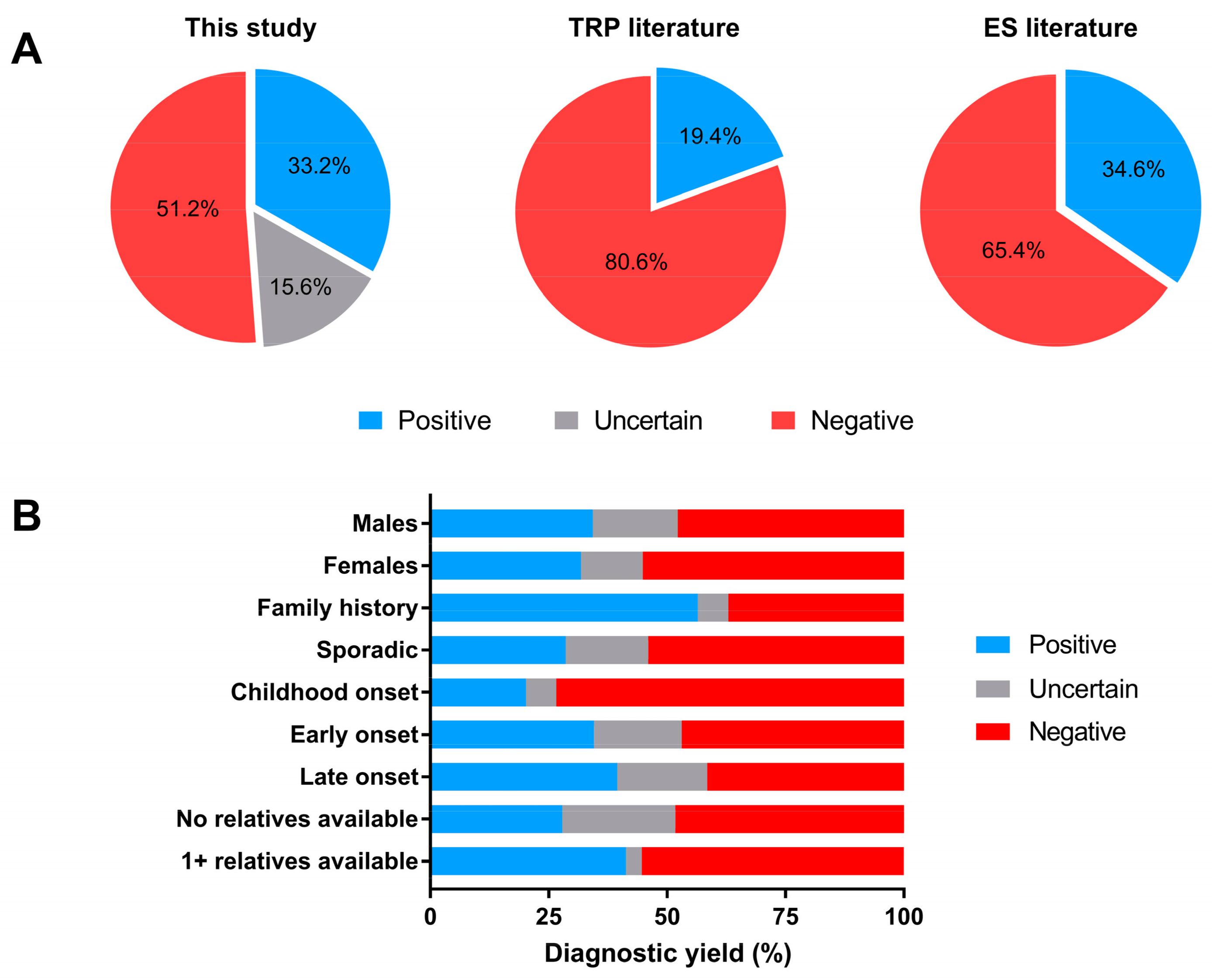
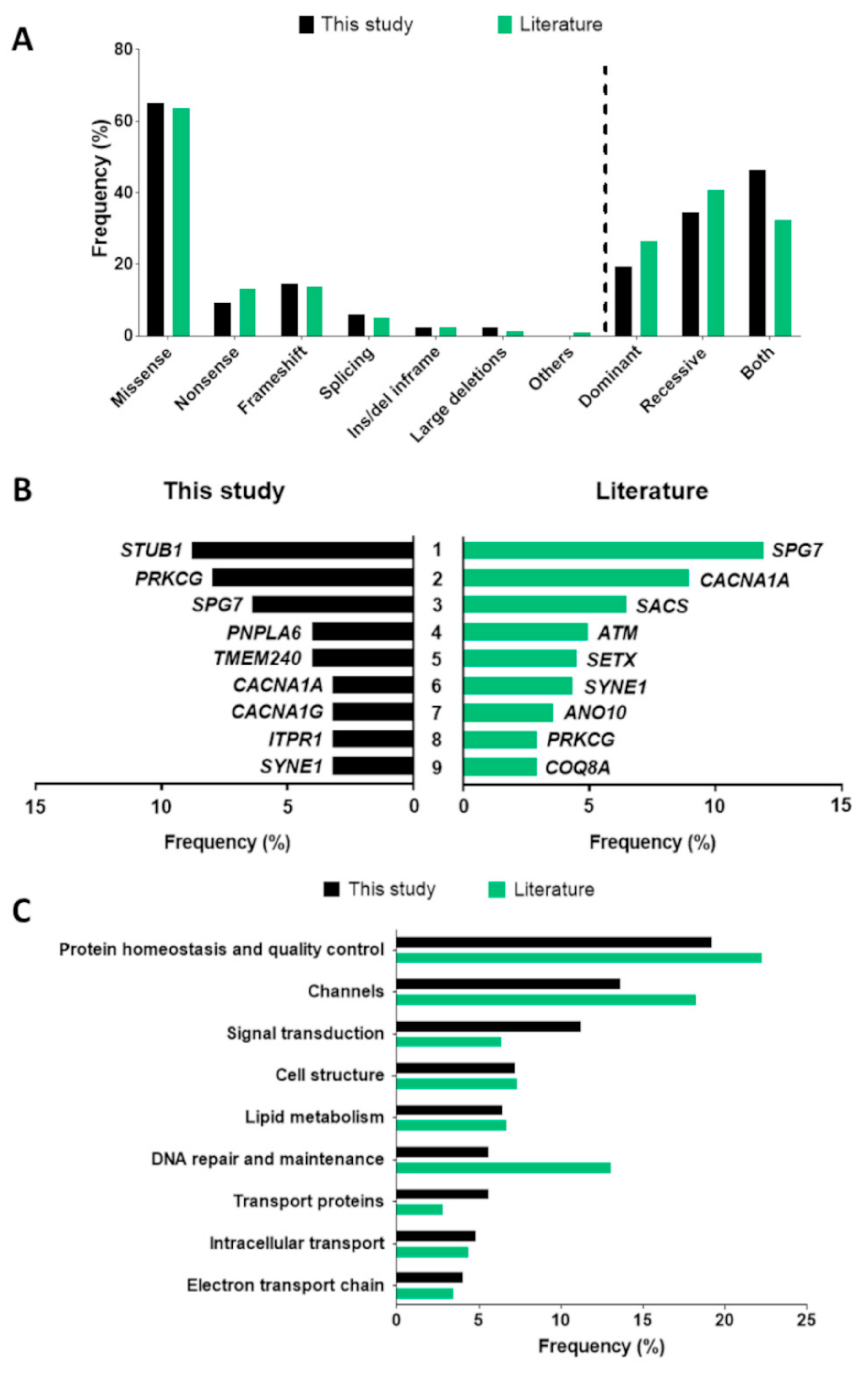

{kind=link}
{kind=link}
{kind=link}
| Index Case | Gender, Age | Transmission/Onset | Gene (Ref_Seq) | cDNA Variant | Protein Variant | Zygosity | Type | Reference |
|---|---|---|---|---|---|---|---|---|
| Pt1 | F, 36 | AR/Early | AARS2 (NM_020745.4) | c.385A > C | p.Thr129Pro | Het | Missense | [8] |
| c.446G > A | p.Cys149Tyr | Het | Missense | |||||
| Pt2 | M, 37 | SP/Early | ABCD1 (NM_000033.4) | c.1661G > A | p.Arg554His | Hem | Missense | [9] |
| Pt3 | M, 50 | SP/Late | ABCD1 (NM_000033.4) | c.2087A > T | p.Lys696Met | Hem | Missense | This study |
| Pt4 | M, 11 | SP/Childhood | AFG3L2 (NM_006796.3) | c.634dupG | p.Val212GlyfsTer4 | Het | Frameshift | [10] |
| c.2167G > A | p.Val723Met | Het | Missense | [11] | ||||
| Pt5 | F, 26 | AD/Childhood | AFG3L2 (NM_006796.3) | c.2105G > A | p.Arg702Gln | Het | Missense | [12] |
| Pt6 | M, 65 | SP/Early | AFG3L2 (NM_006796.3) | c.1712T > G | p.Val571Gly | Het | Missense | This study |
| SPG7 (NM_003119.4) | c.1529C > T | p.Ala510Val | Het | Missense | [13] | |||
| Pt7 | M, 58 | AR/Early | ANO10 (NM_018075.5) | c.289delA | p.Met97Ter | Het | Nonsense | This study |
| c.1009T > G | p.Phe337Val | Het | Missense | [14] | ||||
| Pt8 | F, 52 | SP/Late | ANO10 (NM_018075.5) | c.289delA | p.Met97Ter | Hom | Nonsense | This study |
| Pt9 | F, 43 | SP/Early | ANO10 (NM_018075.5) | c.206T > A | p.Leu69Ter | Hom | Nonsense | This study |
| Pt10 | M, 31 | SP/Early | APTX (NM_001195248.2) | c.544-1G > C | NA | Het | Splicing | This study |
| c.668T > C | p.Leu223Pro | Het | Missense | [15] | ||||
| Pt11 | M, 2 | SP/Childhood | ATM (NM_000051.3) | c.2152dupT | p.Cys718LeufsTer20 | Het | Frameshift | This study |
| c.2929T > C | p.Cys977Arg | Het | Missense | |||||
| P12 | M, 7 | SP/Childhood | ATM (NM_000051.3) | c.3802delG | p.Val1268Ter | Het | Nonsense | [16] |
| c.3894dupT | p.Ala1299CysfsTer3 | Het | Frameshift | [17] | ||||
| Pt13 | M, 50 | SP/Childhood | ATM (NM_000051.3) | c.6650_6657delTTAGTTTT | p.Phe2217SerfsTer29 | Het | Frameshift | This study |
| c.8147T > C | p.Val2716Ala | Het | Missense | [18] | ||||
| Pt14 | M, 77 | AR/Early | ATP13A2 (NM_022089.4) | c.1205C > T | p.Thr402Met | Hom | Missense | [19] |
| Pt15 | F, 12 | SP/Childhood | CACNA1A (NM_001127222.2) | c.4897G > A | p.Asp1633Asn | Het | Missense | This study |
| Pt16 | F, 43 | SP/Early | CACNA1A (NM_001127222.2) | c.3310_3315dupGGCCCC | p.Gly1104_Pro1105dup | Het | Inframe dup | This study |
| Pt17 | F, 18 | SP/Childhood | CACNA1A (NM_001127222.2) | c.4927G > A | p.Asp1643Asn | Het | Missense | This study |
| Pt18 | F, 49 | SP/Late | CACNA1A (NM_001127222.2) | c.4466T > C | p.Ile1489Thr | Het | Missense | This study |
| Pt19 | F, 62 | SP/Late | CACNA1G (NM_018896.5) | c 481A > T | p.Ile161Phe | Het | Missense | This study |
| Pt20 | F, 59 | AD/Early | CACNA1G (NM_018896.5) | c.5144G > A | p.Arg1715His | Het | Missense | [20] |
| Pt21 | M, 48 | SP/Late | CACNA1G (NM_018896.5) | c.5960_5961delCGinsAC | p.Thr1987Asn | Het | Missense | This study |
| Pt22 | M, 55 | SP/Late | CACNA1G (NM_018896.5) | c.3835G > A | p.Asp1279Asn | Het | Missense | This study |
| Pt23 | M, 5 | SP/Childhood | COQ4 (NM_016035.5) | c.577C > T | p.Pro193Ser | Het | Missense | [21] |
| c.718C > T | p.Arg240Cys | Het | Missense | [22] | ||||
| Pt24 | F, 20 | SP/Early | COQ4 (NM_016035.5) | c.284G > A | p.Gly95Asp | Het | Missense | [21] |
| c.305G > A | p.Arg102His | Het | Missense | |||||
| Pt25 | F, 10 | SP/Childhood | COQ8A (NM_020247.5) | c.589-3C > G | NA | Het | Splicing | [23] |
| c.1844G > A | p.Gly615Asp | Het | Missense | |||||
| Pt26 | M, 55 | SP/Early | COQ8A (NM_020247.5) | c.1042C > T | p.Arg348Ter | Hom | Nonsense | [24] |
| Pt27 | M, 65 | SP/Late | COQ8A (NM_020247.5) | c.127delC | p.Leu43CysfsTer166 | Het | Frameshift | [25] |
| c.1376T > C | p.Leu459Pro | Het | Missense | This study | ||||
| Pt28 | F, 61 | SP/Late | DNMT1 (NM_001130823.3) | c.1709C > T | p.Ala570Val | Het | Missense | [26] |
| Pt29 | M, 53 | SP/Late | ERCC4 (NM_005236.3) | c.1730dupA | p.Tyr577Ter | Het | Nonsense | This study |
| c.2248C > T | p.Arg750Cys | Het | Missense | |||||
| Pt30 | M, 27 | AR/Childhood | EXOSC3 (NM_016042.4) | c.395A > C | p.Asp132Ala | Hom | Missense | [27] |
| Pt31 | F, 48 | SP/Late | GJC2 (NM_020435.4) | c.219_220delCC | p.Leu74ValfsTer33 | Het | Frameshift | This study |
| c.254T > C | p.Val85Ala | Het | Missense | |||||
| Pt32 | M, 11 | SP/Childhood | HARS1 (NM_002109.6) | c.616G > T | p.Asp206Tyr | Het | Missense | [28] |
| c.730delG | p.Val244CysfsTer6 | Het | Frameshift | |||||
| Pt33 | F, 39 | AR/Childhood | HARS1 (NM_002109.6) | c.910_912dupTTG | p.Leu305dup | Het | In-frame dup | [28] |
| c.1393A > C | p.Ile465Leu | Het | Missense | |||||
| Pt34 | M, 32 | SP/Early | HSD17B4 (NM_001199291.3) | c.727G > T | p.Val243Leu | Het | Missense | [29] |
| c.2191C > T | p.Gln731Ter | Het | Nonsense | This study | ||||
| Pt35 | M, 6 | SP/Childhood | ITPR1 (NM_001168272.1) | c.722G > A | p.Arg241Lys | Het | Missense | [30] |
| Pt36 | M, 31 | SP/Childhood | ITPR1 (NM_001168272.1) | c.7748T > C | p.Ile2583Thr | Het | Missense | [31] |
| Pt37 | M, 2 | SP/Childhood | ITPR1 (NM_001168272.1) | c.805C > T | p.Arg269Trp | Het | Missense | [30] |
| Pt38 | M, 74 | SP/Late | ITPR1 (NM_001099952.3) | c.2816G > A | p.Gly939Glu | Het | Missense | This study |
| Pt39 | M, 24 | SP/Early | KCNA2 (NM_004974.4) | c.890G > A | p.Arg297Gln | Het | Missense | [32] |
| Pt40 | M, 19 | SP/Early | KCNA2 (NM_004974.4) | c.881G > A | p.Arg294His | Het | Missense | [33] |
| Pt41 | F, 63 | AD/Childhood | KCNC3 (NM_004977.3) | c.1268G > A | p.Arg423His | Het | Missense | [34] |
| Pt42 | M, 50 | SP/Late | KCND3 (NM_004980.4) | c.1646G > A | p.Arg549His | Het | Missense | [35] |
| Pt43 | M, 80 | AD/Early | KCND3 (NM_004980.4) | c.680_682delTCT | p.Phe227del | Het | In-frame del | [36] |
| Pt44 | M, 28 | SP/Childhood | KCND3 (NM_004980.4) | c.611C > T | p.Thr204Met | Het | Missense | This study |
| Pt45 | M, 42 | SP/Childhood | KIF1A (NM_001244008.1) | c.609_610delGAinsAAAAG | p.Arg203_Thr204delins | Het | In-frame indel | This study |
| GlyLysAla | ||||||||
| Pt46 | F, 57 | SP/Late | KIF1A (NM_001244008.1) | c.32G > A | p.Arg11Gln | Het | Missense | [37] |
| Pt47 | M, 29 | SP/Childhood | KIF1C (NM_006612.6) | c.765delC | p.Asp256ThrfsTer10 | Hom | Frameshift | This study |
| Pt48 | M, 7 | SP/Childhood | LAMA1 (NM_005559.4) | c.184C > T | p.Arg62Ter | Het | Nonsense | This study |
| c.1404_1405delAG | p.Gly469AlafsTer5 | Het | Frameshift | [38] | ||||
| Pt49 | F, 32 | SP/Early | MFN2 (NM_001127660.1) | c.1987C > T | p.Arg663Cys | Het | Missense | [39] |
| Pt50 | F, 83 | AD/Late | MMACHC (NM_015506.3) | c.271dupA | p.Arg91LysfsTer14 | Het | Frameshift | [40] |
| c.472T > C | p.Phe158Leu | Het | Missense | [41] | ||||
| Pt51 | F, 56 | SP/Late | MME (NM_007288.3) | c.2154G > T | p.Arg718Ser | Het | Missense | This study |
| Pt52 | M, 39 | SP/Early | OPA1 (NM_130837.2) | c.885C > G | p.Asn295Lys | Het | Missense | This study |
| Pt53 | M, 65 | SP/Late | OPA1 (NM_130837.2) | c.2873_2876delTTAG | p.Val958GlyfsTer3 | Het | Frameshift | [42] |
| Pt54 | F, 5 | SP/Childhood | PLA2G6 (NM_003560.4) | c.1111G > A | p.Val371Met | Het | Missense | [43] |
| c.1703T > C | p.Phe568Ser | Het | Missense | This study | ||||
| Pt55 | M, 33 | AR/Childhood | PMM2 (NM_000303.3) | c.323C > T | p.Ala108Val | Het | Missense | [44] |
| c.422G > A | p.Arg141His | Het | Missense | |||||
| Pt56 | F, 42 | AR/Early | PNPLA6 (NM_001166111.2) | c.1880C > T | p.Ala627Val | Hom | Missense | This study |
| Pt57 | F, 57 | SP/Late | PNPLA6 (NM_001166111.2) | c.2264A > C | p.Gln755Pro | Het | Missense | This study |
| c.3388C > T | p.His1130Tyr | Het | Missense | |||||
| Pt58 | M, 56 | SP/Late | PNPLA6 (NM_001166111.2) | c.3023A > G | p.Asp1008Gly | Het | Missense | This study |
| c.4075C > T | p.Arg1359Trp | Het | Missense | [45] | ||||
| Pt59 | F, 64 | AR/Late | PNPLA6 (NM_001166111.2) | c.3385G > A | p.Gly1129Arg | Hom | Missense | [46] |
| Pt60 | M, 33 | SP/Early | PNPLA6 (NM_001166111.2) | c.3365C > T | p.Pro1122Leu | Het | Missense | [47] |
| c.4081C > T | p.Arg1361Ter | Het | Nonsense | [48] | ||||
| Pt61 | F, 70 | SP/Late | POLG (NM_001126131.2) | c.752C > T | p.Thr251Ile | Het | Missense | [49] |
| c.1760C > T | p.Pro587Leu | Het | Missense | [50] | ||||
| c.2243G > C | p.Trp748Ser | Het | Missense | [51] | ||||
| Pt62 | F, 65 | AR/Late | POLR3A (NM_007055.4) | c.1909 + 22G > A | NA | Het | Splicing | [52] |
| c.4073G > A | p.Gly1358Glu | Het | Missense | This study | ||||
| Pt63 | M, 35 | SP/Childhood | POLR3A (NM_007055.4) | c.1909 + 22G > A | NA | Het | Splicing | [52] |
| Deletion ex. 14-18 | NA | Het | Large deletion | This study | ||||
| Pt64 | M, 65 | AR/Late | POLR3A (NM_007055.4) | c.1909 + 22G > A | NA | Het | Splicing | [52] |
| c.3839dupT | p.Met1280IlefsTer20 | Het | Frameshift | This study | ||||
| Pt65 | M, 69 | AD/Late | PRKCG (NM_002739.5) | c.230G > A | p.Cys77Tyr | Het | Missense | [53] |
| Pt66 | M, 50 | AD/Late | PRKCG (NM_002739.5) | c.358C > T | p.Leu120Phe | Het | Missense | [53] |
| Pt67 | F, 63 | SP/Late | PRKCG (NM_002739.5) | c.1928T > G | p.Phe643Cys | Het | Missense | [53] |
| Pt68 | F, 47 | SP/Late | PRKCG (NM_002739.5) | c.1381G > A | p.Ala461Thr | Het | Missense | [53] |
| Pt69 | M, 40 | AD/Childhood | PRKCG (NM_002739.5) | c.466G > A | p.Glu156Lys | Het | Missense | [53] |
| Pt70 | M, 7 | SP/Childhood | PRKCG (NM_002739.5) | c.1308C > G | p.Tyr436Ter | Het | Nonsense | [53] |
| Pt71 | F, 35 | SP/Early | PRKCG (NM_002739.5) | c.413T > A | p.Val138Glu | Het | Missense | [54] |
| Pt72 | M, 41 | SP/Early | PRKCG (NM_002739.5) | c.380A > C | p.Gln127Pro | Het | Missense | [55] |
| Pt73 | F, 49 | SP/Late | PRKCG (NM_002739.5) | c.230G > A | p.Cys77Tyr | Het | Missense | [53] |
| Pt74 | F, 14 | SP/Childhood | PRKCG (NM_002739.5) | c.419G > A | p.Arg140Gln | Het | Missense | [53] |
| Pt75 | F, 66 | AD/Late | PRNP (NM_001080123.3) | c.305C > T | p.Pro102Leu | Het | Missense | This study |
| Pt76 | M, 45 | SP/Late | PSEN1 (NM_000021.4) | c.300dupT | p.Lys101Ter | Het | Nonsense | This study |
| Pt77 | F, 10 | SP/Childhood | RARS2 (NM_020320.5) | c.517G > A | p.Asp173Asn | Het | Missense | This study |
| c.1037C > T | p.Thr346Ile | Het | Missense | |||||
| Pt78 | F, 27 | SP/Childhood | RNF170 (NM_030954.4) | c.566T > G | p.Phe189Cys | Het | Missense | This study |
| Pt79 | F, 45 | SP/Early | RNF216 (NM_207111.4) | c.1849A > G | p.Met617Val | Het | Missense | [56] |
| c.2061 + 3A > G | NA | Het | Splicing | |||||
| Pt80 | M, 52 | SP/Childhood | SETX (NM_015046.7) | c.7292dupA | p.Asn2431LysfsTer19 | Hom | Frameshift | This study |
| Pt81 | M, 87 | SP/Late | SETX (NM_015046.7) | c.5591A > C | p.Gln1864Pro | Het | Missense | This study |
| Pt82 | F, 5 | SP/Childhood | SLC2A1 (NM_006516.3) | c.136C > T | p.Gln46Ter | Het | Nonsense | [57] |
| Pt83 | F, 15 | SP/Childhood | SLC2A1 (NM_006516.3) | c.985G > A | p.Glu329Lys | Het | Missense | This study |
| Pt84 | M, 6 | SP/Childhood | SLC9A6 (NM_001177651) | Deletion ex. 4-7 | NA | Hem | Large deletion | This study |
| Pt85 | F, 71 | SP/Late | SPG7 (NM_003119.4) | c.679C > T | p.Arg227Ter | Het | Nonsense | [58] |
| c.1231G > A | p.Asp411Asn | Het | Missense | |||||
| Pt86 | F, 57 | SP/Early | SPG7 (NM_003119.4) | c.1529C > T | p.Ala510Val | Het | Missense | [13] |
| c.1940C > A | p.Ala647Glu | Het | Missense | This study | ||||
| Pt87 | M, 72 | AR/Late | SPG7 (NM_003119.4) | c.1529C > T | p.Ala510Val | Hom | Missense | [13] |
| Pt88 | M, 65 | AR/Late | SPG7 (NM_003119.4) | c.1529C > T | p.Ala510Val | Hom | Missense | [13] |
| Pt89 | M, 25 | AR/Early | SPG7 (NM_003119.4) | Deletion ex. 2 | NA | Hom | Large deletion | This study |
| Pt90 | M, 69 | AR/Early | SPG7 (NM_003119.4) | c.73_80delCCAGGCCC | p.Pro25GlyfsTer46 | Het | Frameshift | This study |
| c.1940C > A | p.Ala647Glu | Het | Missense | |||||
| Pt91 | M, 46 | SP/Early | SPG7 (NM_003119.4) | Deletion ex. 2 | NA | Hom | Large deletion | This study |
| Pt92 | F, 63 | AR/Late | SPG7 (NM_003119.4) | c.1529C > T | p.Ala510Val | Het | Missense | [13] |
| c.1972G > A | p.Ala658Thr | Het | Missense | [59] | ||||
| Pt93 | F, 36 | SP/Childhood | SPTAN1 (NM_001363759.2) | c.4870C > T | p.Arg1624Cys | Het | Missense | This study |
| Pt94 | M, 60 | SP/Late | SPTBN2 (NM_006946.3) | c.5066G > A | p.Arg1689His | Het | Missense | This study |
| Pt95 | M, 26 | SP/Early | SPTBN2 (NM_006946.3) | c.1438C > T | p.Arg480Trp | Het | Missense | [60] |
| Pt96 | M, 54 | AR/Late | SPTBN2 (NM_006946.3) | c.157 + 1G > A | NA | Het | Splicing | This study |
| c.1843C > T | p.Arg615Trp | Het | Missense | |||||
| Pt97 | M, 63 | AD/Late | STUB1 (NM_005861.4) | c.97G > A | p.Gly33Ser | Het | Missense | [61] |
| Pt98 | M, 52 | AD/Early | STUB1 (NM_005861.4) | c.689_692delACCT | p.Tyr230CysfsTer9 | Het | Frameshift | [62] |
| Pt99 | M, 61 | AD/Late | STUB1 (NM_005861.4) | c.682C > T | p.Pro228Ser | Het | Missense | [61] |
| Pt100 | F, 53 | SP/Early | STUB1 (NM_005861.4) | c.199G > A | p.Ala67Thr | Het | Missense | [63] |
| Pt101 | F, 48 | AD/Late | STUB1 (NM_005861.4) | c.673C > T | p.Arg225Ter | Het | Nonsense | [63] |
| Pt102 | F, 55 | SP/Late | STUB1 (NM_005861.4) | c.721C > T | p.Arg241Trp | Het | Missense | [63] |
| Pt103 | M, 48 | SP/Late | STUB1 (NM_005861.4) | c.433A > C | p.Lys145Gln | Het | Missense | [62] |
| Pt104 | M, 52 | AD/Late | STUB1 (NM_005861.4) | c.170C > T | p.Pro57Leu | Het | Missense | [63] |
| Pt105 | F, 67 | AD/Late | STUB1 (NM_005861.4) | c.818_819dupGC | p.Pro274AlafsTer3 | Het | Frameshift | [63] |
| Pt106 | F, 70 | AD/Late | STUB1 (NM_005861.4) | c.791_792delTG | p.Val264GlyfsTer4 | Het | Frameshift | [63] |
| Pt107 | F, 60 | SP/Early | STUB1 (NM_005861.4) | c.823_824delCT | p.Leu275AspfsTer16 | Het | Frameshift | [64] |
| Pt108 | F, NA | SP/NA | STXBP1 (NM_003165.5) | c.874C > T | p.Arg292Cys | Het | Missense | [65] |
| Pt109 | F, 9 | SP/Childhood | STXBP1 (NM_003165.5) | c.434A > G | p.Tyr145Cys | Het | Missense | This study |
| Pt110 | F, 48 | SP/Late | STXBP1 (NM_003165.5) | c.298C > T | p.Arg100Trp | Het | Missense | This study |
| Pt111 | M, 32 | SP/Early | SYNE1 (NM_182961.4) | c.15049C > T | p.Gln5017Ter | Hom | Nonsense | This study |
| Pt112 | M, 61 | SP/Late | SYNE1 (NM_182961.4) | c.6724-1G > A | NA | Het | Splicing | This study |
| c.7085dupA | p.Asn2362LysfsTer4 | Het | Frameshift | |||||
| Pt113 | F, 36 | AR/Early | SYNE1 (NM_182961.4) | c.4609C > T | p.Arg1537Ter | Hom | Nonsense | This study |
| Pt114 | F, 38 | AR/Early | SYNE1 (NM_182961.4) | c.3130C > T | p.Arg1044Ter | Het | Nonsense | This study |
| c.7911G > A | p.Trp2637Ter | Het | Nonsense | |||||
| Pt115 | M, 3 | SP/Childhood | SYNE2 (NM_182914.2) | c.2970C > A | p.Tyr990Ter | Het | Nonsense | This study |
| Pt116 | M, 42 | SP/Early | TMEM240 (NM_001114748.1) | c.509C > T | p.Pro170Leu | Het | Missense | [66] |
| Pt117 | M, 46 | AD/Late | TMEM240 (NM_001114748.1) | c.509C > T | p.Pro170Leu | Het | Missense | [66] |
| Pt118 | M, 7 | SP/Childhood | TMEM240 (NM_001114748.1) | c.196G > A | p.Gly66Arg | Het | Missense | [67] |
| Pt119 | F, 64 | SP/Early | TMEM240 (NM_001114748.1) | c.419T > A | p.Leu140Gln | Het | Missense | This study |
| Pt120 | M, 12 | SP/Childhood | TMEM240 (NM_001114748.1) | c.239C > T | p.Thr80Met | Het | Missense | [66] |
| Pt121 | M, 6 | SP/Childhood | TPP1 (NM_000391.4) | c.225A > G | p.Gln75= | Hom | Splicing | [68] |
| Pt122 | F, 64 | SP/Late | TRPC3 (NM_001130698.2) | c.1419delT | p.Val474CysfsTer29 | Het | Frameshift | This study |
| Pt123 | F, 73 | SP/Late | TTBK2 (NM_173500.4) | c.239T > A | p.Phe80Tyr | Het | Missense | This study |
| Pt124 | F, 74 | SP/Late | TTPA (NM_000370.3) | c.553-1G > T | NA | Hom | Splicing | This study |
| Pt125 | M, 53 | SP/Late | WFS1 (NM_006005.3) | c.1291G > C | p.Glu431Gln | Het | Missense | This study |
| c.1523A > G | p.Tyr508Cys | Het | Missense | [69] |
| Study | NGS Application | Index Cases | Main Cohort Feature(s) | Diagnostic Yield (%) |
|---|---|---|---|---|
| Nemeth et al., 2013 [6] | TRP (118 genes) | 50 | Heterogeneous | 16 |
| Ohba et al., 2013 [70] | ES | 23 | Childhood onset, sporadic | 39 |
| Sawyer et al., 2014 [71] | ES | 28 | Pediatric onset | 39 |
| Fogel et al., 2014 [72] | ES | 76 | Heterogeneous | 21 |
| Pyle et al., 2015 [73] | ES | 22 | Heterogeneous | 41 |
| Keogh et al., 2015 [74] | ES | 12 | Late onset | 33 |
| Mallaret et al., 2016 [75] | TRP (57 genes) | 145 | Onset < 60 years | 16 |
| van de Warrenburg et al., 2016 [76] | ES | 28 | Heterogeneous | 32 |
| Marelli et al., 2016 [77] | ES | 33 | Onset < 50 years | 42 |
| Kuperberg et al., 2016 [78] | ES | 21 | Pediatric onset | 57 |
| Hadjivassiliou et al., 2016 [79] | TRP (57 genes) | 146 | Progressive ataxia | 24 |
| Coutelier et al., 2017 [25] | TRP (65 genes) | 412 | Dominant inheritance | 11 |
| Iqbal et al., 2017 [80] | TRP (159 genes) | 58 | Heterogeneous | 14 |
| Nibbeling et al., 2017 [81] | ES | 20 | Dominant inheritance | 35 |
| Nibbeling et al., 2017 [81] | TRP (42 genes) | 96 | Dominant inheritance | 15 |
| Coutelier et al., 2018 [59] | ES | 319 | Heterogeneous | 28 |
| Montaut et al., 2018 [82] | ES | 23 | Heterogeneous | 43 |
| Dong et al., 2019 [83] | TRP (56 genes) + mDNA | 33 | Heterogeneous | 15 |
| Kang et al., 2019 [84] | TRP (46 and 98 genes) | 32 | Heterogeneous | 25 |
| Shakya et al., 2019 [85] | ES | 16 | Early onset | 56 |
| Shakya et al., 2019 [85] | TRP (41 genes) | 82 | Early onset | 17 |
| Sun et al., 2019 [86] | ES | 170 | Heterogeneous | 52 |
| Arslan et al., 2020 [87] | TRP (111 genes) | 84 | Pediatric onset | 25 |
| Ngo et al., 2020 [88] | ES | 184 | Heterogeneous | 24 |
| Mutlu-Albayrak et al., 2020 [89] | TRP (13 genes) | 40 | Consanguinity, childhood | 82 |
| onset, recessive inheritance | ||||
| Gauquelin et al., 2020 [90] | ES | 66 | Heterogeneous | 53 |
| Ignatius et al., 2020 [91] | ES | 50 | Onset < 5 years | 40 |
| Kim et al., 2020 [92] | ES | 68 | Heterogeneous | 26 |
| Bogdanova-Mihaylova et al., 2021 [93] | ES | 20 | Progressive ataxia | 20 |
| Bogdanova-Mihaylova et al., 2021 [93] | TRP (87 genes) * | 84 | Progressive ataxia | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galatolo, D.; De Michele, G.; Silvestri, G.; Leuzzi, V.; Casali, C.; Musumeci, O.; Antenora, A.; Astrea, G.; Barghigiani, M.; Battini, R.; et al. NGS in Hereditary Ataxia: When Rare Becomes Frequent. Int. J. Mol. Sci. 2021, 22, 8490. https://doi.org/10.3390/ijms22168490
Galatolo D, De Michele G, Silvestri G, Leuzzi V, Casali C, Musumeci O, Antenora A, Astrea G, Barghigiani M, Battini R, et al. NGS in Hereditary Ataxia: When Rare Becomes Frequent. International Journal of Molecular Sciences. 2021; 22(16):8490. https://doi.org/10.3390/ijms22168490
Chicago/Turabian StyleGalatolo, Daniele, Giovanna De Michele, Gabriella Silvestri, Vincenzo Leuzzi, Carlo Casali, Olimpia Musumeci, Antonella Antenora, Guja Astrea, Melissa Barghigiani, Roberta Battini, and et al. 2021. "NGS in Hereditary Ataxia: When Rare Becomes Frequent" International Journal of Molecular Sciences 22, no. 16: 8490. https://doi.org/10.3390/ijms22168490
APA StyleGalatolo, D., De Michele, G., Silvestri, G., Leuzzi, V., Casali, C., Musumeci, O., Antenora, A., Astrea, G., Barghigiani, M., Battini, R., Battisti, C., Caputi, C., Cioffi, E., De Michele, G., Dotti, M. T., Fico, T., Fiorillo, C., Galosi, S., Lieto, M., ... Santorelli, F. M. (2021). NGS in Hereditary Ataxia: When Rare Becomes Frequent. International Journal of Molecular Sciences, 22(16), 8490. https://doi.org/10.3390/ijms22168490