
A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine

 ,
,  ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
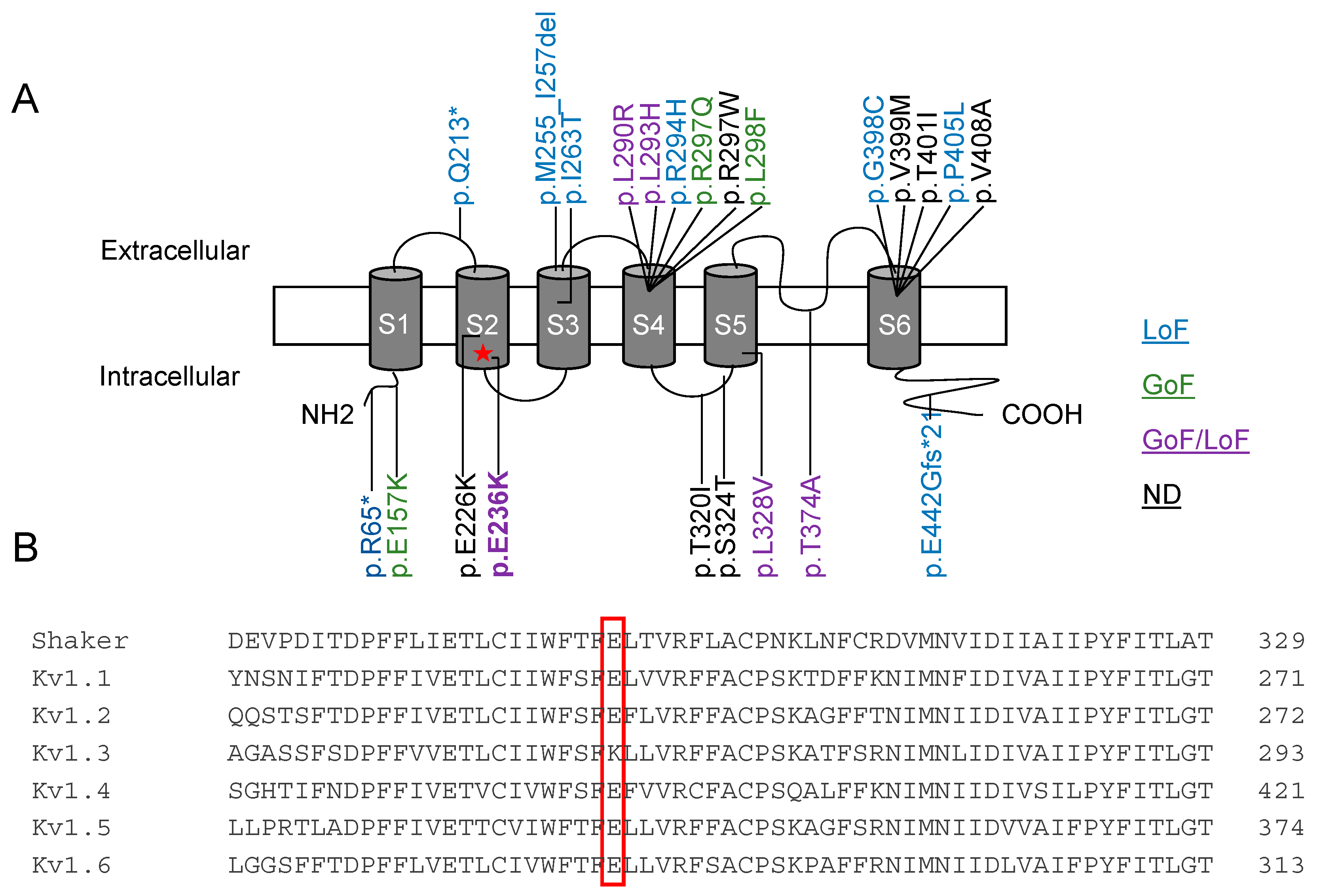
2.1. Clinical Description
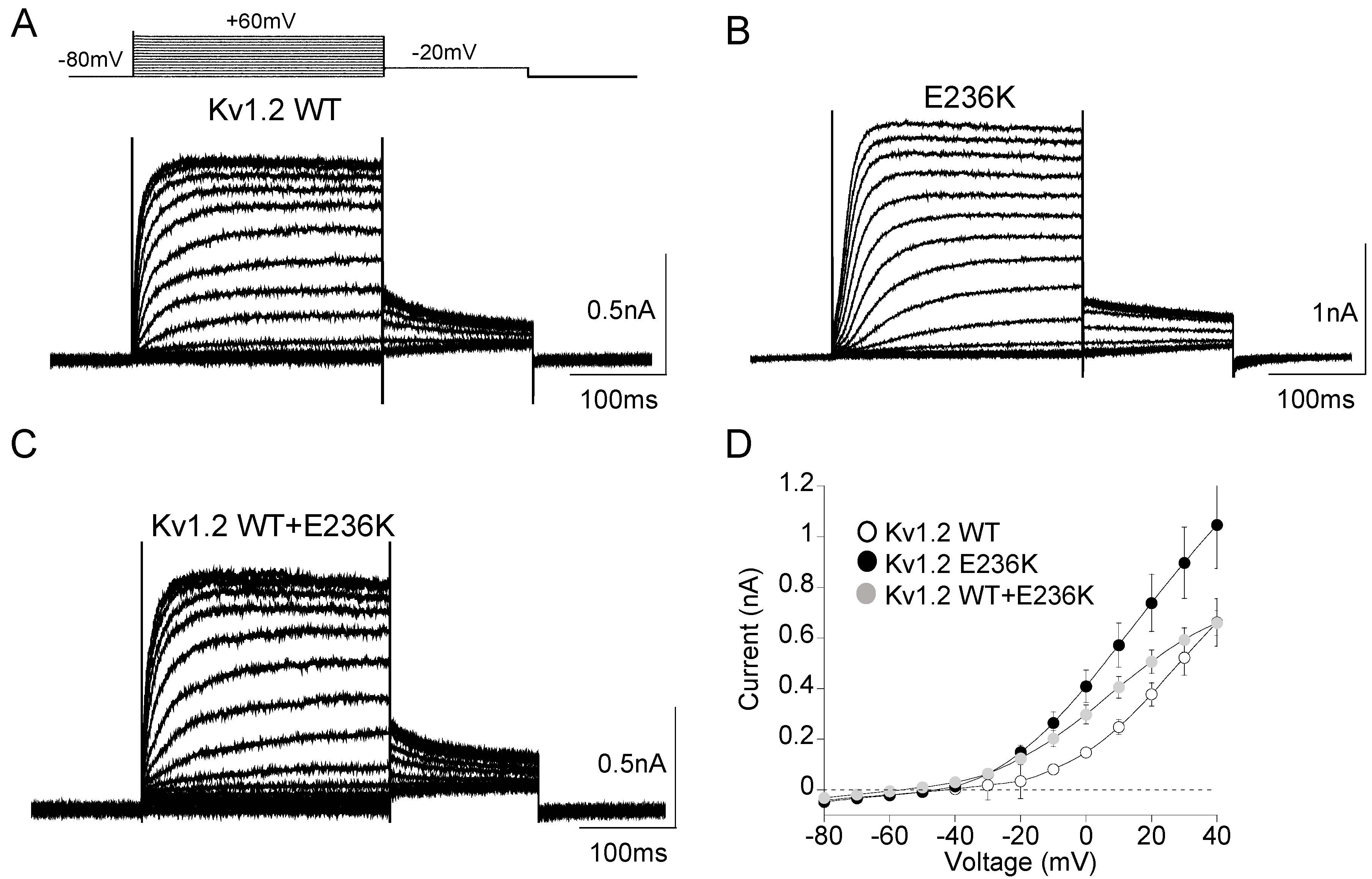
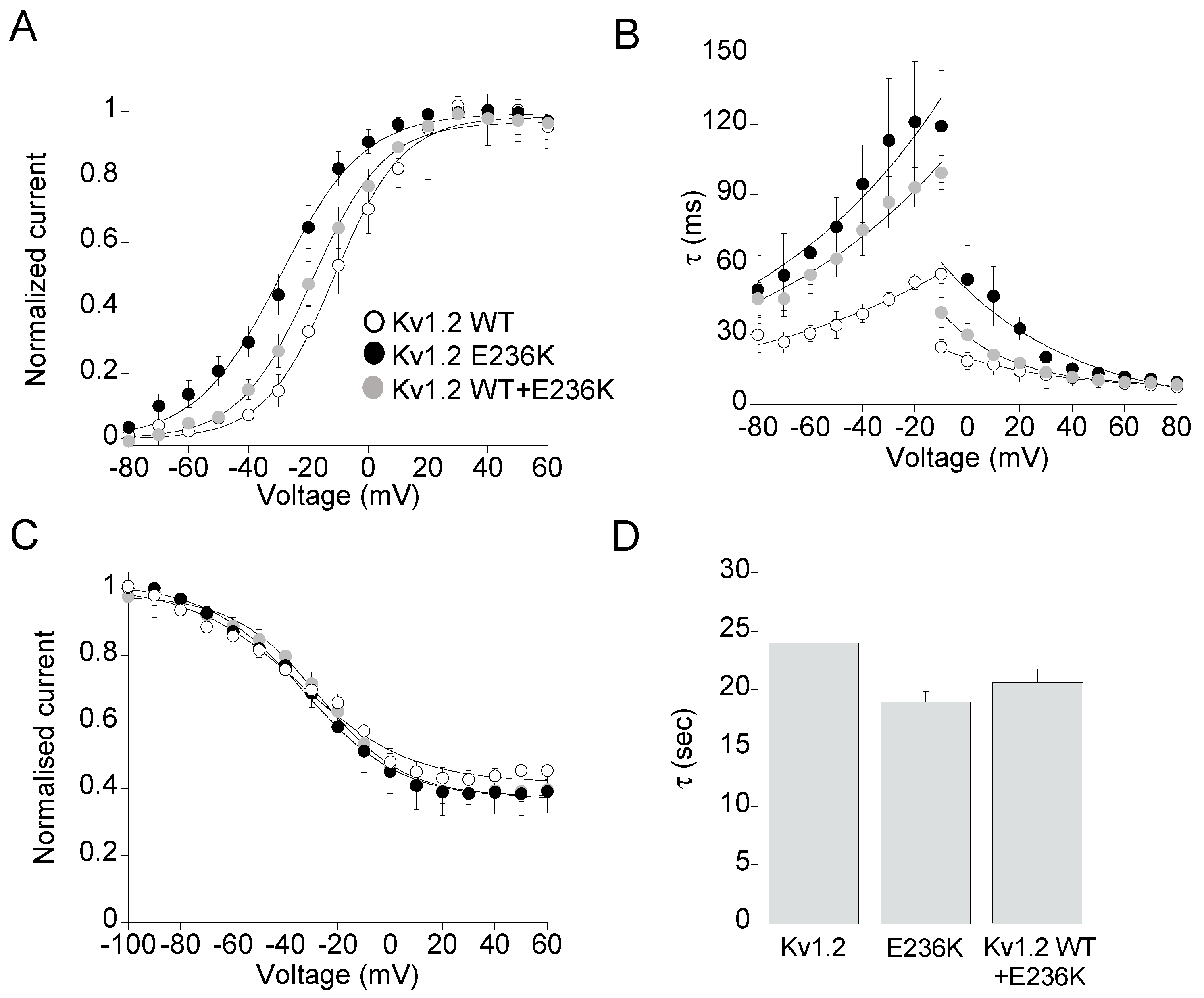
2.2. Functional Characterization of Kv1.2E236K Channels
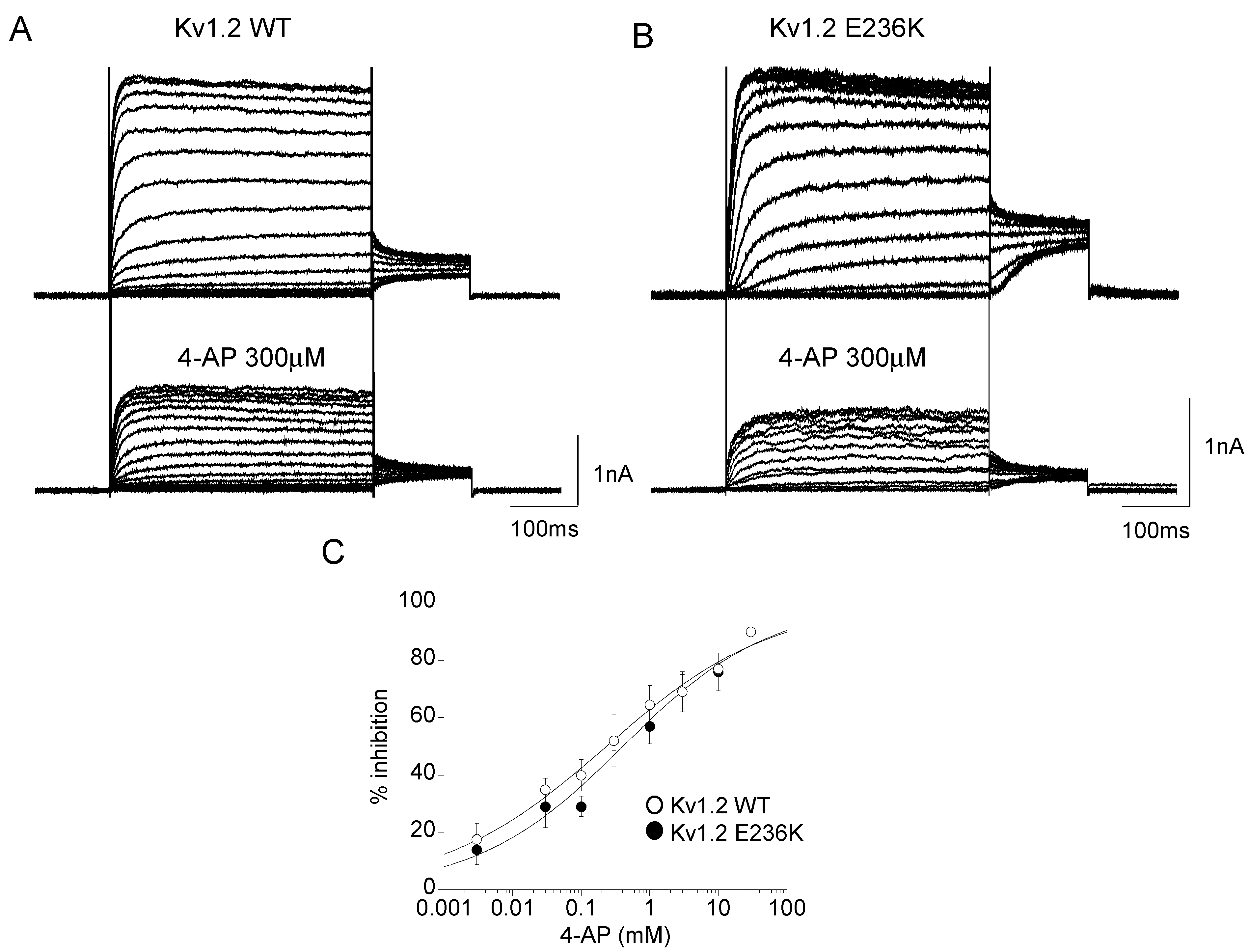
2.3. Effect of 4-Aminopyridine (4-AP) on Kv1.2 WT and E236K Channel
2.4. Molecular Dynamic Simulations
3. Discussion
3.1. Genotype-Phenotype Correlation and Mechanistic Hypotheses
3.2. Towards Precision Medicine for KCNA2-Disorders
4. Materials and Methods
4.1. Clinical Diagnosis
4.2. Genetic Testing
4.3. Mutagenesis and Expression of Kv1.2 WT and Mutant Channel
4.4. Electrophysiology
4.5. Homology Modeling and Molecular Dynamic Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trimmer, J.S. Subcellular localization of K+ channels in mammalian brain neurons: Remarkable precision in the midst of extraordinary complexity. Neuron 2015, 85, 238–256. [Google Scholar] [CrossRef] [Green Version]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Liantonio, A.; Conte, E.; Pessia, M.; Imbrici, P. Ion Channels Involvement in Neurodevelopmental Disorders. Neuroscience 2020, 440, 337–359. [Google Scholar] [CrossRef] [PubMed]
- Dodson, P.D.; Forsythe, I.D. Presynaptic K+ channels: Electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004, 27, 210–217. [Google Scholar] [CrossRef]
- Robbins, C.A.; Tempel, B.L. Kv1.1 and Kv1.2: Similar channels, different seizure models. Epilepsia 2012, 53 (Suppl. 1), 134–141. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.; Leitner, I.; Seppi, K.; Trieb, M.; Wietzorrek, G.; Marksteiner, J.; Knaus, H.G. Shaker-related voltage-gated potassium channels Kv1 in human hippocampus. Brain Struct. Funct. 2018, 223, 2663–2671. [Google Scholar] [CrossRef]
- Smart, S.L.; Lopantsev, V.; Zhang, C.L.; Robbins, C.A.; Wang, H.; Chiu, S.Y.; Schwartzkroin, P.A.; Messing, A.; Tempel, B.L. Deletion of the Kv1.1 potassium channel causes epilepsy in mice. Neuron 1998, 20, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Brew, H.M.; Gittelman, J.X.; Silverstein, R.S.; Hanks, T.D.; Demas, V.P.; Robinson, L.C.; Robbins, C.A.; McKee-Johnson, J.; Chiu, S.Y.; Messing, A.; et al. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J. Neurophysiol. 2007, 98, 1501–1525. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Harrison, J.; Clapcote, S.J.; Huang, Y.; Zhang, Y.-Y.; Wang, L.-Y.; Roder, J.C. A new Kv1.2 channelopathy underlying cerebellar ataxia. J. Biol. Chem. 2010, 285, 32160–32173. [Google Scholar] [CrossRef] [Green Version]
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, J.H.; Schröter, J.; Jüngling, J.; Biskup, S.; Klotz, K.A.; Bast, T.; Dietel, T.; Korenke, G.C.; Christoph, S.; Brennenstuhl, H.; et al. Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 2824. [Google Scholar] [CrossRef]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djémié, T.; Müller, S.; Møller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Loboda, A. A model for 4-aminopyridine action on K channels: Similarities to tetraethylammonium ion action. Biophys. J. 2001, 81, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, U.B.S.; Lauxmann, S.; Wolff, M.; Synofzik, M.; Bast, T.; Binelli, A.; Serratosa, J.M.; Martínez-Ulloa, P.; Allen, N.M.; King, M.D.; et al. 4-aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Arnold, R.; Huynh, W.; Kiernan, M.C.; Krishnan, A.V. Ion Channel Modulation as a Therapeutic Approach in Multiple Sclerosis. Curr. Med. Chem. 2015, 22, 4366–4378. [Google Scholar] [CrossRef]
- Kalla, R.; Strupp, M. Aminopyridines and Acetyl-DL-leucine: New Therapies in Cerebellar Disorders. Curr. Neuropharmacol. 2019, 17, 7–13. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Grissmer, S.; Nguyen, A.N.; Aiyar, J.; Hanson, D.C.; Mather, R.J.; Gutman, G.A.; Karmilowicz, M.J.; Auperin, D.D.; Chandy, K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994, 45, 1227–1234. [Google Scholar] [PubMed]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the strength of salt bridges: A comparison of current biomolecular force fields. J. Phys. Chem. B 2014, 118, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Baronas, V.A.; McGuinness, B.R.; Brigidi, G.S.; Gomm Kolisko, R.N.; Vilin, Y.Y.; Kim, R.Y.; Lynn, F.C.; Bamji, S.X.; Yang, R.; Kurata, H.T. Use-dependent activation of neuronal Kv1.2 channel complexes. J. Neurosci. 2015, 35, 3515–3524. [Google Scholar] [CrossRef] [Green Version]
- Baronas, V.A.; Runying, Y.R.; Vilin, Y.Y.; Kurata, H.T. Determinants of frequency-dependent regulation of Kv1.2-containing potassium channels. Channels 2016, 10, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palani, D.; Baginskas, A.; Raastad, M. Bursts and hyperexcitability in non-myelinated axons of the rat hippocampus. Neuroscience 2010, 167, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Fleming, K.L.; Raymond, S.; Wong, A.Y.C.; Bergeron, R. The sigma-1 receptor behaves as an atypical auxiliary subunit to modulate the functional characteristics of Kv1.2 channels expressed in HEK293 cells. Physiol. Rep. 2019, 7, e14147. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.H.; Spain, W.J. Kv1 channels control spike threshold dynamics and spike timing in cortical pyramidal neurones. J. Physiol. 2011, 589 Pt 21, 5125–5142. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Weckhuysen, S.; Gorman, K.; King, M.D.; Lerche, H. Genetic potassium channel associated epilepsies: Clinical review of the Kv family. Eur. J. Paediatr. Neurol. 2020, 24, 105–116. [Google Scholar] [CrossRef]
- Niday, Z.; Tzingounis, A.V. Potassium channel gain of function in epilepsy: An unresolved paradox. Neuroscientist 2018, 24, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, A.; Kaneko, M.; Angelini, M.; Steccanella, F.; Westerlund, A.M.; Lindström, S.H.; Nilsson, M.; Delemotte, L.; Saitta, S.C.; Olcese, R. Tracking the motion of the KV 1.2 voltage sensor reveals the molecular perturbations caused by a de novo mutation in a case of epilepsy. J. Physiol. 2020, 598, 5245–5269. [Google Scholar] [CrossRef]
- Shore, A.N.; Colombo, S.; Tobin, W.F.; Petri, S.; Cullen, E.R.; Dominguez, S.; Bostick, C.D.; Beaumont, M.A.; Williams, D.; Khodagholy, D.; et al. Reduced GABAergic Neuron Excitability, Altered Synaptic Connectivity, and Seizures in a KCNT1 Gain-of-Function Mouse Model of Childhood Epilepsy. Cell Rep. 2020, 33, 108303. [Google Scholar] [CrossRef]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion channels in genetic epilepsy: From genes and mechanisms to disease-targeted therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef]
- Alviña, K.; Khodakhah, K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 2010, 30, 7258–7268. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, K.J.; Strassle, B.W.; Monaghan, M.M.; Bekele-Arcuri, Z.; Matos, M.F.; Trimmer, J.S. Association and colocalization of the Kvbeta1 and Kvbeta2 beta-subunits with Kv1 alpha-subunits in mammalian brain K+ channel complexes. J. Neurosci. 1997, 17, 8246–8258. [Google Scholar] [CrossRef] [Green Version]
- Baronas, V.A.; Yang, R.Y.Y.; Morales, L.C.; Sipione, S.; Kurata, H.T. Slc7a5 regulates Kv1.2 channels and modifies functional outcomes of epilepsy-linked channel mutations. Nat. Commun. 2018, 9, 4417. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, L.; Su, L.D.; Cao, S.L.; Xie, Y.J.; Wang, N.; Shao, C.Y.; Wa, Y.N.; Zhou, J.H.; Cowell, J.K.; et al. Celecoxib Ameliorates Seizure Susceptibility in Autosomal Dominant Lateral Temporal Epilepsy. J. Neurosci. 2018, 38, 3346–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seagar, M.; Russier, M.; Caillard, O.; Maulet, Y.; Fronzaroli-Moliniere, L.; De San Feliciano, M.; Boumedine-Guignon, N.; Rodriguez, L.; Zbili, M.; Usseglio, F.; et al. LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels. Proc. Natl. Acad. Sci. USA 2017, 114, 7719–7724. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Arsenault, J.; Bah, A.; Krzeminski, M.; Fekete, A.; Chao, O.Y.; Pacey, L.K.; Wang, A.; Forman-Kay, J.; Hampson, D.R.; et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol. Psychiatry 2020, 25, 2017–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrini, S.; Chiarello, D.; Gogou, M.; Silvennoinen, K.; Puvirajasinghe, C.; Jones, W.D.; Reif, P.; Klein, K.M.; Rosenow, F.; Weber, Y.G.; et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J. Neurol. Neurosurg. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Uysal, B.; Rosa, F.; Löffler, H.; Mau-Holzmann, U.A.; Liebau, S.; Lerche, H. Establishment of a human induced pluripotent stem cell (iPSC) line (HIHDNEi002-A) from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p.Arg297Gln) mutation. Stem. Cell. Res. 2019, 7, 101445. [Google Scholar] [CrossRef]
- Uysal, B.; Löffler, H.; Rosa, F.; Lerche, H.; Schwarz, N. Generation of an induced pluripotent stem cell (iPSC) line (HIHDNEi003-A) from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p.Thr374Ala) mutation. Stem. Cell Res. 2019, 40, 101543. [Google Scholar] [CrossRef]
- Wacker, S.J.; Jurkowski, W.; Simmons, K.J.; Fishwick, C.W.; Johnson, A.P.; Madge, D.; Lindahl, E.; Rolland, J.F.; de Groot, B.L. Identification of selective inhibitors of the potassium channel Kv1.1-1.2((3)) by high-throughput virtual screening and automated patch clamp. ChemMedChem 2012, 7, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Meythaler, J.M.; Brunner, R.C.; Peduzzi, J. Phase IIB Randomized Trial on the Use of 4-Aminopyridine in Guillain-Barré Syndrome. Arch. Rehabil. Res. Clin. Transl. 2021, 3, 100123. [Google Scholar] [CrossRef]
- Simmons, R.L.; Li, H.; Alten, B.; Santos, M.S.; Jiang, R.; Paul, B.; Lalani, S.J.; Cortesi, A.; Parks, K.; Khandelwal, N.; et al. Overcoming presynaptic effects of VAMP2 mutations with 4-aminopyridine treatment. Hum. Mutat. 2020, 41, 1999–2011. [Google Scholar] [CrossRef]
- Summa, S.; Schirinzi, T.; Bernava, G.M.; Romano, A.; Favetta, M.; Valente, E.M.; Bertini, E.; Castelli, E.; Petrarca, M.; Pioggia, G.; et al. Development of SaraHome: A novel, well-accepted, technology-based assessment tool for patients with ataxia. Comput. Methods Programs Biomed. 2020, 188, 105257. [Google Scholar] [CrossRef] [PubMed]
- Vasco, G.; Gazzellini, S.; Petrarca, M.; Lispi, M.L.; Pisano, A.; Zazza, M.; Della Bella, G.; Castelli, E.; Bertini, E. Functional and Gait Assessment in Children and Adolescents Affected by Friedreich’s Ataxia: A One-Year Longitudinal Study. PLoS ONE 2016, 11, e0162463. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rangel, S.; Bravin, A.D.; Ramos-Torres, K.M.; Brugarolas, P.; Sánchez-Rodríguez, J.E. Structure-activity relationship studies of four novel 4-aminopyridine K(+) channel blockers. Sci. Rep. 2020, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, O.Y.; Marron Fernandez de Velasco, E.; Pathak, S.S.; Maitra, S.; Zhang, H.; Duvick, L.; Wickman, K.; Orr, H.T.; Hirai, H.; Yang, Y.M. Targeting inhibitory cerebellar circuitry to alleviate behavioral deficits in a mouse model for studying idiopathic autism. Neuropsychopharmacology 2020, 45, 1159–1170. [Google Scholar] [CrossRef]
- Glasscock, E.; Qian, J.; Yoo, J.W.; Noebels, J.L. Masking epilepsy by combining two epilepsy genes. Nat. Neurosci. 2007, 10, 1554–1558. [Google Scholar] [CrossRef] [PubMed]
- Indumathy, J.; Pruitt, A.; Gautier, N.M.; Crane, K.; Glasscock, E. Kv1.1 deficiency alters repetitive and social behaviors in mice and rescues autistic-like behaviors due to Scn2a haploinsufficiency. Brain Behav. 2021, 11, e02041. [Google Scholar] [CrossRef]
- Imbrici, P.; Altamura, C.; Gualandi, F.; Mangiatordi, G.F.; Neri, M.; De Maria, G.; Ferlini, A.; Padovani, A.; D’Adamo, M.C.; Nicolotti, O.; et al. A novel KCNA1 mutation in a patient with paroxysmal ataxia, myokymia, painful contractures and metabolic dysfunctions. Mol. Cell. Neurosci. 2017, 83, 6–12. [Google Scholar] [CrossRef]
- Rezazadeh, S.; Kurata, H.T.; Claydon, T.W.; Kehl, S.J.; Fedida, D. An activation gating switch in Kv1.2 is localized to a threonine residue in the S2–S3 linker. Biophys. J. 2007, 93, 4173–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Current Density +40 mV/−10 mV | Voltage Dependence of Activation | Kinetic of Activation | Kinetic of Deactivation | Steady-State Inactivation | |||
|---|---|---|---|---|---|---|---|
| nA | V1/2 (mV) | k (mV) | τV1/2 (ms) | τV1/2 (ms) | V1/2 (mV) | k (mV) | |
| Kv1.2 WT | 0.7 ± 0.2 0.08 ± 0.01 (28) | −11.5 ± 0.9 (9) | 11.5 ± 0.8 | 18.5 ± 1.05 (9) | 55.5 ± 1.5 (13) | −34.0 ± 3.0 (8) | 20.0 ± 3.0 |
| E236K | 1.0 ± 0.3 0.27 ± 0.04 (28) | −29.4 ± 1.0 * (15) | 13.7 ± 1.0 | 96.1 ± 8.5 * (16) | 101.4 ± 3.3 * (10) | −32.9 ± 1.3 (7) | 18.4 ± 1.2 |
| Kv1.2 WT+E236K | 0.7 ± 0.9 0.20 ± 0.03 (23) | −18.8 ± 0.9 * (18) | 12.0 ± 0.6 | 45.5 ± 0.9 * (16) | 93.1 ± 2.0 * (14) | −27.1 ± 0.8 (n = 13) | 16.3 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbrici, P.; Conte, E.; Blunck, R.; Stregapede, F.; Liantonio, A.; Tosi, M.; D’Adamo, M.C.; De Luca, A.; Brankovic, V.; Zanni, G. A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine. Int. J. Mol. Sci. 2021, 22, 9913. https://doi.org/10.3390/ijms22189913
Imbrici P, Conte E, Blunck R, Stregapede F, Liantonio A, Tosi M, D’Adamo MC, De Luca A, Brankovic V, Zanni G. A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine. International Journal of Molecular Sciences. 2021; 22(18):9913. https://doi.org/10.3390/ijms22189913
Chicago/Turabian StyleImbrici, Paola, Elena Conte, Rikard Blunck, Fabrizia Stregapede, Antonella Liantonio, Michele Tosi, Maria Cristina D’Adamo, Annamaria De Luca, Vesna Brankovic, and Ginevra Zanni. 2021. "A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine" International Journal of Molecular Sciences 22, no. 18: 9913. https://doi.org/10.3390/ijms22189913
APA StyleImbrici, P., Conte, E., Blunck, R., Stregapede, F., Liantonio, A., Tosi, M., D’Adamo, M. C., De Luca, A., Brankovic, V., & Zanni, G. (2021). A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine. International Journal of Molecular Sciences, 22(18), 9913. https://doi.org/10.3390/ijms22189913