
Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
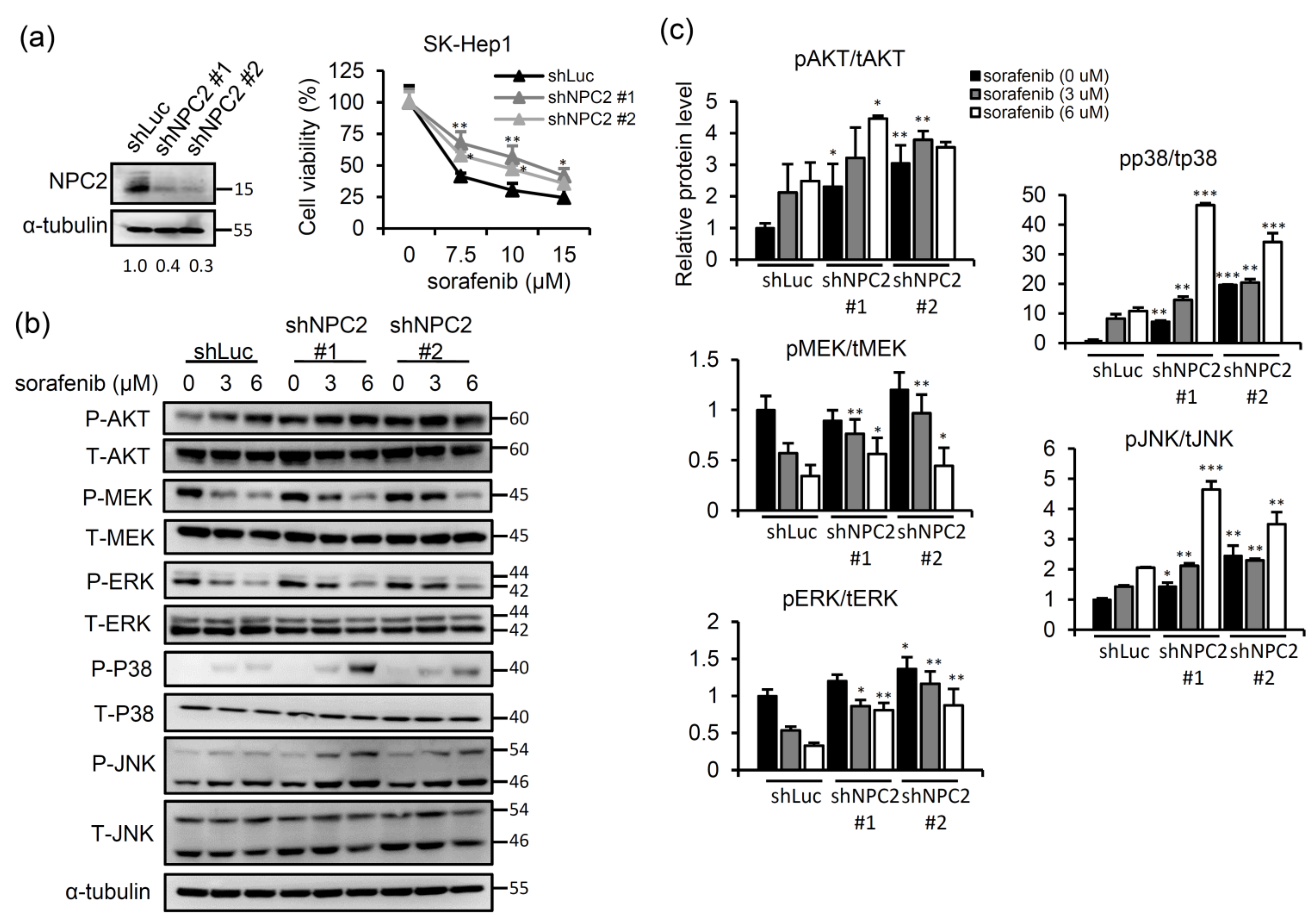
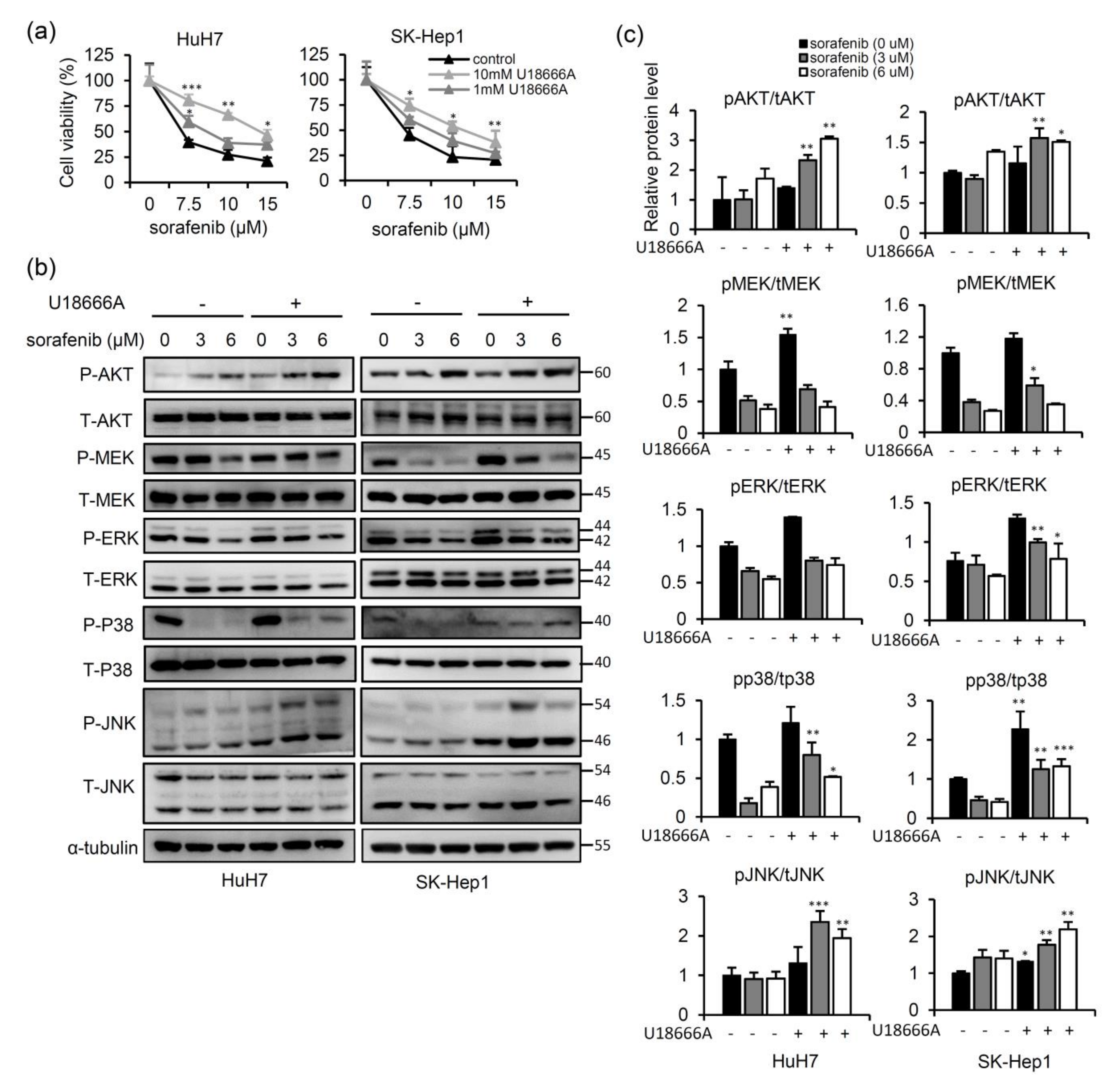
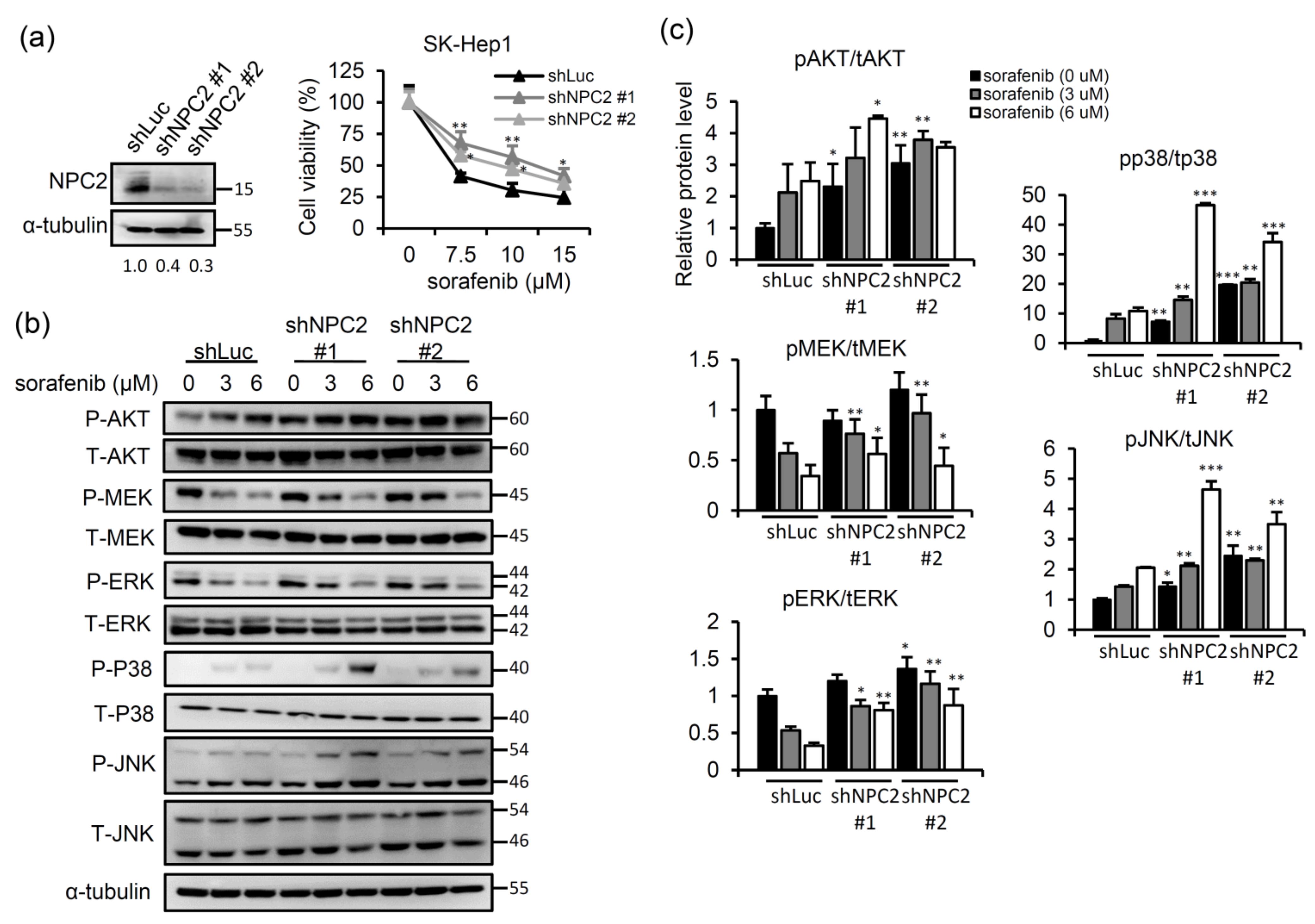
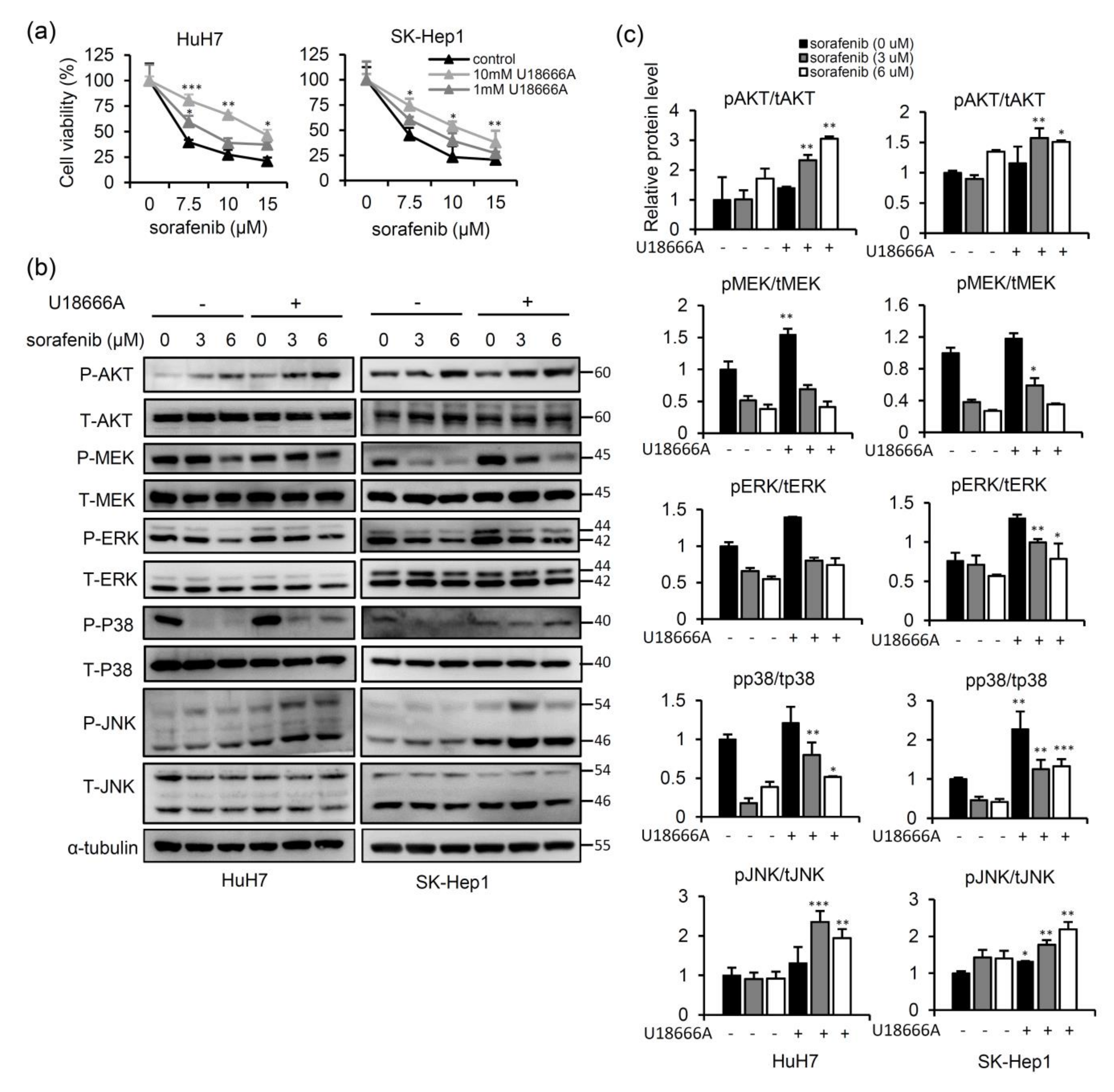
2.1. NPC2 Downregulation Facilitates Free Cholesterol Accumulation which Weakened Sorafenib Efficacy through Enhancing MAPK/AKT Signaling in HCC Cells
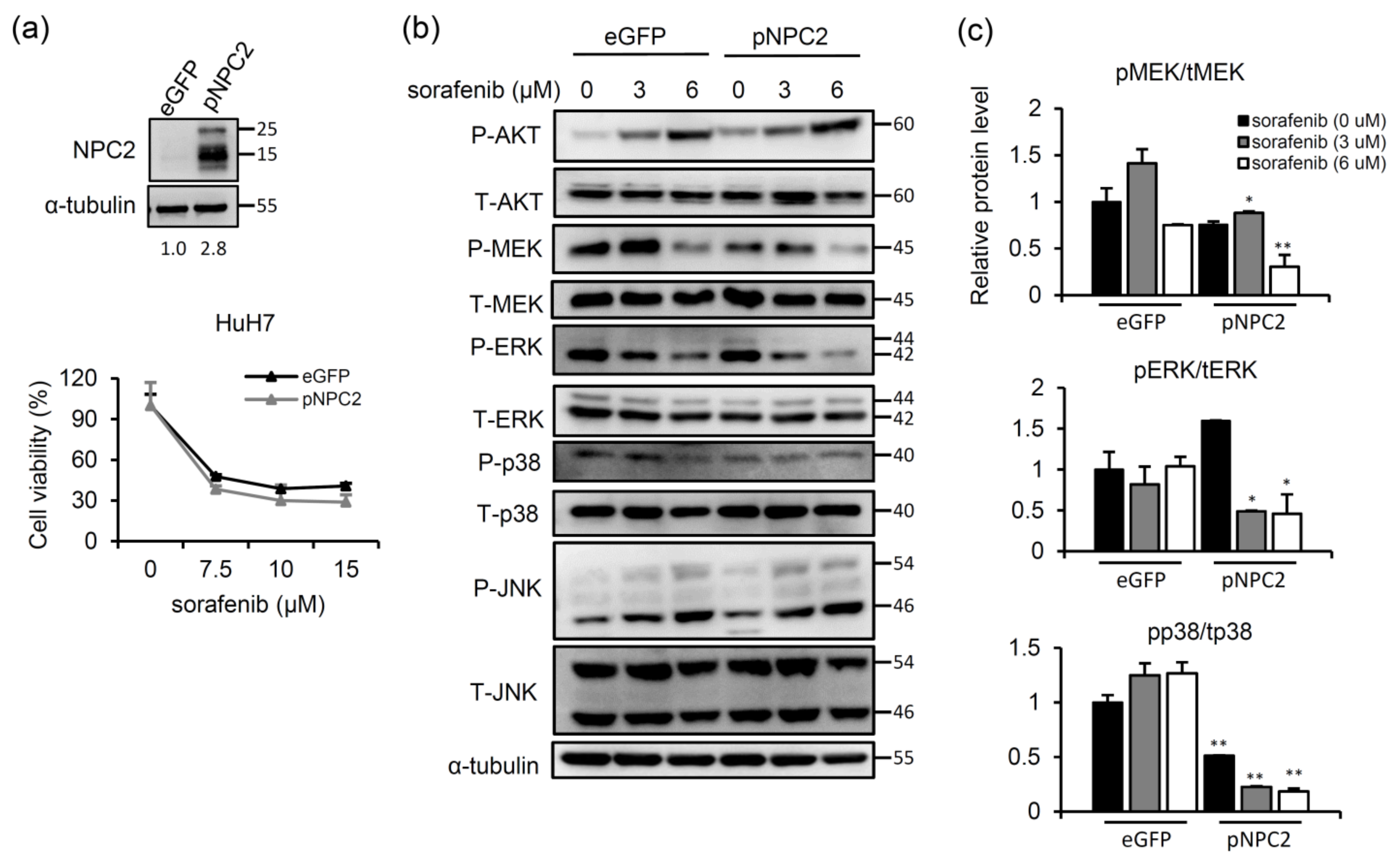
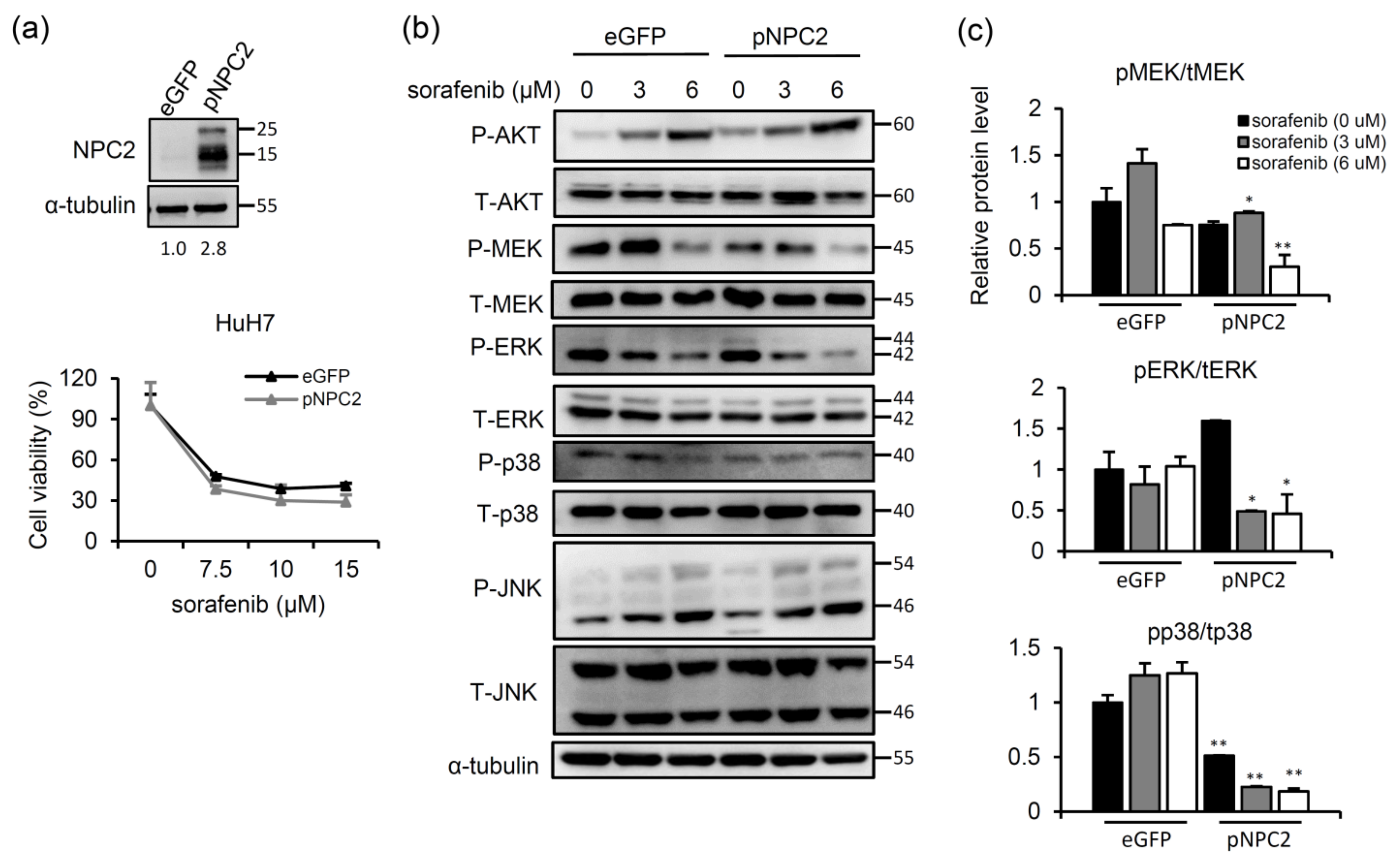
2.2. NPC2 Overexpression Inhibits MAPK/ERK Signaling and Slightly Enhances Sorafenib-Induced Cytotoxicity
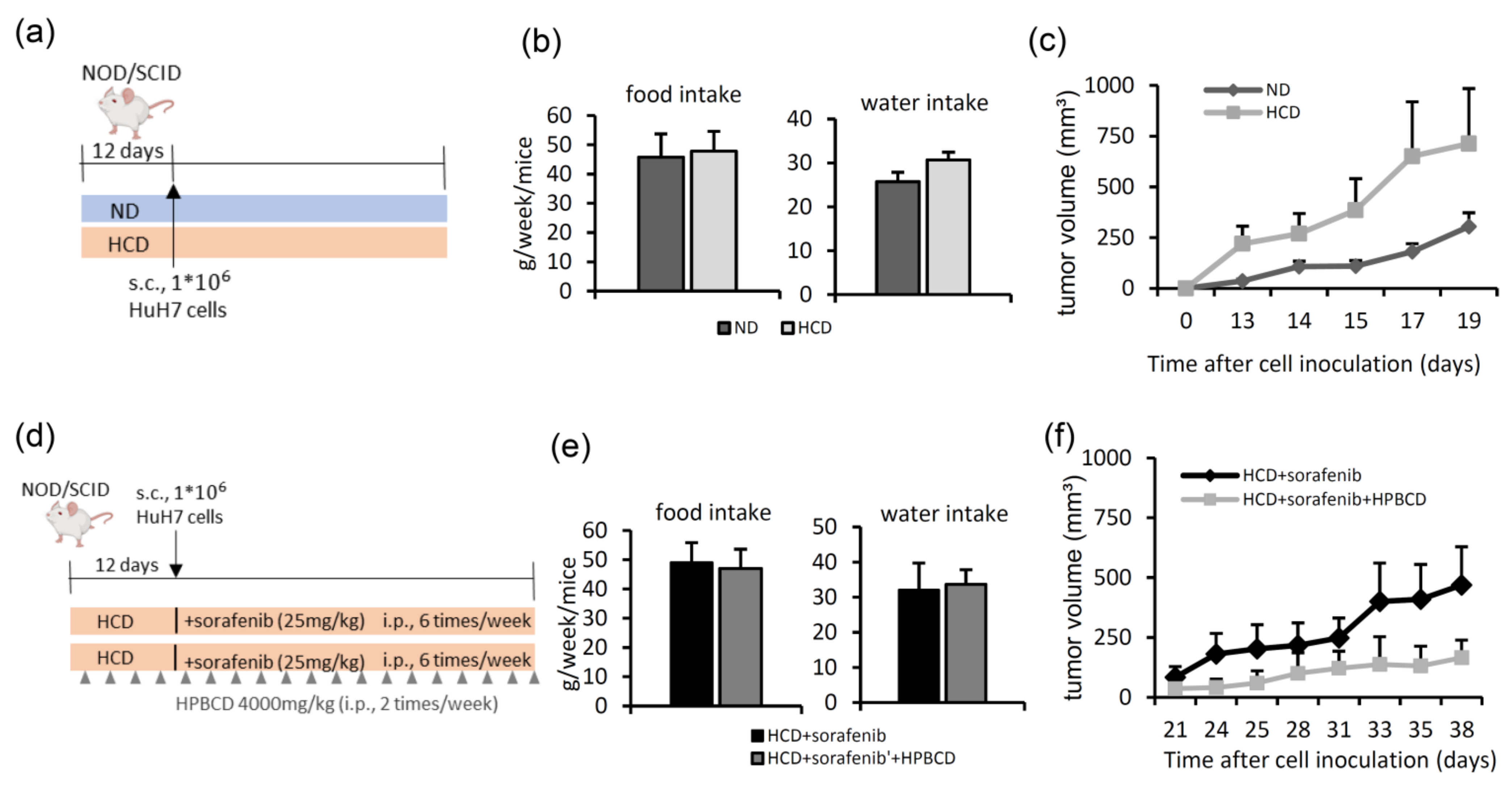
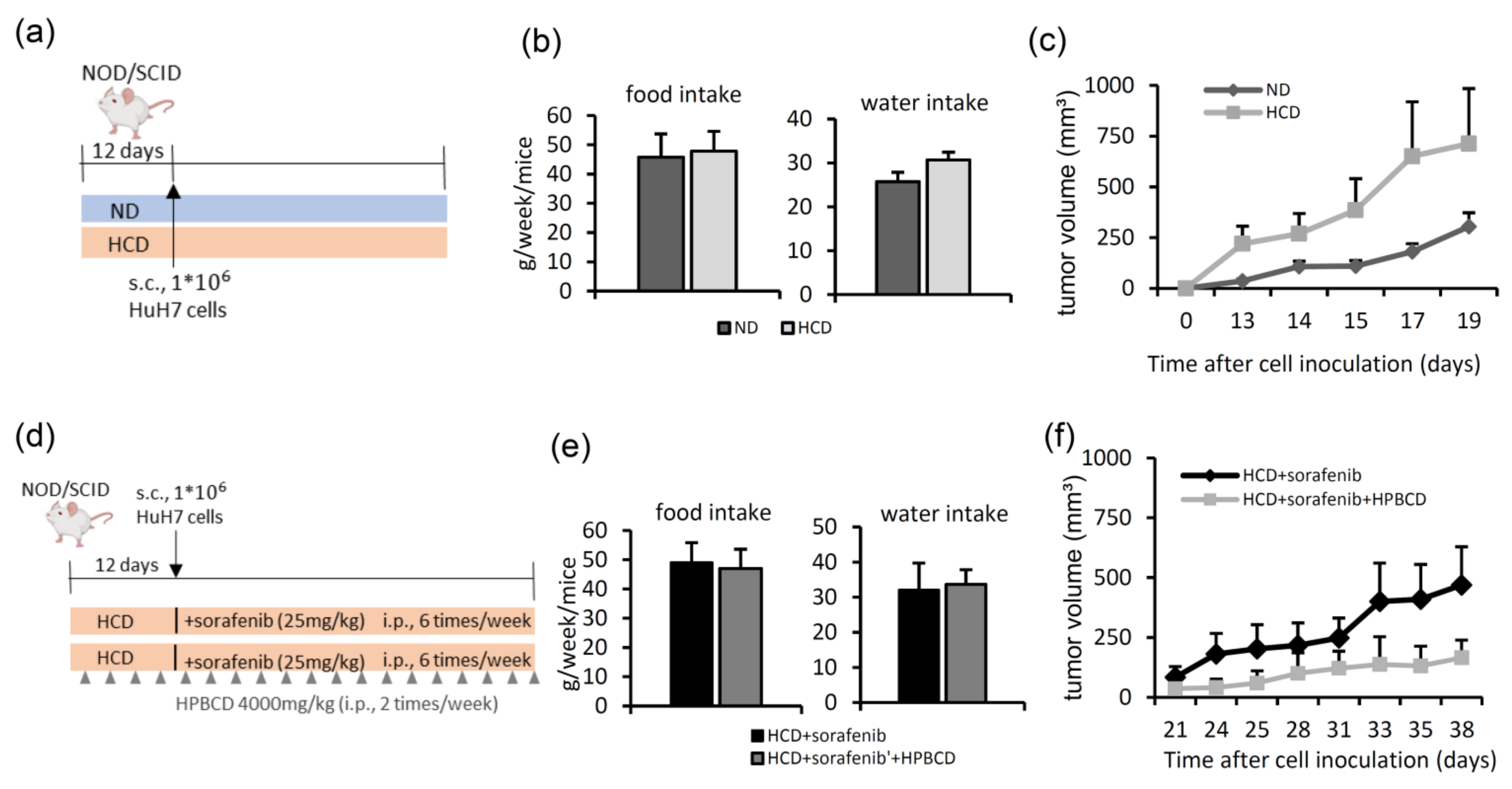
2.3. Treatment with a Free Cholesterol-Lowering Agent Improves Sorafenib’s Ability to Inhibit Tumors in High-Cholesterol Diet-Fed Groups
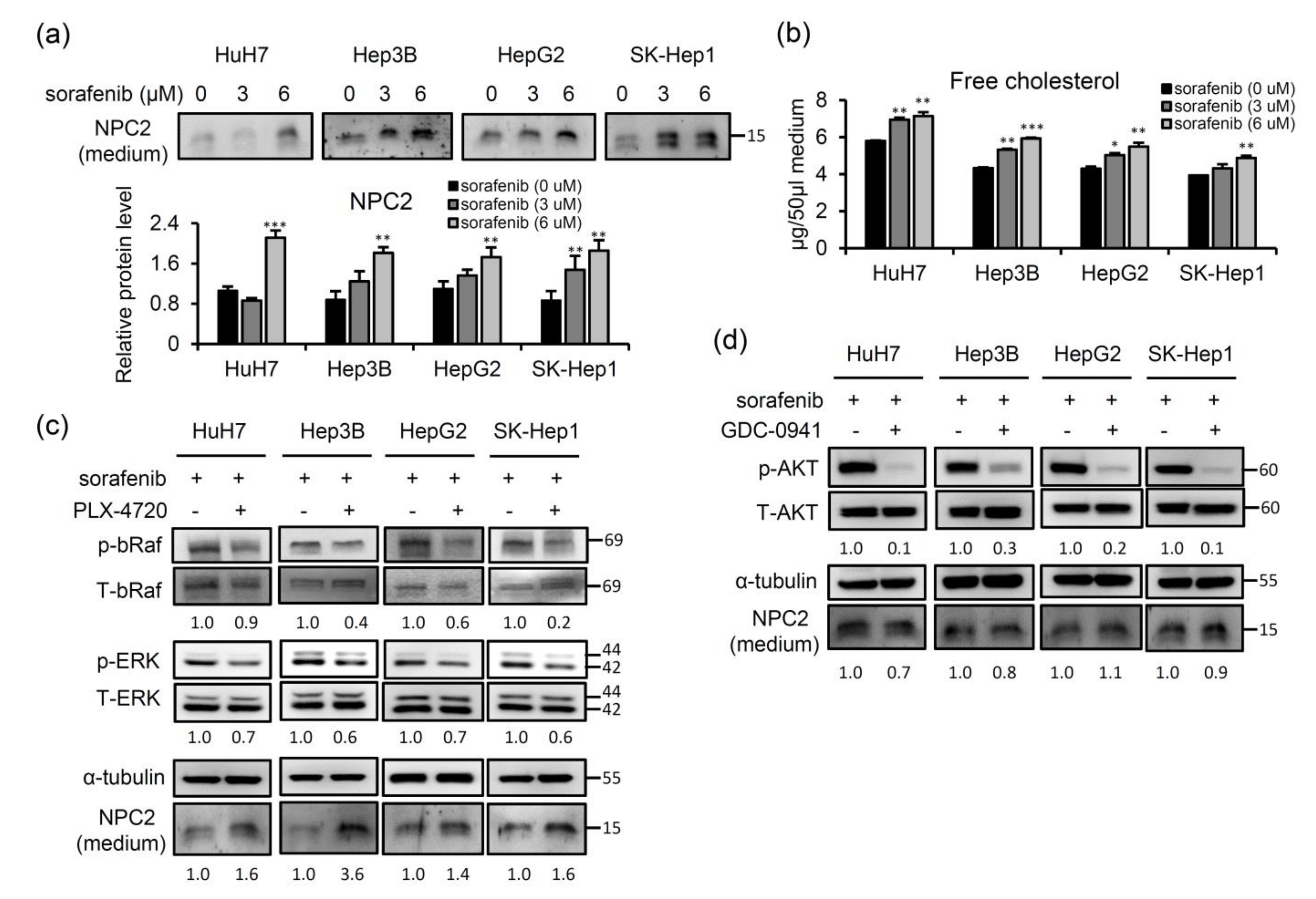
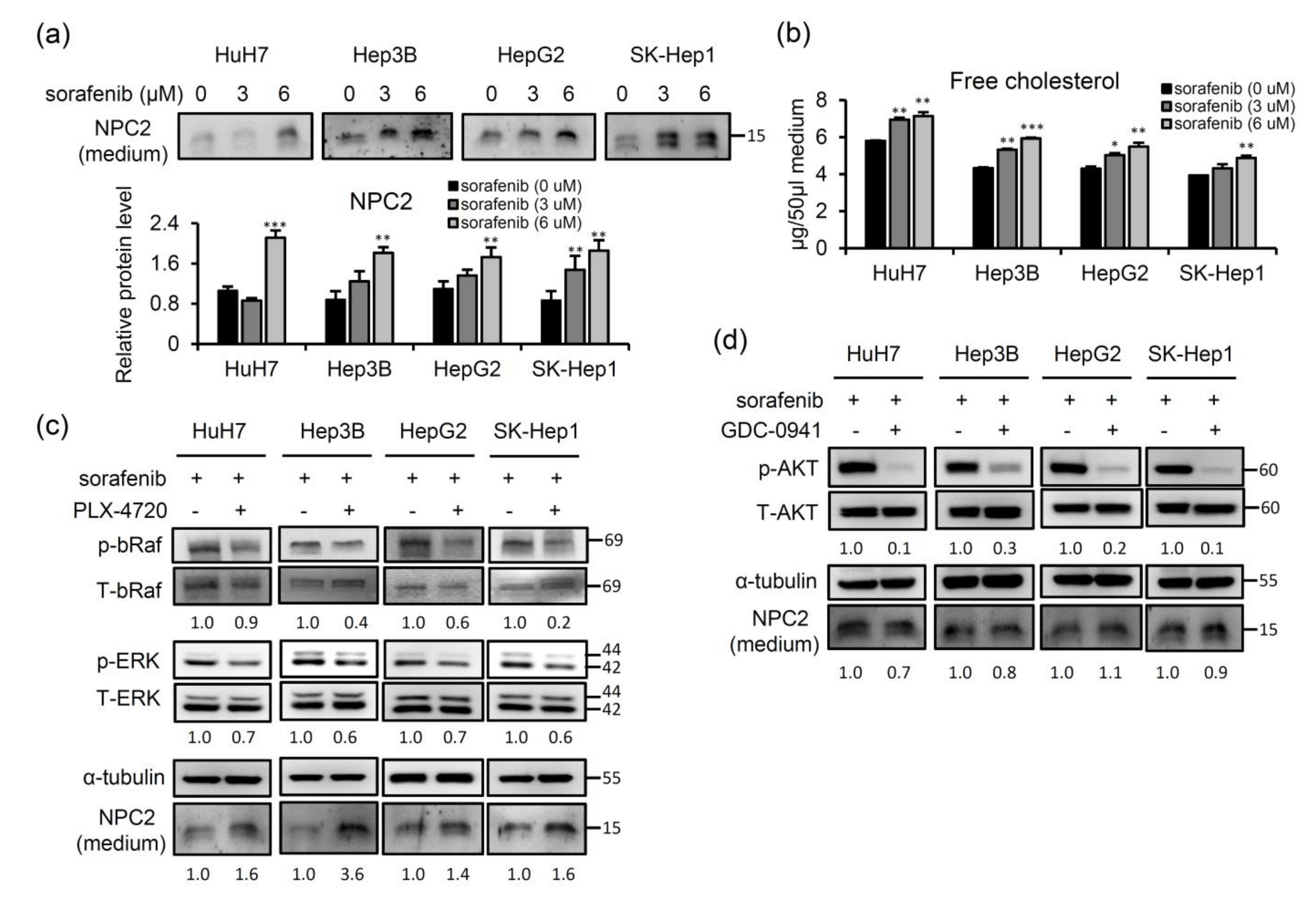
2.4. Sorafenib-Inhibited Raf Signaling Pathway Promotes Secretion of NPC2 and Free Cholesterol in Cell Culture Supernatant
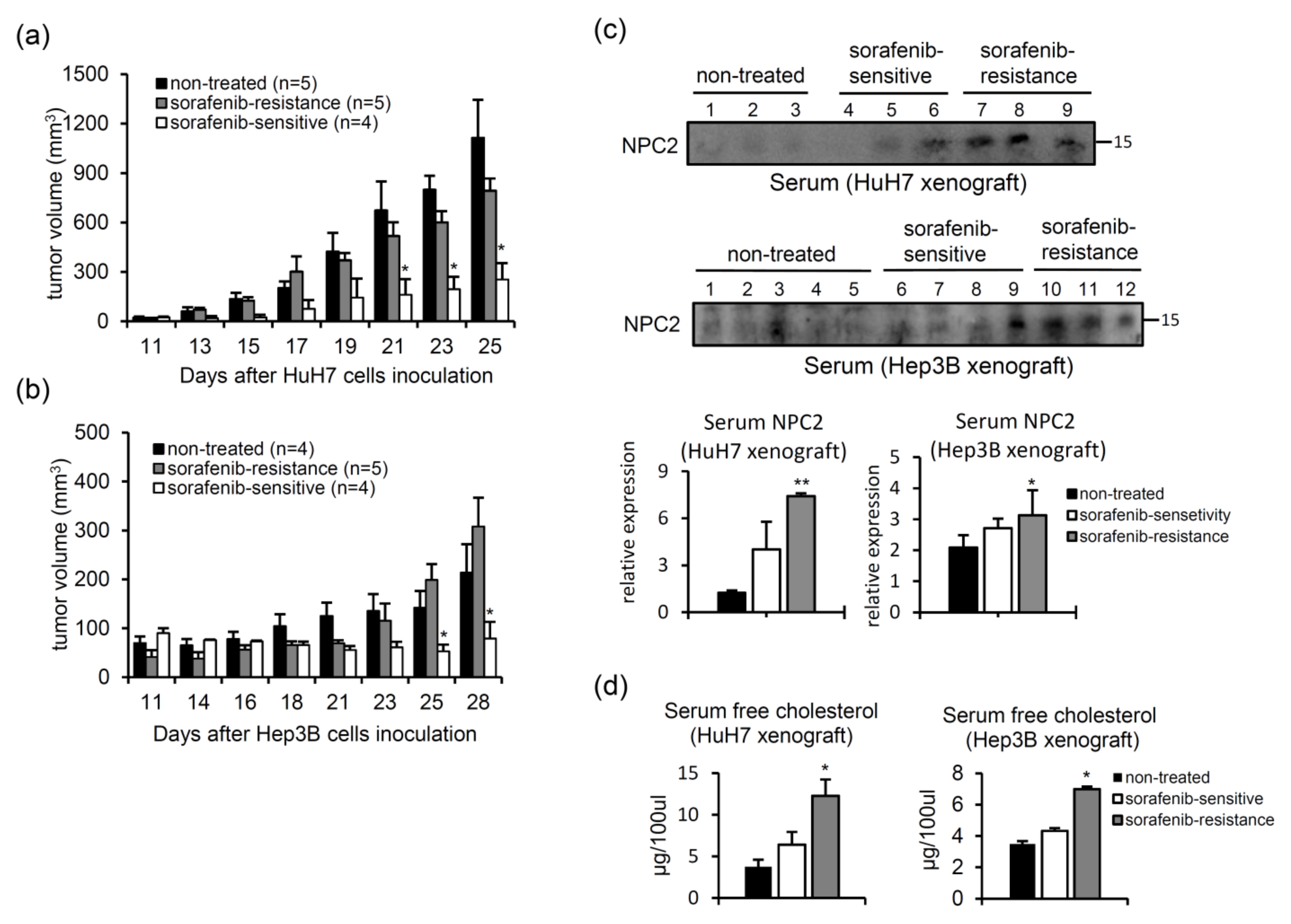
2.5. NPC2 and Free Cholesterol Levels Increase in the Culture Supernatant of Cells with Sorafenib Treatment-Induced Acquisition of Sorafenib Resistance
2.6. Serum NPC2 and Free Cholesterol Levels Increase in Xenografts with Acquired Sorafenib Resistance
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Generation of Sorafenib-Resistant (SR) Cells
4.3. Western Blot Experiments
4.4. Cell Viability Assay
4.5. Free Cholesterol Quantification
4.6. Animal Experiments
- (A)
- In the first HCC xenograft model, we compared the tumor growth rates between animals fed a normal diet and those fed a 2% high-cholesterol diet (Table 1). Six–seven-week-old female NOD/SCID mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan). Mice were randomly divided into two groups: a normal diet and a high-cholesterol diet for pre-administration for 12 days, after which HuH7 cells (106) were subcutaneously inoculated into each mouse (Figure 4a). The tumor volume was measured three times per week using Vernier calipers.
- (B)
- Next, we studied the effects of a high-cholesterol diet plus a free cholesterol-lowering drug in a sorafenib-treated HCC xenograft model (Figure 4d). After 12 days of feeding mice a high-cholesterol diet, HuH7 cells (106) were subcutaneously injected into NOD/SCID mice. Then, the mice were divided into two groups: (1) sorafenib treatment (intraperitoneal injection 25 mg/kg, six times per week) and (2) sorafenib (intraperitoneal injection 25 mg/kg, six days per week) combined with 2-hydroxypropyl-β-cyclodextrin (HPBCD) treatment (intraperitoneal injection 4000 mg/kg, twice weekly, Sigma-Aldrich). HPBCD is currently in phase 2 clinical trials for evaluation of its effectiveness in treating NPC2 deficit-associated free cholesterol accumulation in Niemann-Pick type C disease [27,56,57]. The tumor volume was measured three times per week using Vernier calipers. Water intake and food intake were measured twice a week.
- (C)
- Xenograft HCC model of acquired sorafenib resistance. HuH7 and Hep3B cells (106) were subcutaneously injected into NOD/SCID mice, and sorafenib (25 mg/kg) was intraperitoneally injected every day. The tumor volume was measured three times per week using Vernier calipers. After 25 (HuH7) or 28 days (Hep3B), animals were sacrificed to collect tumor tissues and serum. Serum samples (100 μg) were subjected to a western blot analysis.
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, S.; Ward, J.W.; Watson, M.; Momin, B.; Richardson, L.C. Centers for Disease Control and Prevention. Hepatocellular carcinoma—United States, 2001−2006. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 517–520. [Google Scholar]
- Balogh, J.; Victor, D., 3rd; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Sergio Ricci, M.D.; Vincenzo Mazzaferro, M.D.; Philip Hilgard, M.D.; Edward Gane, M.D.; Jean-Frédéric Blanc, M.D.; Andre Cosme de Oliveira, M.D.; Armando Santoro, M.D.; Jean-Luc Raoul, M.D.; Alejandro Forner, M.D.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Bertot, L.C.; Sato, M.; Tateishi, R.; Yoshida, H.; Koike, K. Mortality and complication rates of percutaneous ablative techniques for the treatment of liver tumors: A systematic review. Eur. Radiol. 2011, 21, 2584–2596. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Ben Mousa, A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi. J. Gastroenterol. 2008, 14, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal. Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Trapani, L.; Segatto, M.; Pallottini, V.M. Regulation and deregulation of cholesterol homeostasis: The liver as a metabolic “power station”. World J. Hepatol. 2012, 4, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Q.; Teoh, N.; Xu, L.X.; Pok, S.; Li, X.C.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [Green Version]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basanez, G.; Antonsson, B.; Prieto, J.; Garcia-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer. Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.H.; Yang, Z.S.; Li, M.; Chen, Y.; Zhao, X.F.; Qin, Y.Y.; Song, J.Q.; Wang, B.B.; Yuan, B.; Cui, X.L.; et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology 2020, 158, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Qin, W.; Chen, Y.; Yuan, B.; Song, X.; Wang, B.; Shen, F.; Fu, J.; Wang, H. Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts. Cancer. Lett. 2018, 429, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, Y.M.; Oh, T.I.; Shin, D.H.; Kim, G.H.; Kan, S.Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kan, S.Y.; Kang, H.; Lee, S.; Ko, H.M.; Kim, J.H.; Lim, J.H. Ursolic Acid Suppresses Cholesterol Biosynthesis and Exerts Anti-Cancer Effects in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. U S A 2004, 101, 5886–5891. [Google Scholar] [CrossRef] [Green Version]
- Storch, J.; Xu, Z. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta 2009, 1791, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Maxfield, F.R. Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta 2004, 1685, 28–37. [Google Scholar] [CrossRef]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J Mol. Sci. 2016, 17, 1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Twu, Y.C.; Wang, C.K.; Lin, F.Z.; Lee, C.Y.; Liao, Y.J. Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function. Int. J. Mol. Sci. 2018, 19, 1678. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.J.; Fang, C.C.; Yen, C.H.; Hsu, S.M.; Wang, C.K.; Huang, S.F.; Liang, Y.C.; Lin, Y.Y.; Chu, Y.T.; Arthur Chen, Y.M. Niemann-Pick type C2 protein regulates liver cancer progression via modulating ERK1/2 pathway: Clinicopathological correlations and therapeutical implications. Int. J. Cancer 2015, 137, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Chen, T.L.; Lee, T.S.; Wang, H.A.; Wang, C.K.; Liao, L.Y.; Liu, R.S.; Huang, S.F.; Chen, Y.M. Glycine N-methyltransferase deficiency affects Niemann-Pick type C2 protein stability and regulates hepatic cholesterol homeostasis. Mol. Med. 2012, 18, 412–422. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [Green Version]
- Cenedella, R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009, 44, 477–487. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.T.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1-2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.; Amigo, L.; Retamal, M.J.; Morales, M.G.; Miquel, J.F.; Rigotti, A.; Zanlungo, S. NPC2 is expressed in human and murine liver and secreted into bile: Potential implications for body cholesterol homeostasis. Hepatology 2006, 43, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cholesterol metabolism and cancer: The good, the bad and the ugly. Curr. Opin. Pharmacol. 2012, 12, 673–676. [Google Scholar] [CrossRef]
- Esau, L.; Sagar, S.; Bangarusamy, D.; Kaur, M. Identification of CETP as a molecular target for estrogen positive breast cancer cell death by cholesterol depleting agents. Genes. Cancer 2016, 7, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Saha, S.T.; Thomas, J.; Kaur, M. Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019, 286, 4192–4208. [Google Scholar] [CrossRef]
- Mohammad, N.; Malvi, P.; Meena, A.S.; Singh, S.V.; Chaube, B.; Vannuruswamy, G.; Kulkarni, M.J.; Bhat, M.K. Cholesterol depletion by methyl-beta-cyclodextrin augments tamoxifen induced cell death by enhancing its uptake in melanoma. Mol. Cancer 2014, 13, 204. [Google Scholar] [CrossRef] [Green Version]
- Haralampiev, I.; Scheidt, H.A.; Abel, T.; Luckner, M.; Herrmann, A.; Huster, D.; Muller, P. The interaction of sorafenib and regorafenib with membranes is modulated by their lipid composition. Biochim. Biophys. Acta 2016, 1858, 2871–2881. [Google Scholar] [CrossRef]
- Jian, C.; Fu, J.; Cheng, X.; Shen, L.J.; Ji, Y.X.; Wang, X.; Pan, S.; Tian, H.; Tian, S.; Liao, R.; et al. Low-Dose Sorafenib Acts as a Mitochondrial Uncoupler and Ameliorates Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 1206. [Google Scholar] [CrossRef]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Personeni, N.; Bozzarelli, S.; Pressiani, T.; Rimassa, L.; Tronconi, M.C.; Sclafani, F.; Carnaghi, C.; Pedicini, V.; Giordano, L.; Santoro, A. Usefulness of alpha-fetoprotein response in patients treated with sorafenib for advanced hepatocellular carcinoma. J. Hepatol. 2012, 57, 101–107. [Google Scholar] [CrossRef]
- Saito, K.; Ikeda, M.; Kojima, Y.; Hosoi, H.; Saito, Y.; Kondo, S. Lipid profiling of pre-treatment plasma reveals biomarker candidates associated with response rates and hand-foot skin reactions in sorafenib-treated patients. Cancer Chemoth. Pharm. 2018, 82, 677–684. [Google Scholar] [CrossRef]
- Takaki, S.; Fukuhara, T.; Mori, N.; Tsuji, K. High cholinesterase predicts tolerance to sorafenib treatment and improved prognosis in patients with transarterial chemoembolization refractory intermediate stage hepatocellular carcinoma. Mol. Clin. Oncol. 2020, 12, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Dietschy, J.M.; Turley, D.; Spady, D.K. Role of Liver in the Maintenance of Cholesterol and Low-Density-Lipoprotein Homeostasis in Different Animal Species, Including Humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, L.; Zuo, H.; Le, W.; Hu, J.; Zhang, T.; Li, M.; Yuan, Y. Overriding sorafenib resistance via blocking lipid metabolism and Ras by sphingomyelin synthase 1 inhibition in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2021, 87, 217–228. [Google Scholar] [CrossRef]
- Kim, M.J.; Choi, Y.K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.G.; Jeon, H.J.; Kim, J.H.; Kim, J.G.; et al. PPAR delta Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Sun, M.; Dwyer, N.K.; Comly, M.E.; Patel, S.C.; Sundaram, R.; Hanover, J.A.; Blanchette-Mackie, E.J. Differential trafficking of the Niemann-Pick C1 and 2 proteins highlights distinct roles in late endocytic lipid trafficking. Acta Paediatr. Suppl. 2003, 92, 63–73, discussion 45. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parchure, A.; Vyas, N.; Mayor, S. Wnt and Hedgehog: Secretion of Lipid-Modified Morphogens. Trends Cell Biol. 2018, 28, 157–170. [Google Scholar] [CrossRef]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Liao, Y.-J.; Lin, M.-W.; Yen, C.-H.; Lin, Y.-T.; Wang, C.-K.; Huang, S.-F.; Chen, K.-H.; Yang, C.-P.; Chen, T.-L.; Hou, M.-F.; et al. Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 2017, 26, 2277–2289. [Google Scholar] [CrossRef] [PubMed]
- Formichi, P.; Battisti, C.; De Santi, M.M.; Guazzo, R.; Tripodi, S.A.; Radi, E.; Rossi, B.; Tarquini, E.; Federico, A. Primary cilium alterations and expression changes of Patched1 proteins in niemann-pick type C disease. J. Cell Physiol. 2018, 233, 663–672. [Google Scholar] [CrossRef]
- Incardona, J.P.; Gaffield, W.; Lange, Y.; Cooney, A.; Pentchev, P.G.; Liu, S.; Watson, J.A.; Kapur, R.P.; Roelink, H. Cyclopamine inhibition of Sonic hedgehog signal transduction is not mediated through effects on cholesterol transport. Dev. Biol. 2000, 224, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Canterini, S.; Dragotto, J.; Dardis, A.; Zampieri, S.; De Stefano, M.E.; Mangia, F.; Erickson, R.P.; Fiorenza, M.T. Characterization of Niemann-Pick Type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS ONE 2013, 8, e77586. [Google Scholar] [CrossRef] [PubMed]
- Ebner, L.; Glaser, A.; Brauer, A.; Witt, M.; Wree, A.; Rolfs, A.; Frank, M.; Vollmar, B.; Kuhla, A. Evaluation of Two Liver Treatment Strategies in a Mouse Model of Niemann-Pick-Disease Type C1. Int. J. Mol. Sci. 2018, 19, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, S.; Scott, R.C. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): A toxicology review. Food Chem. Toxicol. 2005, 43, 1451–1459. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | Normal Diet g/kg | High Cholesterol Diet g/kg |
|---|---|---|
| Cornstarch | 465 | 440 |
| Maltodextrin | 155 | 155 |
| Sucrose | 100 | 100 |
| Casein | 140 | 140 |
| L-Cysteine | 2 | 2 |
| Fresh soybean oil | 40 | 40 |
| Cellulose | 50 | 50 |
| Mineral mix (AIN-93M-MI) | 35 | 35 |
| Vitamin mix (AIN-93-VX) | 10 | 10 |
| Choline bitartrate | 3 | 3 |
| Cholesterol | 20 | |
| Sodium cholate | 5 | |
| Total | 1000 | 1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suk, F.-M.; Wang, Y.-H.; Chiu, W.-C.; Liu, C.-F.; Wu, C.-Y.; Chen, T.-L.; Liao, Y.-J. Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 8567. https://doi.org/10.3390/ijms22168567
Suk F-M, Wang Y-H, Chiu W-C, Liu C-F, Wu C-Y, Chen T-L, Liao Y-J. Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(16):8567. https://doi.org/10.3390/ijms22168567
Chicago/Turabian StyleSuk, Fat-Moon, Yuan-Hsi Wang, Wan-Chun Chiu, Chiao-Fan Liu, Chien-Ying Wu, Tzu-Lang Chen, and Yi-Jen Liao. 2021. "Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 16: 8567. https://doi.org/10.3390/ijms22168567
APA StyleSuk, F.-M., Wang, Y.-H., Chiu, W.-C., Liu, C.-F., Wu, C.-Y., Chen, T.-L., & Liao, Y.-J. (2021). Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(16), 8567. https://doi.org/10.3390/ijms22168567