
Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca2+ Homeostasis

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
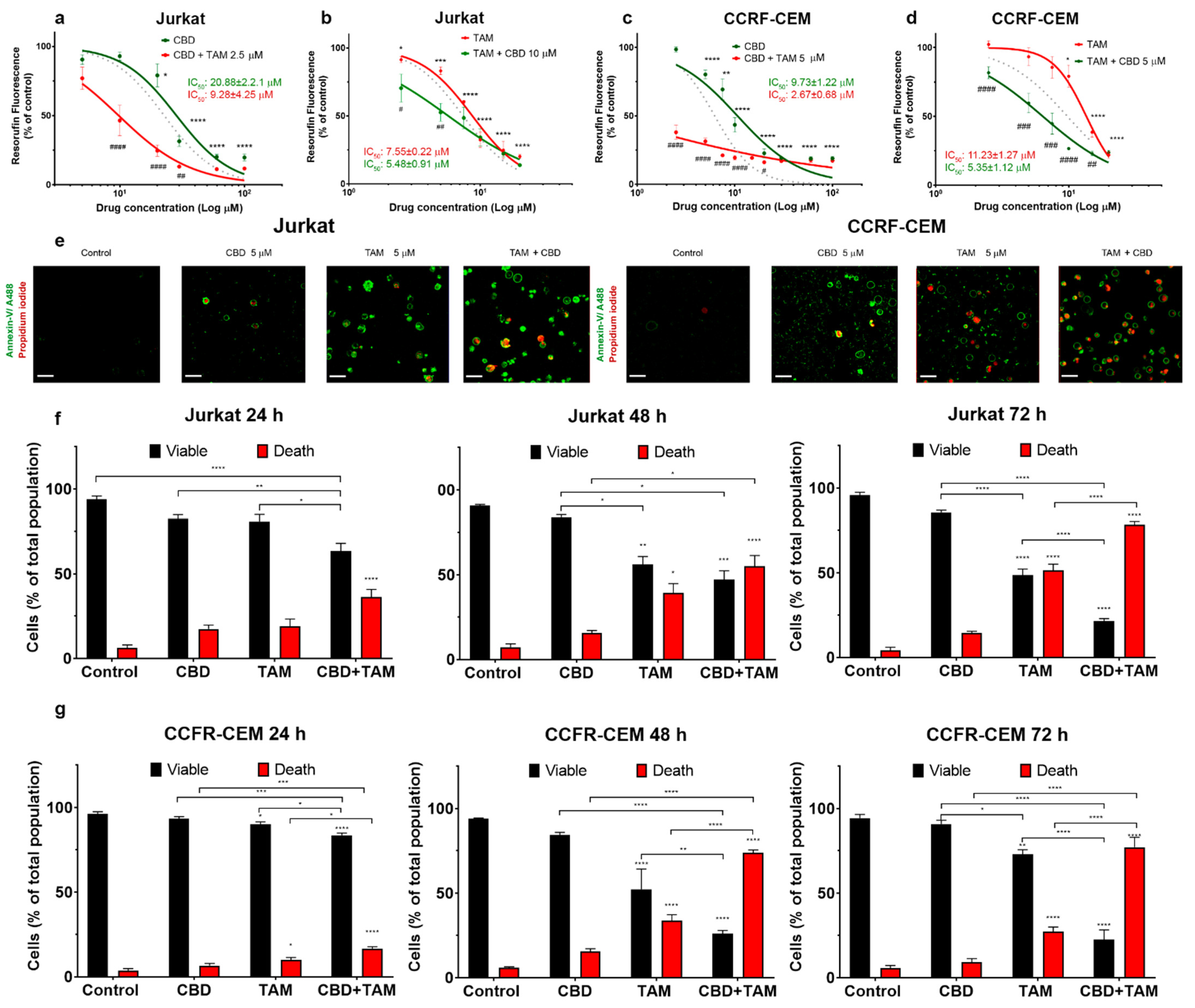
2.1. CBD and TAM Act Synergistically to Decrease T-ALL Viability
2.2. CBD and TAM Induce Cell Death
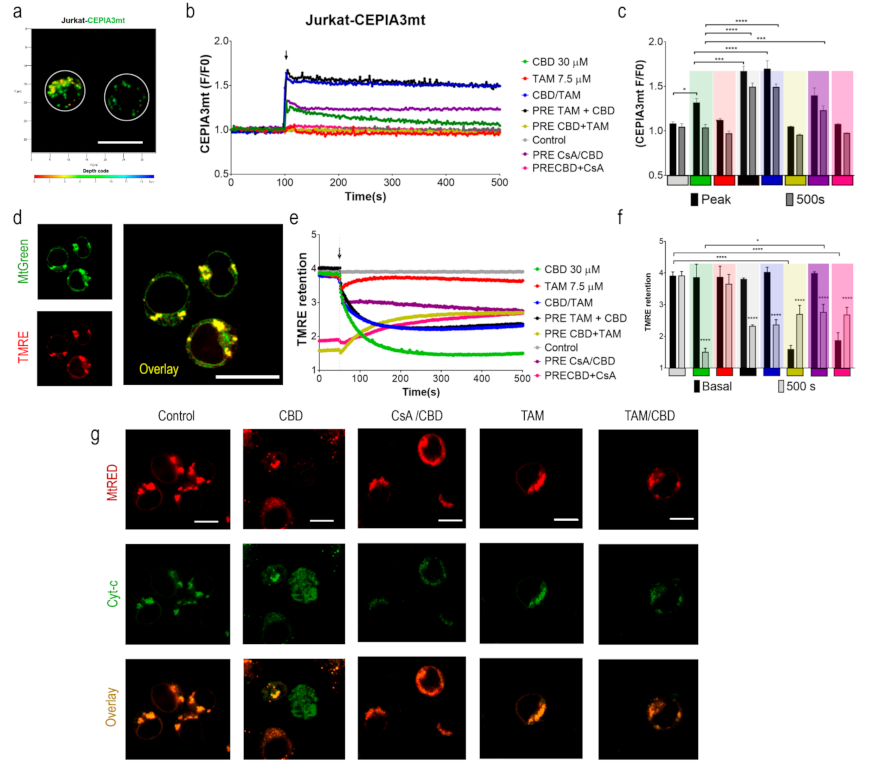
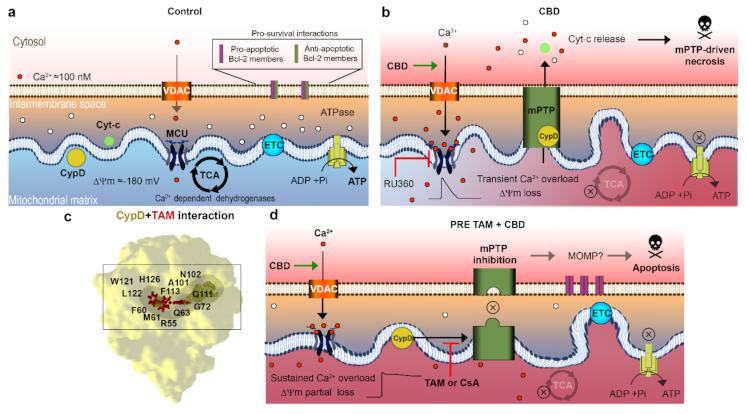
2.3. TAM Modifies the CBD Effect on Mitochondria
2.4. TAM Limits the mPTP-Mediated Cyt-c Release
2.5. TAM Can Interact with CypD to Inhibit the mPTP: In Silico Evidence
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Culture Conditions
4.3. Viability Assay
4.4. Cell Death Analysis
4.5. Leukemic Cell Transfection with CEPIA3mt or EYFP-Cyt-c
4.6. Mitochondrial Ca2+ Measurements
4.7. Evaluation of the Mitochondrial Membrane Potential
4.8. In Silico Protein-Ligand Interaction
4.9. Cyt-c Release Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphoblastic Leukemia |
| CBD | Cannabidiol |
| CsABD | Cyclosporine A Binding Domain |
| CypD | Cyclophilin D |
| Cyt-c | Cytochrome c |
| mPTP | Mitochondrial Permeability Transition Pore |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| TAM | Tamoxifen |
| [Ca2+]m | Mitochondrial matrix free calcium concentration |
| ΔΨm | Mitochondrial inner membrane potential |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Pui, C.H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef]
- Aster, J.C.; DeAngelo, D.J. Resistance revealed in acute lymphoblastic leukemia. Nat. Med. 2013, 19, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Masselli, M.; De Lorenzo, E.; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D’Amico, M.; Basso, G.; et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood 2011, 117, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Follini, E.; Marchesini, M.; Roti, G. Strategies to overcome resistance mechanisms in T-cell acute lymphoblastic leukemia. Int. J. Mol. Sci. 2019, 20, 3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Overcoming glucocorticoid resistance in acute lymphoblastic leukemia: Repurposed drugs can improve the protocol. Front. Oncol. 2021, 11, 647. [Google Scholar] [CrossRef] [PubMed]
- Lato, M.W.; Przysucha, A.; Grosman, S.; Zawitkowska, J.; Lejman, M. The new therapeutic strategies in pediatric T-cell acute lymphoblastic leukemia. Int. J. Mol. Sci. 2021, 22, 4502. [Google Scholar] [CrossRef]
- Lee, J.B.; Vasic, D.; Kang, H.; Fang, K.K.; Zhang, L. State-of-art of cellular therapy for acute leukemia. Int. J. Mol. Sci. 2021, 22, 4590. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges, and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Szaflarski, J.P.; Bebin, E.M.; Comi, A.M.; Patel, A.D.; Joshi, C.; Checketts, D.; Beal, J.C.; Laux, L.C.; De Boer, L.M.; Wong, M.H.; et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia 2018, 59, 1540–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.T.; Szaflarski, J.P. The US food and drug administration’s authorization of the first cannabis-derived pharmaceutical: Are we out of the haze? JAMA Neurol. 2019, 76, 135–136. [Google Scholar] [CrossRef]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalenderoglou, N.; Macpherson, T.; Wright, K.L. Cannabidiol reduces leukemic cell size–but is it important? Front. Pharmacol. 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a promising anti-cancer drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as potential anticancer drug. Br. J. Clin. Pharmacol. 2013, 75, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, E.; Mukhopadhyay, P.; Cao, Z.; Erdélyi, K.; Holovac, E.; Liaudet, L.; Lee, W.S.; Haskó, G.; Mechoulam, R.; Pacher, P. Cannabidiol protects against doxorubicin-induced cardiomyopathy by modulating mitochondrial function and biogenesis. Mol. Med. 2015, 21, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.Y.; Kim, S.R.; Kim, D.Y.; Chae, S.W.; Song, J.J. Cannabidiol enhances cytotoxicity of anti-cancer drugs in human head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Ferlini, C.; Scambia, G.; Distefano, M.; Filippini, P.; Isola, G.; Riva, A.; Bombardelli, E.; Fattorossi, A.; Benedetti Panici, P.; Mancuso, S. Synergistic antiproliferative activity of tamoxifen and docetaxel on three oestrogen receptor-negative cancer cell lines is mediated by the induction of apoptosis. Br. J. Cancer 1997, 75, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Hayon, T.; Atlas, L.; Levy, E.; Dvilansky, A.; Shpilberg, O.; Nathan, I. Multifactorial activities of nonsteroidal antiestrogens against leukemia. Cancer Detect. Prev. 2003, 27, 389–396. [Google Scholar] [CrossRef]
- Nagahara, Y.; Shiina, I.; Nakata, K.; Sasaki, A.; Miyamoto, T.; Ikekita, M. Induction of mitochondria-involved apoptosis in estrogen receptor-negative cells by a novel tamoxifen derivative, ridaifen-B. Cancer Sci. 2008, 99, 608–614. [Google Scholar] [CrossRef]
- Adachi, K.; Honma, Y.; Miyake, T.; Kawakami, K.; Takahashi, T.; Suzumiya, J. Tamoxifen enhances the differentiation-inducing and growth-inhibitory effects of all-trans retinoic acid in acute promyelocytic leukemia cells. Int. J. Oncol. 2016, 48, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-López, L.; Maycotte, P.; Liñán-Rico, A.; Liñán-Rico, L.; Donis-Maturano, L.; Delgado-Enciso, I.; Meza-Robles, C.; Vásquez-Jiménez, C.; Hernández-Cruz, A.; Dobrovinskaya, O. Tamoxifen induces toxicity, causes autophagy, and partially reverses dexamethasone resistance in Jurkat T cells. J. Leukoc. Biol. 2019, 105, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Tan, S.F.; Feith, D.J.; Kester, M.; Claxton, D.F.; Loughran, T.P., Jr.; Barth, B.M.; Fox, T.E.; Cabot, M.C. Modification of sphingolipid metabolism by tamoxifen and N-desmethyltamoxifen in acute myelogenous leukemia--Impact on enzyme activity and response to cytotoxics. Biochim. Biophys. Acta 2015, 1851, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Wanitpongpun, C.; Honma, Y.; Okada, T.; Suzuki, R.; Takeshi, U.; Suzumiya, J. Tamoxifen enhances romidepsin-induced apoptosis in T-cell malignant cells via activation of FOXO1 signaling pathway. Leuk. Lymphoma 2021, 28, 1–15. [Google Scholar] [CrossRef]
- de Almeida, D.L.; Devi, L.A. Diversity of molecular targets and signaling pathways for CBD. Pharmacol. Res. Perspect. 2020, 8, e00682. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Zenebe, W.J.; Parihar, A.; Larson, S.K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007, 67, 1282–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custodio, J.B.; Moreno, A.J.; Wallace, K.B. Tamoxifen inhibits induction of the mitochondrial permeability transition by Ca2+ and inorganic phosphate. Toxicol. Appl. Pharmacol. 1998, 152, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Couldwell, W.T.; Song, H.; Takano, T.; Lin, J.H.; Nedergaard, M. Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells. Cancer Res. 2000, 60, 5395–5400. [Google Scholar] [PubMed]
- Ribeiro, M.P.; Santos, A.E.; Custódio, J.B. Mitochondria: The gateway for tamoxifen-induced liver injury. Toxicology 2014, 323, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Phenolic compounds cannabidiol, curcumin and quercetin cause mitochondrial dysfunction and suppress acute lymphoblastic leukemia cells. Int. J. Mol. Sci. 2020, 22, 204. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-cancer agents in proliferation and cell death: The calcium connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1, 4, 5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef]
- Javadov, S.; Kuznetsov, A. Mitochondrial permeability transition and cell death: The role of cyclophilin D. Front. Physiol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Grädler, U.; Schwarz, D.; Blaesse, M.; Leuthner, B.; Johnson, T.L.; Bernard, F.; Jiang, X.; Marx, A.; Gilardone, M.; Lemoine, H.; et al. Discovery of novel Cyclophilin D inhibitors starting from three dimensional fragments with millimolar potencies. Bioorg. Med. Chem. Lett. 2019, 29, 126717. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Londhe, A.M.; Lim, J.W.; Park, B.G.; Jung, S.Y.; Lee, J.Y.; Lim, S.M.; No, K.T.; Lee, J.; Pae, A.N. Discovery of non-peptidic small molecule inhibitors of cyclophilin D as neuroprotective agents in Aβ-induced mitochondrial dysfunction. J. Comput. Aided Mol. Des. 2017, 31, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003, 10, 1485–1506. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Acosta, D., Jr. Mitochondrial Ca2+ overload in primary cultures of rat renal cortical epithelial cells by cytotoxic concentrations of cyclosporine: A digitized fluorescence imaging study. Toxicology 1995, 95, 155–166. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, S.I.; Jeung, E.B.; Yoo, Y.M. Cyclosporine A induces apoptotic and autophagic cell death in rat pituitary GH3 cells. PLoS ONE 2014, 9, e108981. [Google Scholar] [CrossRef]
- Porter, G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.; Wang, L.; Zhang, X.; Fu, Y.; Liu, Y.; Chen, W.; Liu, W.; Shi, Z.; Yin, X. Cyclosporin A induces autophagy in cardiac fibroblasts through the NRP-2/WDFY-1 axis. Biochimie 2018, 148, 55–62. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Gabrusiewicz, K.; Szczepankiewicz, A.A.; Kaminska, B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene 2013, 32, 1518–1529. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, S.J.; Tamura, J.; Kincaid, R.L.; Tocci, M.J.; O’Neill, E.A. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature 1992, 357, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Koyasu, S. Mechanisms of action of cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, Y.; Jaiseng, W.; Hu, L.; Lu, Y.; Ma, Y.; Furuyashiki, T. Characterization of tamoxifen as an antifungal agent using the yeast Schizosaccharomyces pombe model organism. Kobe J. Med. Sci. 2015, 61, E54–E63. [Google Scholar] [PubMed]
- Al-Janabi, A.; Al-Mosawe, H.; AI-Moswai, K. Tamoxifen: From Anti-cancer to Antifungal Drug. Int. J. Med. Rev. 2019, 6, 88–91. [Google Scholar] [CrossRef]
- Butts, A.; Koselny, K.; Chabrier-Roselló, Y.; Semighini, C.P.; Brown, J.C.; Wang, X.; Annadurai, S.; Di Done, L.; Tabroff, J.; Childers, W.E., Jr.; et al. Estrogen receptor antagonists are anti-cryptococcal agents that directly bind EF hand proteins and synergize with fluconazole in vivo. mBio 2014, 5, e00765-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iipe, L.; Jain, S.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Aguirre, M.; Torres-López, L.; Gómez-Sandoval, Z.; Villatoro-Gómez, K.; Pottosin, I.; Dobrovinskaya, O. Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca2+ Homeostasis. Int. J. Mol. Sci. 2021, 22, 8688. https://doi.org/10.3390/ijms22168688
Olivas-Aguirre M, Torres-López L, Gómez-Sandoval Z, Villatoro-Gómez K, Pottosin I, Dobrovinskaya O. Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca2+ Homeostasis. International Journal of Molecular Sciences. 2021; 22(16):8688. https://doi.org/10.3390/ijms22168688
Chicago/Turabian StyleOlivas-Aguirre, Miguel, Liliana Torres-López, Zeferino Gómez-Sandoval, Kathya Villatoro-Gómez, Igor Pottosin, and Oxana Dobrovinskaya. 2021. "Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca2+ Homeostasis" International Journal of Molecular Sciences 22, no. 16: 8688. https://doi.org/10.3390/ijms22168688