
TGF-β/IL-7 Chimeric Switch Receptor-Expressing CAR-T Cells Inhibit Recurrence of CD19-Positive B Cell Lymphoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Characterization of CAR-T Cells
2.2. tTRII Improves TGF-β1-Mediated Inhibition of Ag-Specific Tumor Killing by CAR-T Cells
2.3. CD19 CAR-tTRII-I7R-T Cells Show Increased Anti-Tumor Efficacy In Vivo
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Lines
4.3. Mice
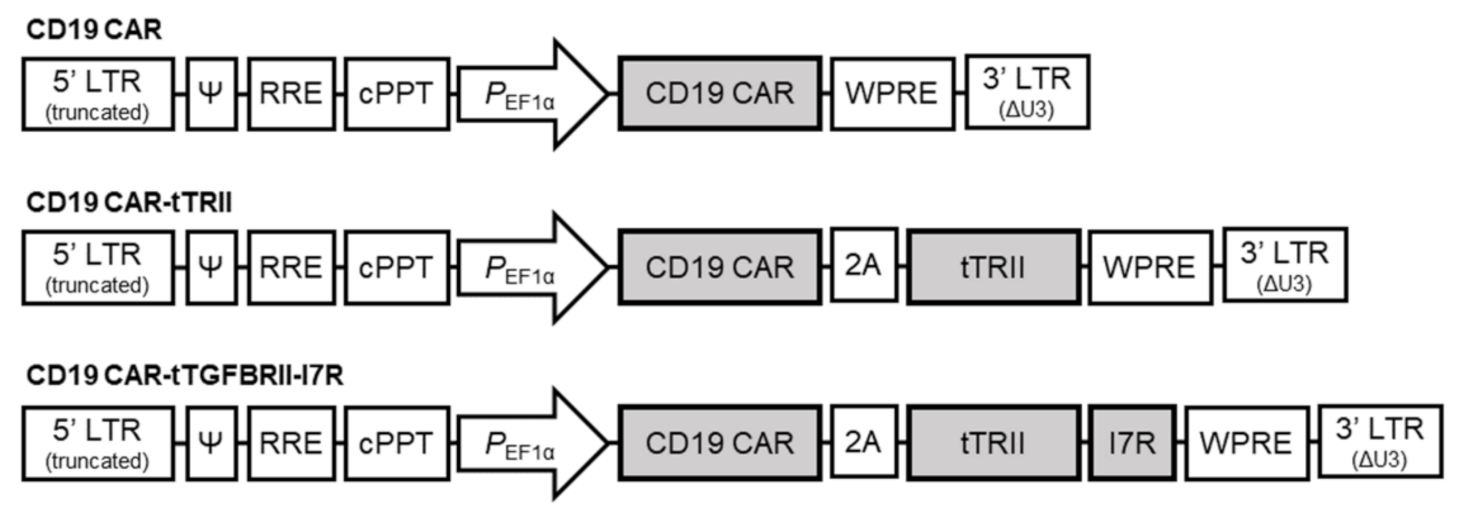
4.4. Construction of Lentivirus-Based Vectors and Vector Design
4.5. Lentiviral Vector Production and Titration
4.6. Generation of CAR-T Cells
4.7. Flow Cytometric Analysis
4.8. Western Blotting
4.9. qRT-PCR
4.10. Cytotoxicity of CAR-T-19 Cells
4.11. Bioluminescence Imaging
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ag | Antigen |
| CAR | Chimeric antigen receptor |
| CD19 CAR | Human anti-CD19-specific scFv CAR |
| CD19 CAR-expressing T cells | CAR-T cells, CD19 CAR-tTRII-T cells, and CD19 CAR-tTRII-I7R-T cells |
| CD19 CAR-tTRII-I7R-T cells | Co-expressing both CD19 CAR and tTRII-I7R-T cells |
| CSR | Chimeric switch receptor |
| Group CAR-19 | Mice receiving CD19 CAR-T cells |
| Group I7R | Mice receiving CD19 CAR-tTRII-I7R-T cells |
| Group tTRII | Mice receiving CD19 CAR-tTRII-T cells |
| IL | Interleukin |
| IU | International unit |
| NHL | Non-Hodgkin’s lymphoma |
| PBS | Phosphate-buffered saline |
| qRT-PCR | Quantitative reverse transcription-polymerase chain reaction |
| rh | Recombinant human |
| SMAD | Mothers against decapentaplegic homologs |
| TGF-β | Transforming growth factor-beta CD19 |
| TME | Tumor microenvironment |
| TR | TGF-β receptor |
| tTRII | Dominant negative TRII |
| tTRII-expressing T cells | CD19 CAR-tTRII-T cells and CD19 CAR-tTRII-I7R-T cells |
| tTRII-I7R | TGF-β/IL-7 CSR |
References
- Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In vitro-transcribed mRNA chimeric antigen receptor t cell (IVT mRNA CAR T) therapy in hematologic and solid tumor management: A preclinical update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newick, K.; O’brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Jindal, V.; Arora, E.; Gupta, S. Challenges and prospects of chimeric antigen receptor T cell therapy in solid tumors. Med. Oncol. 2018, 35, 87. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor t cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ø.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Lanksburg, D.; et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; Mcguirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Birtwistle, L.; Micklethwaite, K. Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells. Mamm. Genome 2018, 29, 739–756. [Google Scholar] [CrossRef]
- Höpken, U.E.; Rehm, A. Targeting the tumor microenvironment of leukemia and lymphoma. Trends Cancer 2019, 5, 351–364. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef]
- Govinden, R.; Bhoola, K. Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacol. Ther. 2003, 98, 257–265. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Rudensky, A.Y. TGFβ signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 2007, 7, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.K.; Kähäri, V.M. Transforming growth factor-β signaling in cancer invasion and metastasis. Int. J. Cancer 2007, 121, 2119–2124. [Google Scholar] [CrossRef]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; Von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Wieser, R.; Attisano, L.; Wrana, J.L.; Massagué, J. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol. Cell. Biol. 1993, 13, 7239–7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacuesta, K.; Buza, E.; Hauser, H.; Granville, L.; Pule, M.; Corboy, G.; Finegold, M.; Weiss, H.; Chen, S.Y.; Brenner, M.K.; et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-β receptor. J. Immunother. 2006, 29, 250–260. [Google Scholar] [CrossRef]
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.-J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor β–related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed Hodgkin lymphoma. J. Clin. Oncol. 2018, 36, 1128. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhu, Z.; Xiao, H.; Wakefield, M.R.; Ding, V.A.; Bai, Q.; Fang, Y. The role of IL-7 in immunity and cancer. Anticancer Res. 2017, 37, 963–967. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, D.; Melão, A.; Barata, J.T. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia. Adv. Biol. Regul. 2013, 53, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Q.; Jiang, Q.; Khaled, A.R.; Keller, J.R.; Durum, S.K. Interleukin-7 inactivates the pro-apoptotic protein bad promoting T cell survival. J. Biol. Chem. 2004, 279, 29160–29166. [Google Scholar] [CrossRef] [Green Version]
- Hand, T.W.; Cui, W.; Jung, Y.W.; Sefik, E.; Joshi, N.S.; Chandele, A.; Liu, Y.; Kaech, S.M. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. USA 2010, 107, 16601–16606. [Google Scholar] [CrossRef] [Green Version]
- Niu, N.; Qin, X. New insights into IL-7 signaling pathways during early and late T cell development. Cell. Mol. Immunol. 2013, 10, 187–189. [Google Scholar] [CrossRef]
- Deiser, K.; Stoycheva, D.; Bank, U.; Blankenstein, T.; Schüler, T. Interleukin-7 modulates anti-tumor CD8+ T cell responses via its action on host cells. PLoS ONE 2016, 11, e0159690. [Google Scholar] [CrossRef]
- Tay, J.C.; Zha, S.; Wang, S. Chimeric switch receptor: Switching for improved adoptive T-cell therapy against cancers. Immunotherapy 2017, 9, 1339–1349. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E. The challenge of the microenvironment in B-cell lymphomas. Histopathology 2011, 58, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-S.; Wasserman, R.; Hayakawa, K.; Hardy, R.R. Identification of the earliest B lineage stage in mouse bone marrow. Immunity 1996, 5, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choi, S.-Y.; Jung, N.-C.; Song, J.-Y.; Seo, H.G.; Lee, H.S.; Lim, D.-S. The effect of the tumor microenvironment and tumor-derived metabolites on dendritic cell function. J. Cancer 2020, 11, 769. [Google Scholar] [CrossRef] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J. Immunol. 2013, 191, 4121–4129. [Google Scholar] [CrossRef] [Green Version]
- Kobold, S.; Grassmann, S.; Chaloupka, M.; Lampert, C.; Wenk, S.; Kraus, F.; Rapp, M.; Düwel, L.P.; Zeng, Y.; Schmollinger, J.C.; et al. Impact of a new fusion receptor on PD-1–mediated immunosuppression in adoptive T cell therapy. J. Natl. Cancer Inst. 2015, 107, djv146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Park, H.B.; Lim, D.P.; Lee, J.E.; Seo, H.H.; Lee, S.F.; Eom, H.S.; Kim, I.H.; Lee, S.H.; Choi, K. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 2012, 119, 5678–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombetti, S.; Lévy, F.; Chapatte, L. IL-7 adjuvant treatment enhances long-term tumor antigen-specific CD8+ T-cell responses after immunization with recombinant lentivector. Blood 2009, 113, 6629–6637. [Google Scholar] [CrossRef]
- Kneissl, S.; Zhou, Q.; Schwenkert, M.; Cosset, F.-L.; Verhoeyen, E.; Buchholz, C.J. CD19 and CD20 targeted vectors induce minimal activation of resting B lymphocytes. PLoS ONE 2013, 8, e79047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneissl, S.; Abel, T.; Rasbach, A.; Brynza, J.; Schneider-Schaulies, J.; Buchholz, C.J. Measles virus glycoprotein-based lentiviral targeting vectors that avoid neutralizing antibodies. PLoS ONE 2012, 7, e46667. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.Y.; Liong, K.H.; Gunalan, M.G.; Li, N.; Lim, D.S.L.; Fisher, D.A.; Macary, P.A.; Leo, Y.S.; Wong, S.C.; Puan, K.J.; et al. Type I IFNs and IL-18 regulate the antiviral response of primary human γδ T cells against dendritic cells infected with Dengue virus. J. Immunol. 2015, 194, 3890–3900. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noh, K.-E.; Lee, J.-H.; Choi, S.-Y.; Jung, N.-C.; Nam, J.-H.; Oh, J.-S.; Song, J.-Y.; Seo, H.G.; Wang, Y.; Lee, H.S.; et al. TGF-β/IL-7 Chimeric Switch Receptor-Expressing CAR-T Cells Inhibit Recurrence of CD19-Positive B Cell Lymphoma. Int. J. Mol. Sci. 2021, 22, 8706. https://doi.org/10.3390/ijms22168706
Noh K-E, Lee J-H, Choi S-Y, Jung N-C, Nam J-H, Oh J-S, Song J-Y, Seo HG, Wang Y, Lee HS, et al. TGF-β/IL-7 Chimeric Switch Receptor-Expressing CAR-T Cells Inhibit Recurrence of CD19-Positive B Cell Lymphoma. International Journal of Molecular Sciences. 2021; 22(16):8706. https://doi.org/10.3390/ijms22168706
Chicago/Turabian StyleNoh, Kyung-Eun, Jun-Ho Lee, So-Yeon Choi, Nam-Chul Jung, Ji-Hee Nam, Ji-Soo Oh, Jie-Young Song, Han Geuk Seo, Yu Wang, Hyun Soo Lee, and et al. 2021. "TGF-β/IL-7 Chimeric Switch Receptor-Expressing CAR-T Cells Inhibit Recurrence of CD19-Positive B Cell Lymphoma" International Journal of Molecular Sciences 22, no. 16: 8706. https://doi.org/10.3390/ijms22168706