
Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. SARS-CoV-2 S Protein Genome/Protein Analysis in Patients with COVID-19
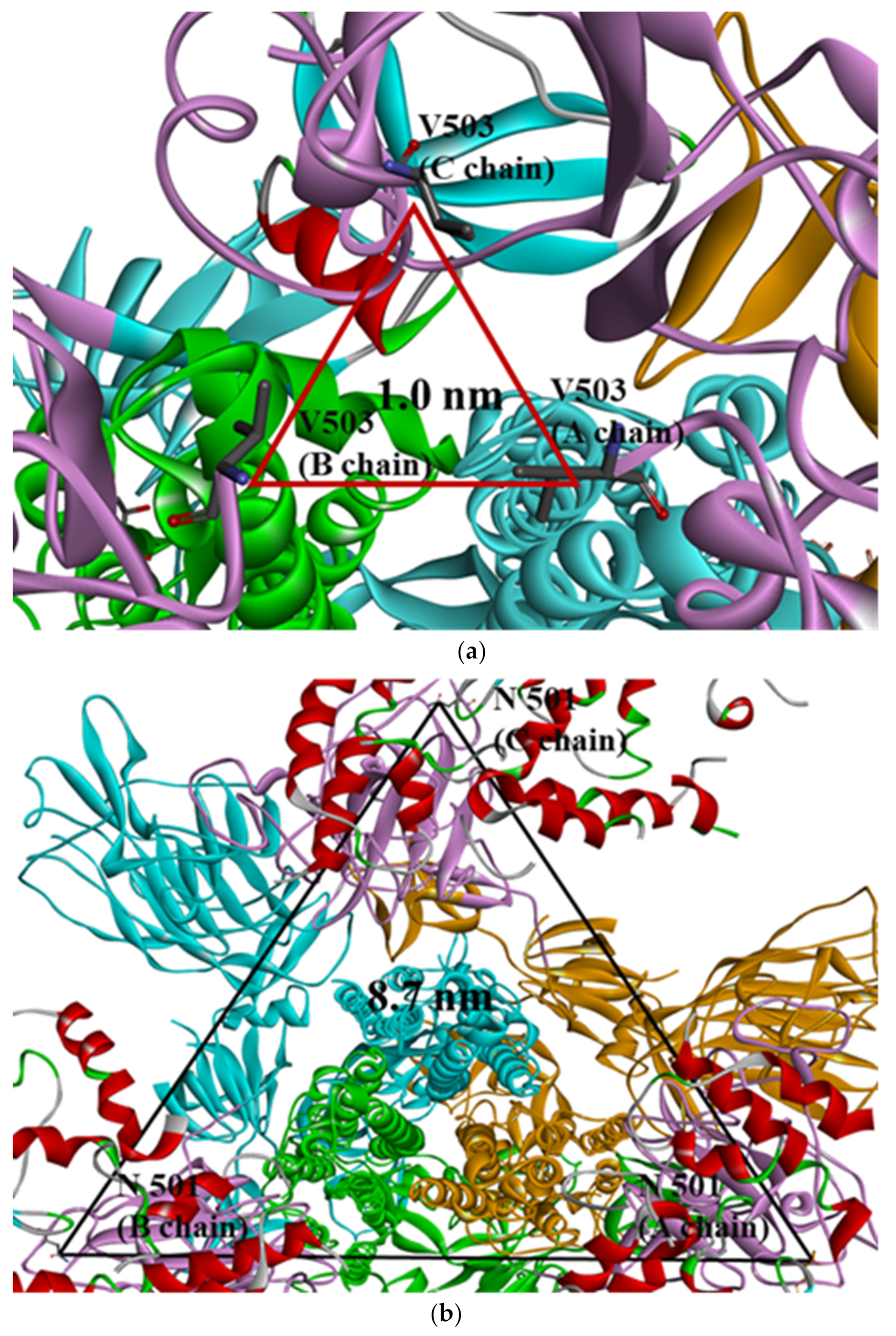
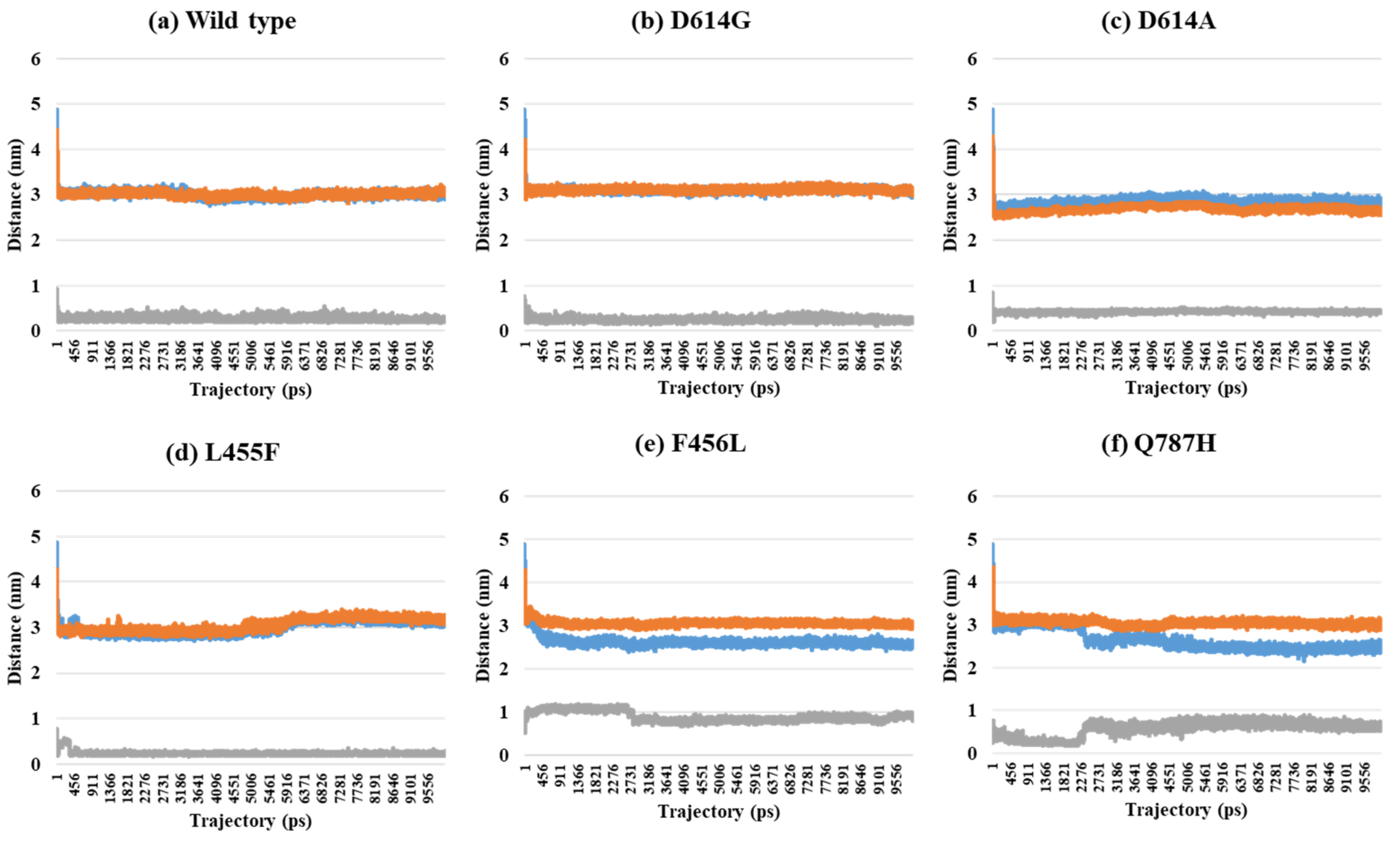
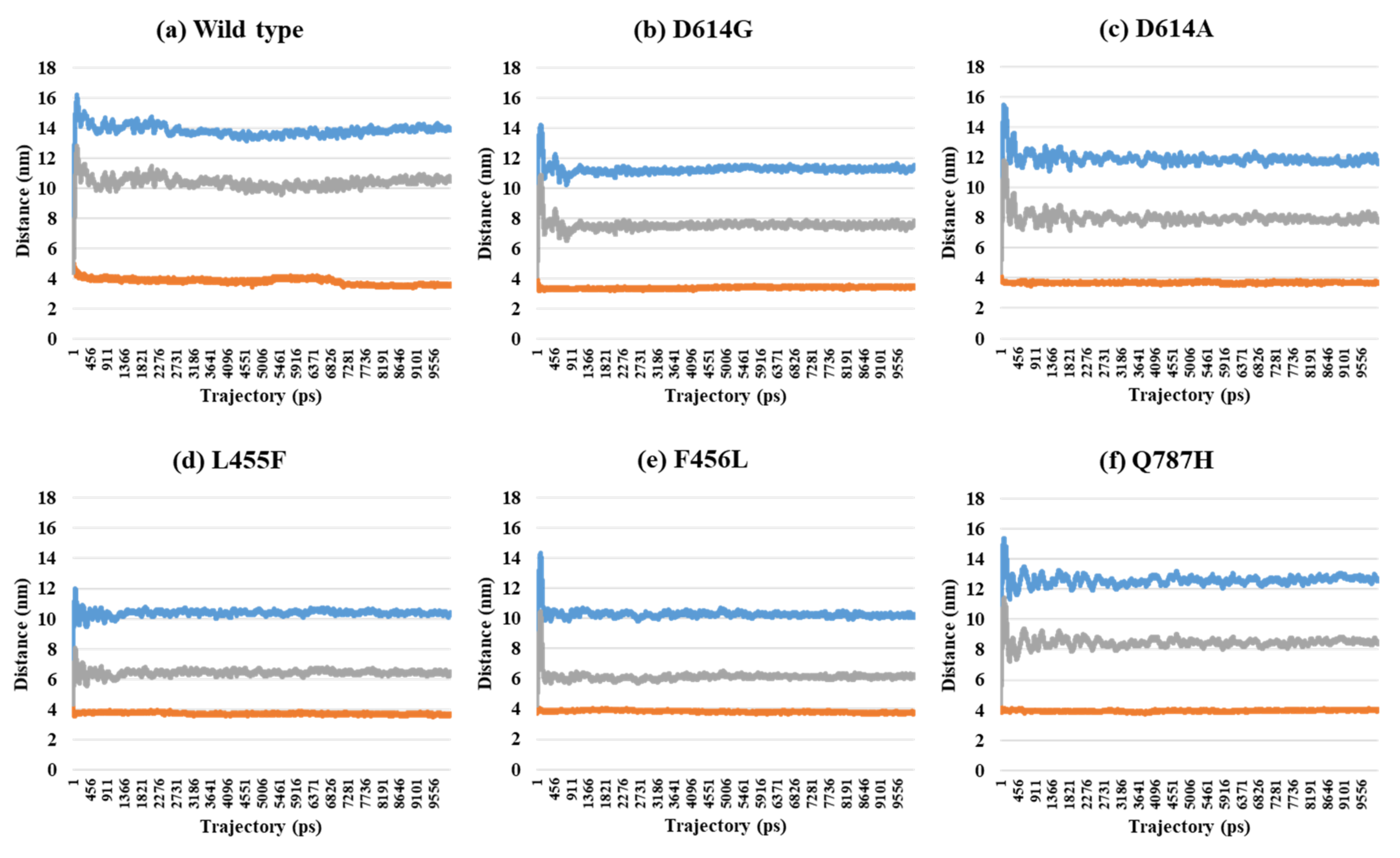
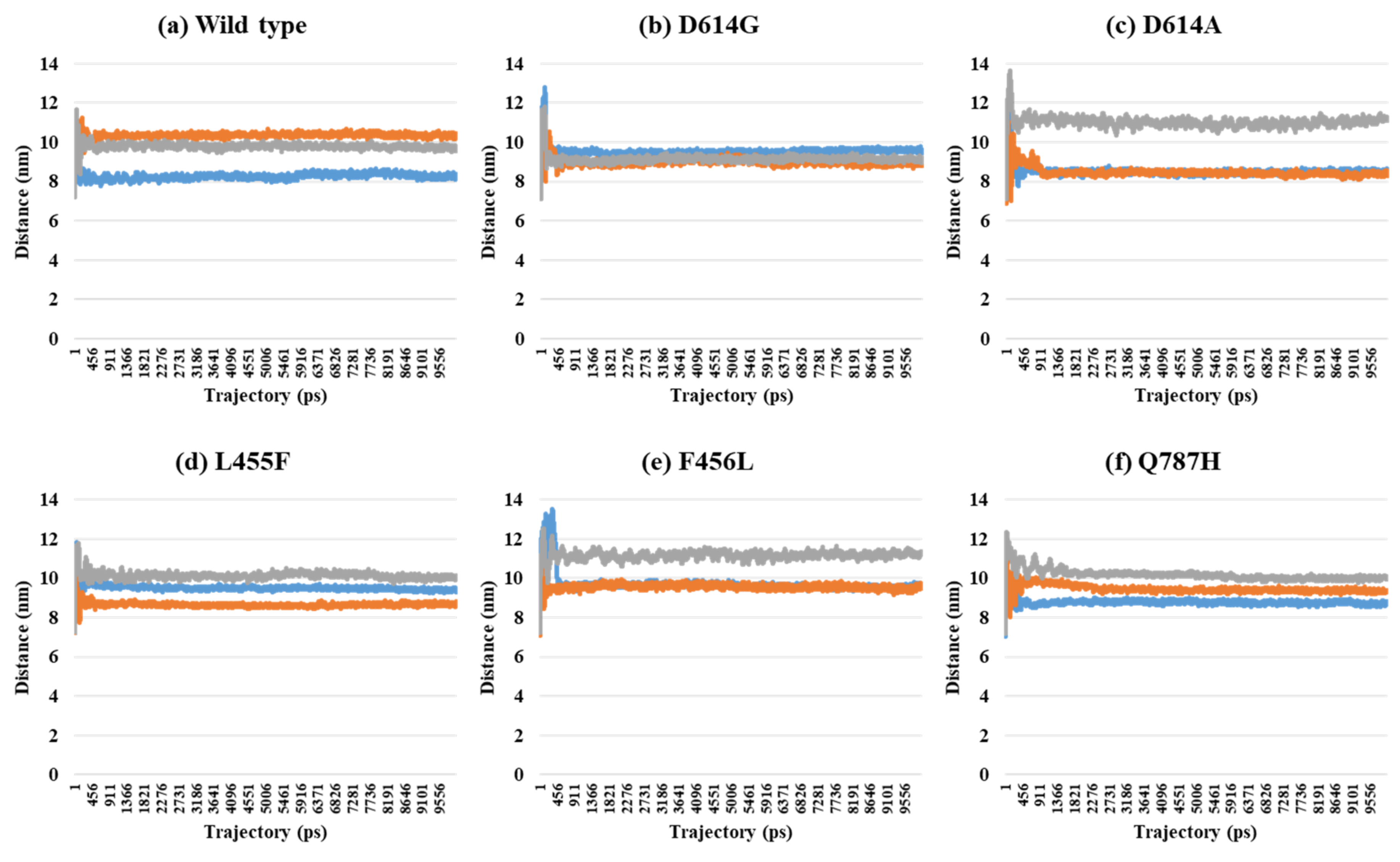
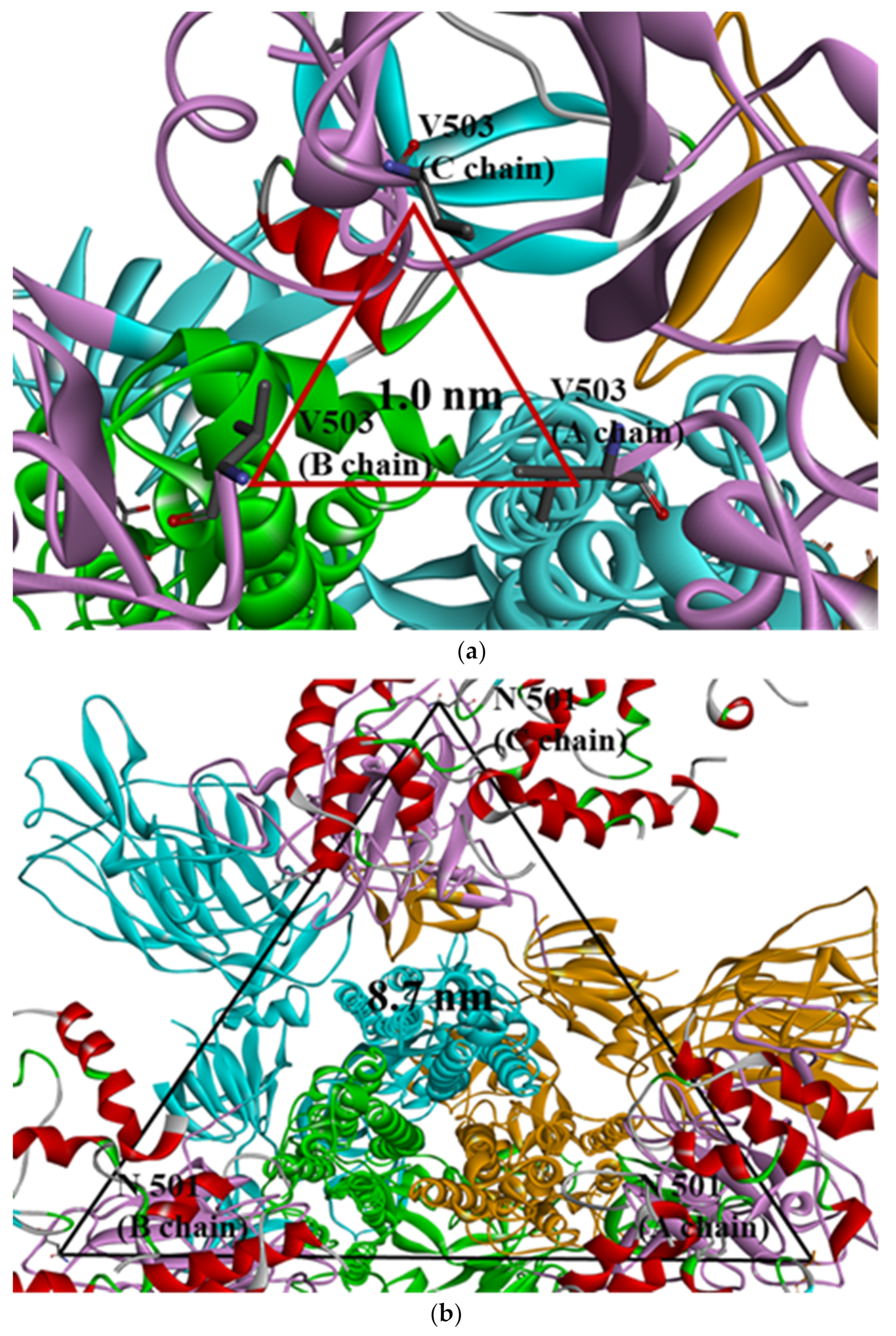
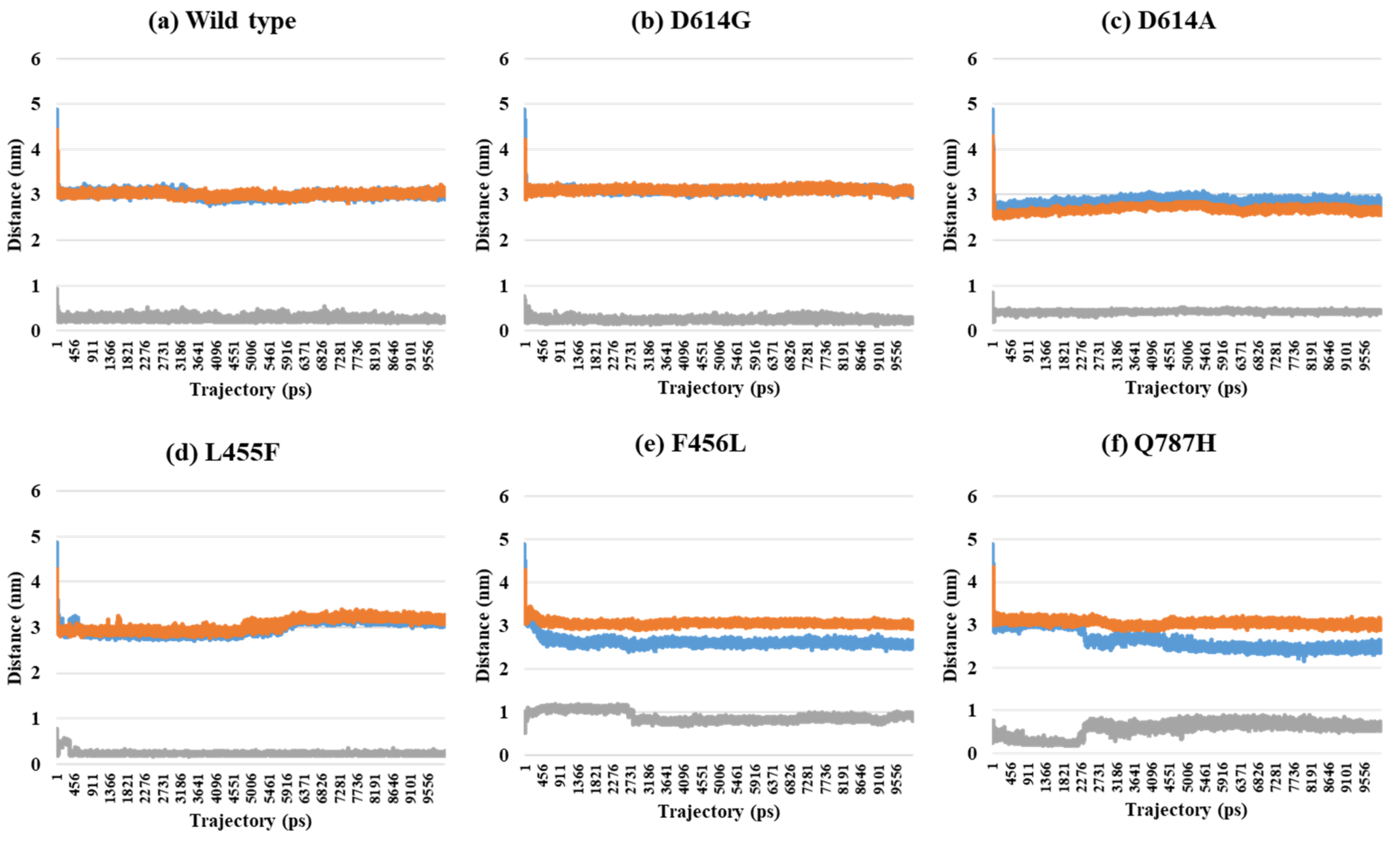
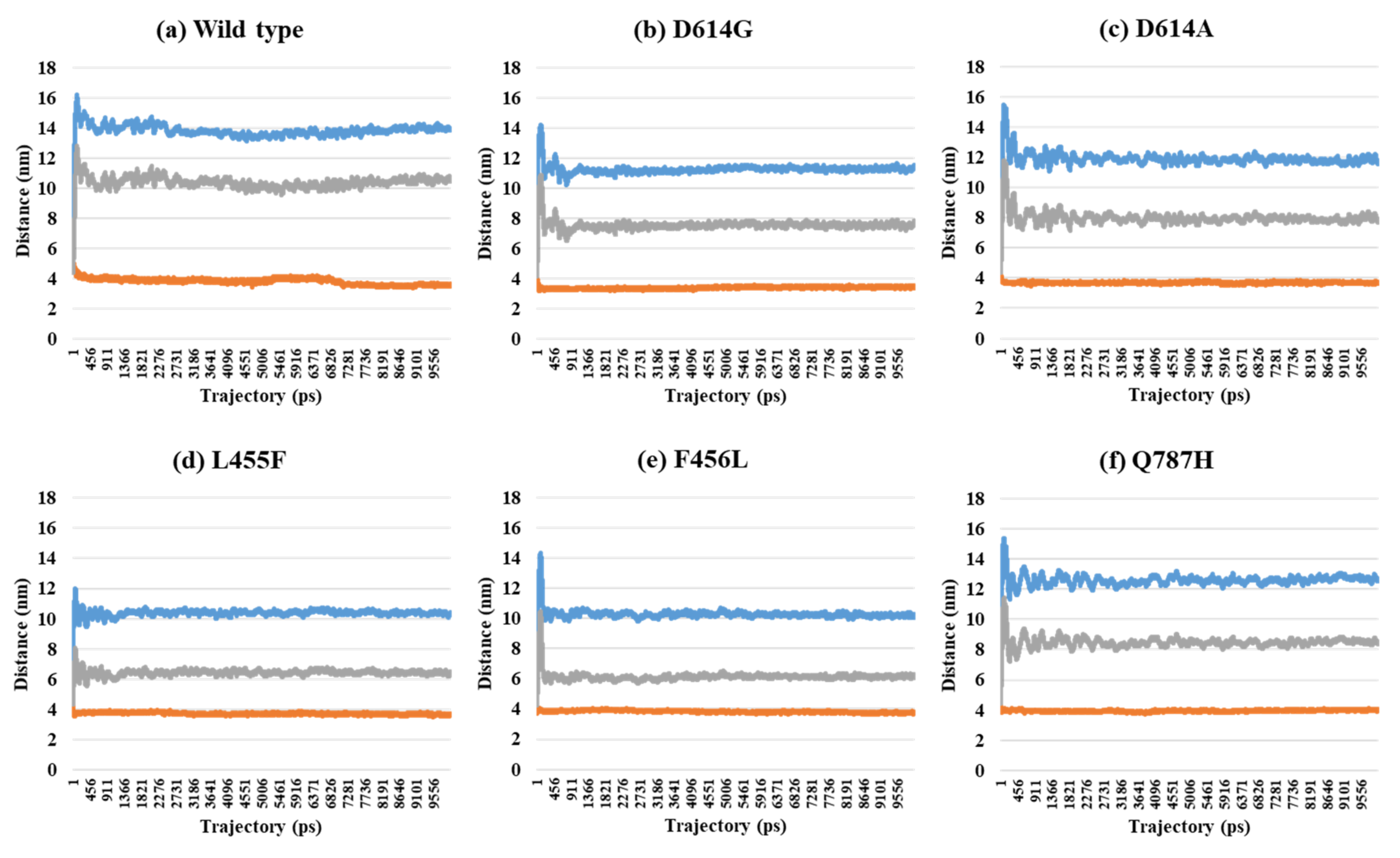
2.2. Distance Analysis of V503 and N501 Residues among Trimeric S Protein Protomers
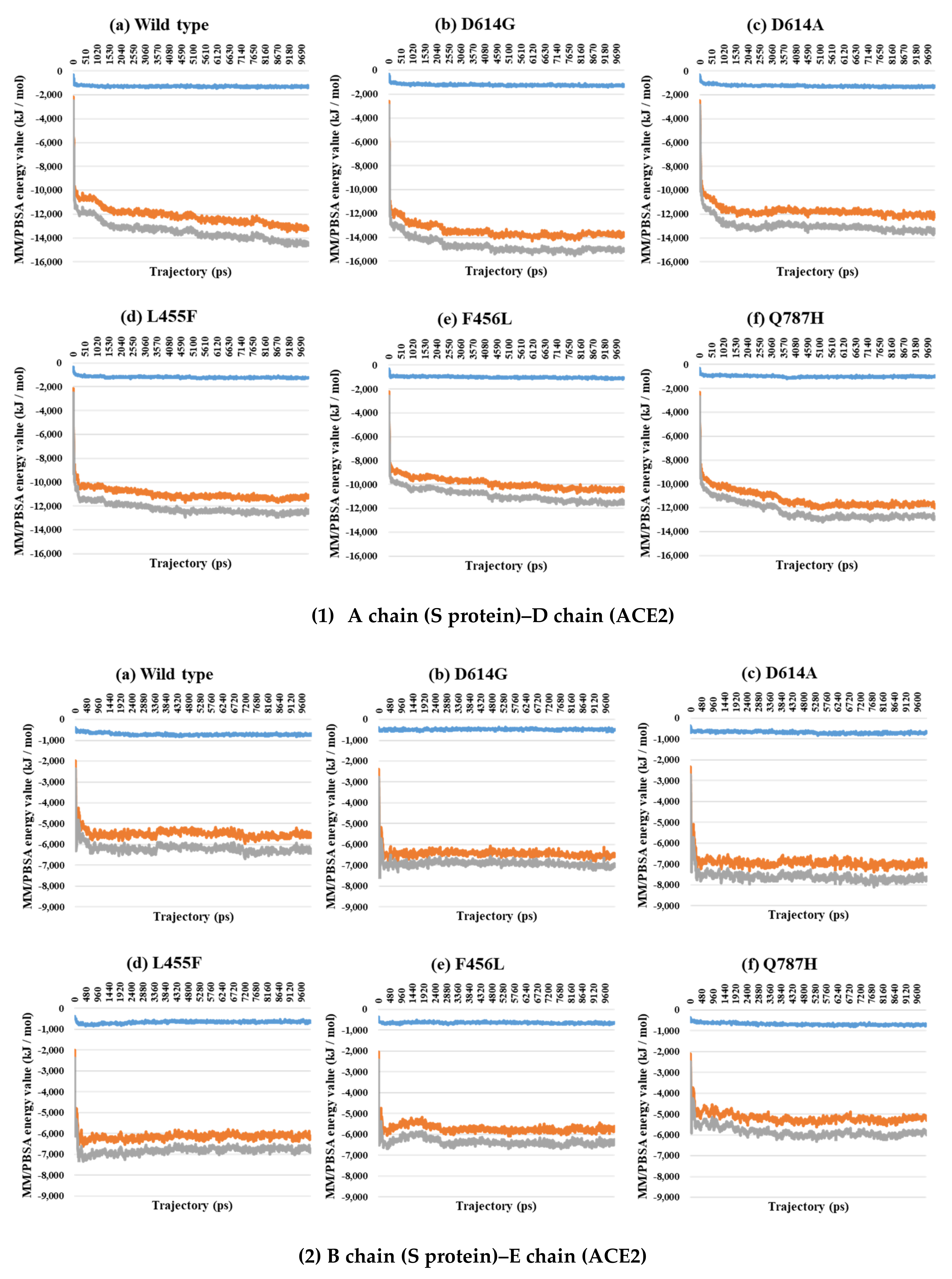
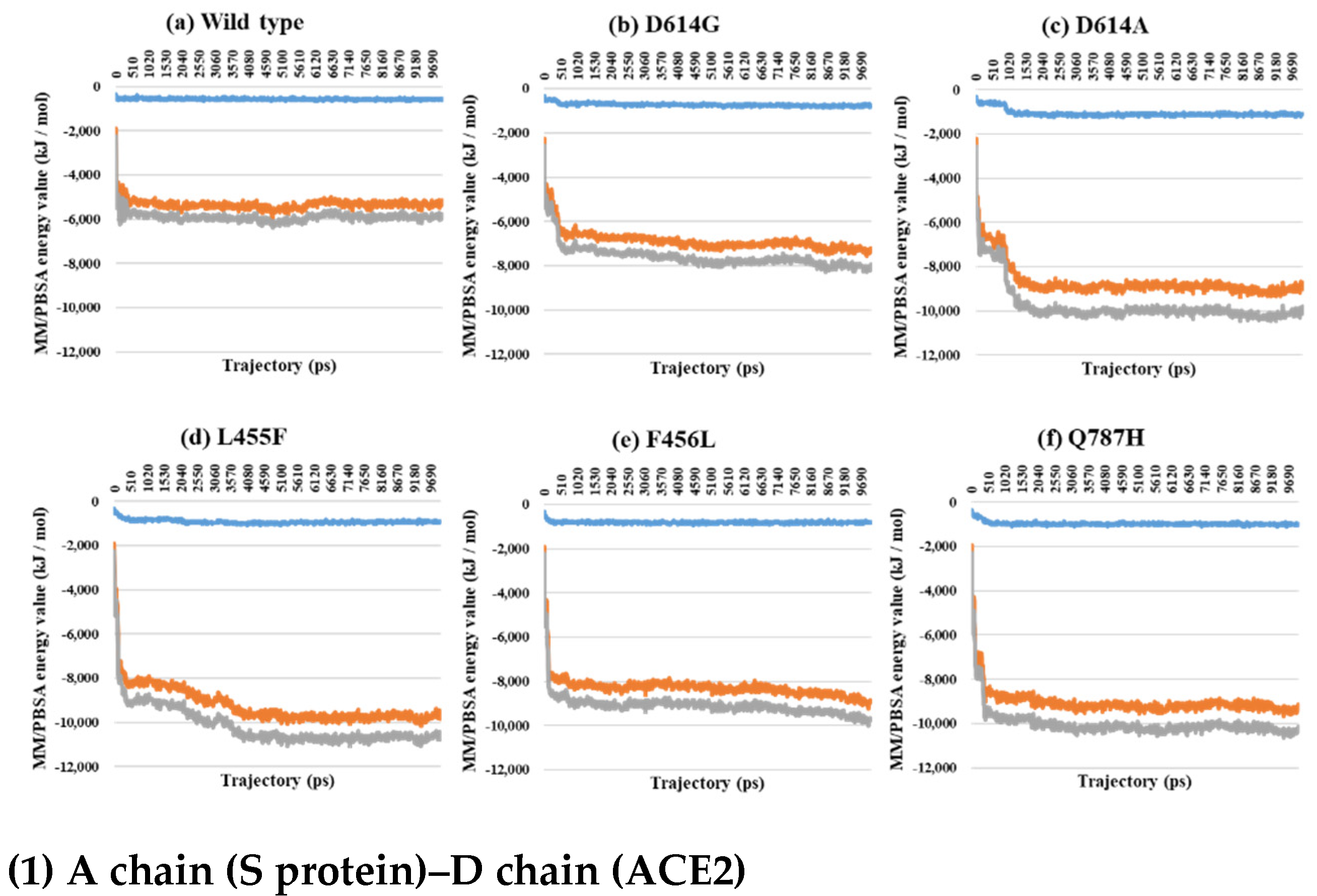
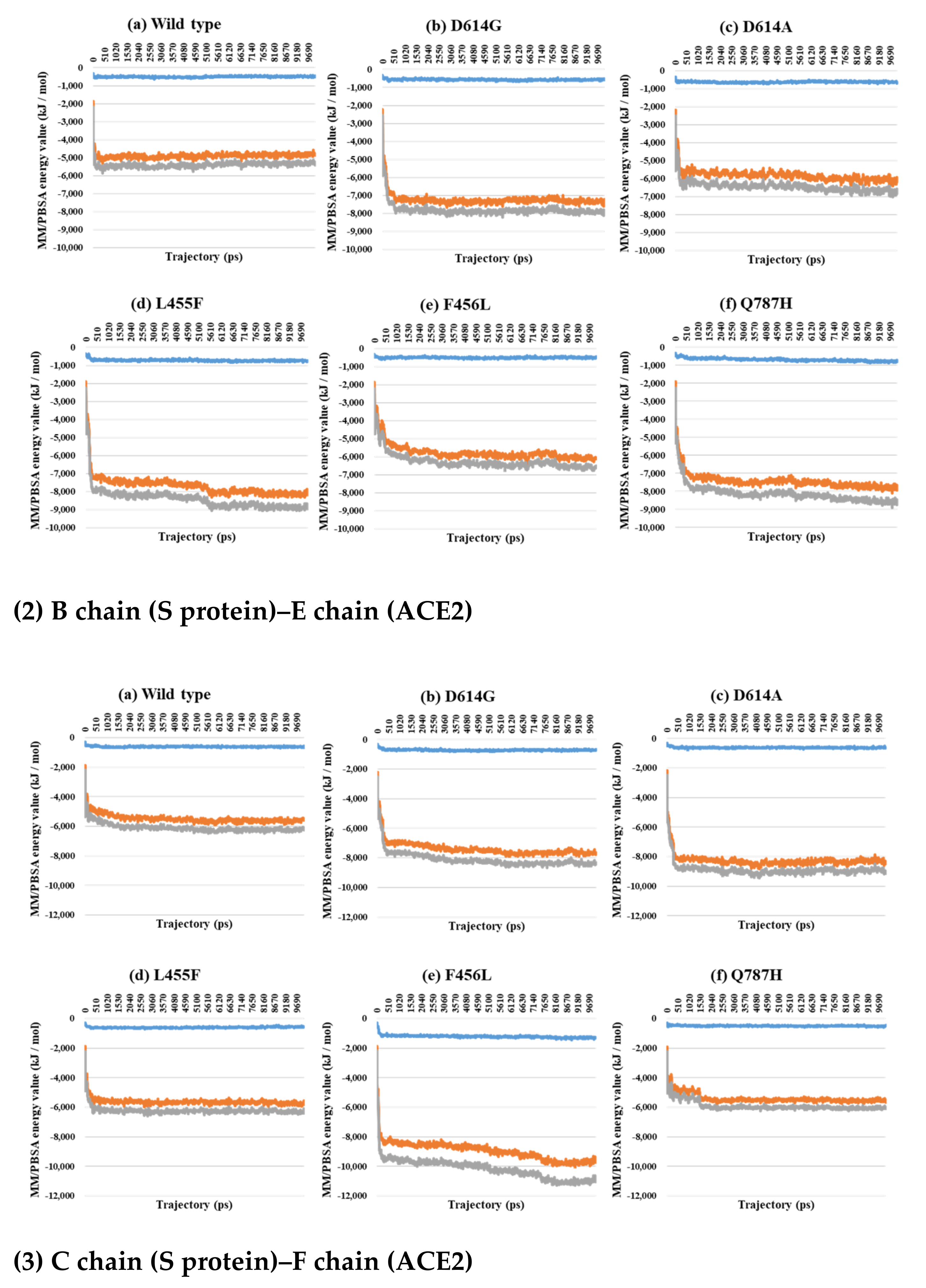
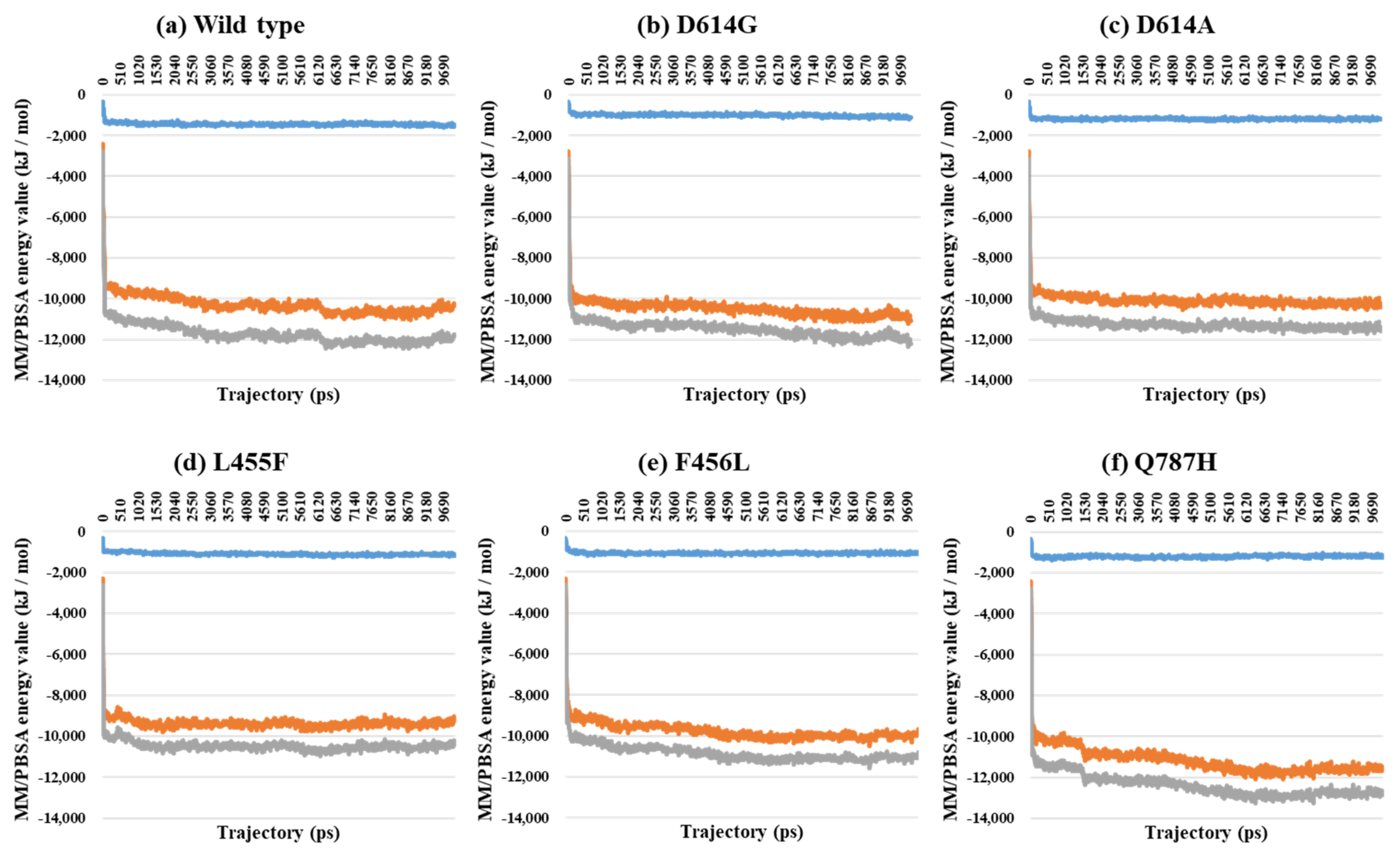
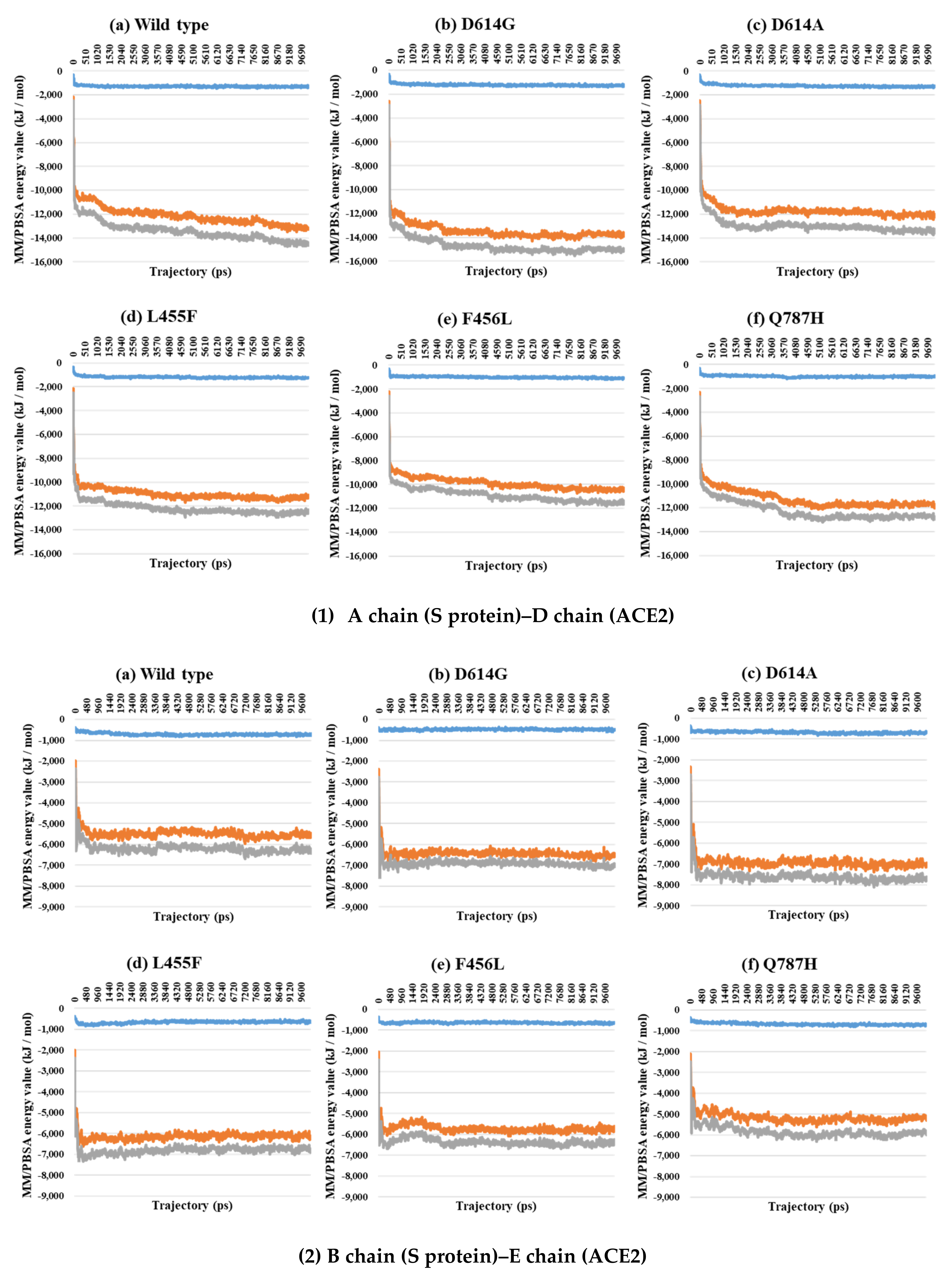
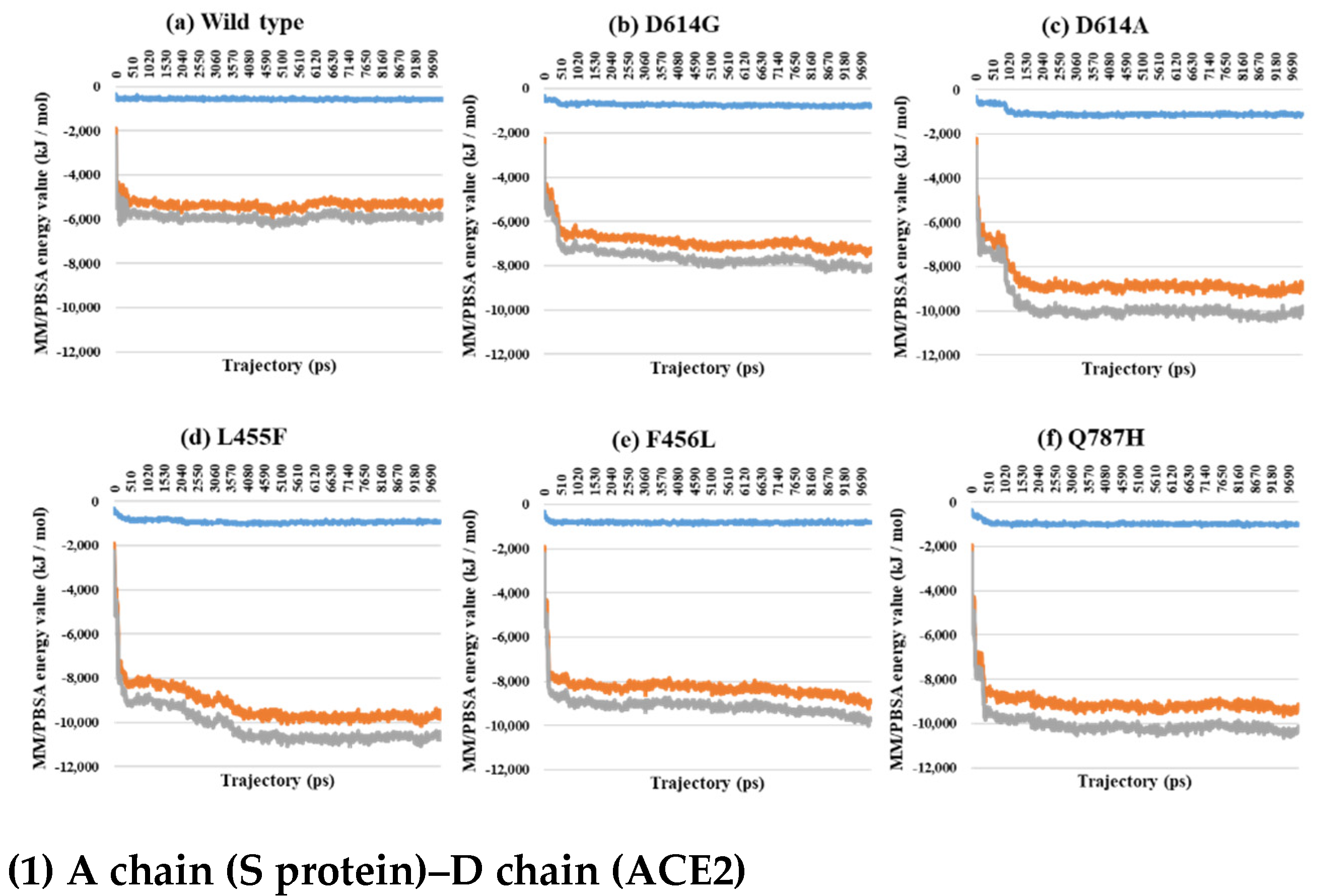
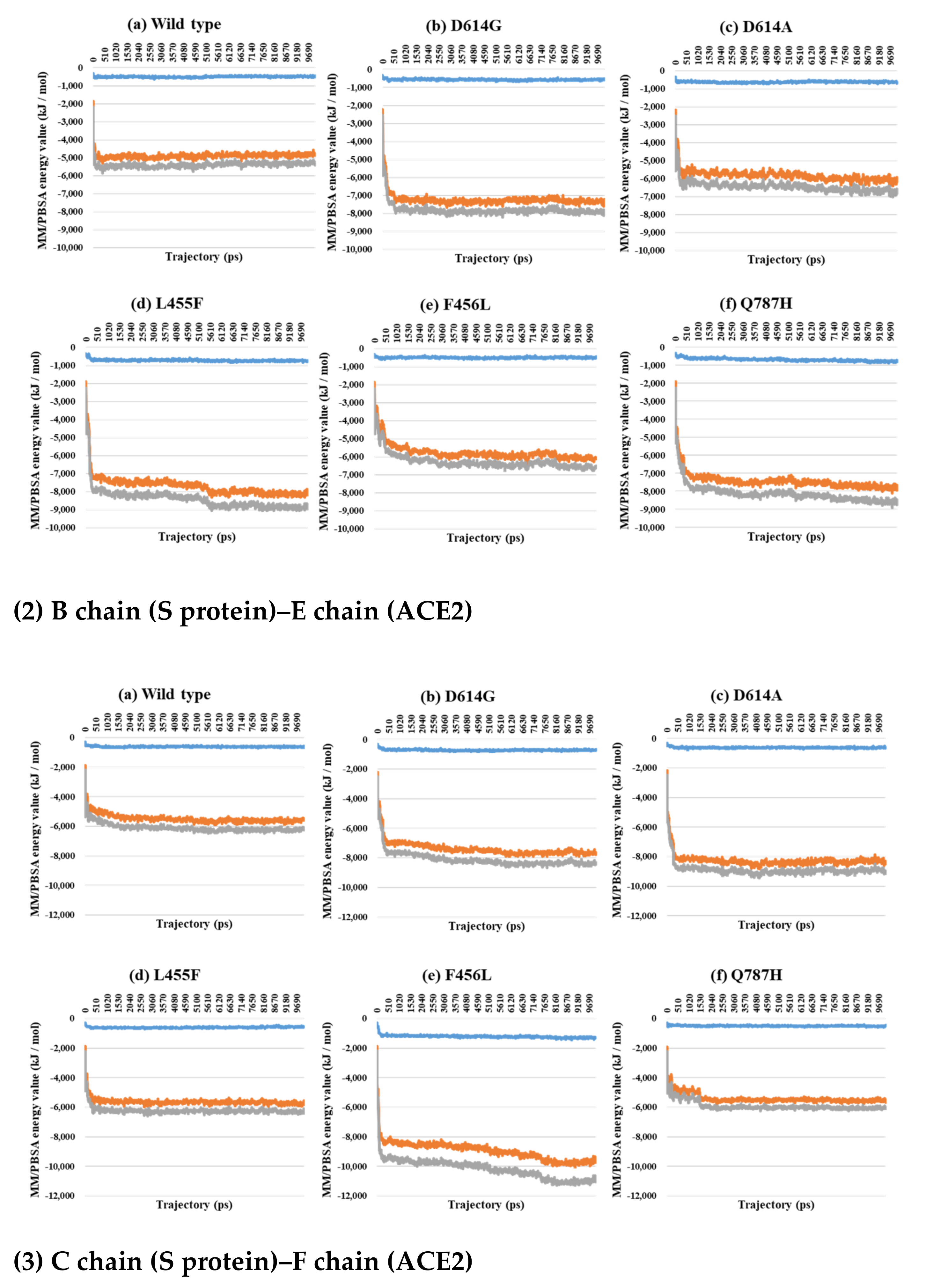
2.3. Binding Free Energy Analysis through MM/PBSA Calculations between the S Protein and ACE2
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Genome/Protein Sequence Investigation of Korean Patients with COVID-19
4.3. Dataset Preparation
4.4. MD Simulation
4.5. V503 and N501 Residue Distance Calculation
4.6. Binding Free Energy Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Vagner, D.; Frumence, É.; Ah-Pine, F.; Guillot, X.; Nobécourt, E.; Raffray, L.; Gasque, P. Deciphering SARS-CoV-2 virologic and immunologic features. Int. J. Mol. Sci. 2020, 21, 5932. [Google Scholar] [CrossRef]
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the era of SARS-CoV-2: Controversies and novel perspectives. Front. Mol. Biosci. 2020, 7, 588618. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Rath, S.L.; Kumar, K. Investigation of the effect of temperature on the structure of SARS-Cov-2 Spike Protein by Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 7, 583523. [Google Scholar] [CrossRef]
- Deganutti, G.; Prischi, F.; Reynolds, C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2020, 35, 195–207. [Google Scholar] [CrossRef]
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arXiv 2020, arXiv:2005.06199. [Google Scholar]
- Nelson, G.; Buzko, O.; Bassett, A.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Millisecond-scale molecular dynamics simulation of spike RBD structure reveals evolutionary adaption of SARS-CoV-2 to stably bind ACE2. bioRxiv 2020, 422055. [Google Scholar]
- Malaspina, D.C.; Faraudo, J. Computer Simulations of the interaction between SARS-CoV-2 spike glycoprotein and different surfaces. Biointerphases 2020, 15, 051008. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Saltalamacchia, A.; Magistrato, A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the hallmark for its enhanced infectivity? Insights from all-atom simulations. J. Phys. Chem. Lett. 2020, 11, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tao, H.; Yan, Y.; Huang, S.-Y.; Xiao, Y. Molecular mechanism of evolution and human infection with SARS-CoV-2. Viruses 2020, 12, 428. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Zhang, X.; Xu, Z.; Zhu, W. Exploring the binding mechanism and accessible angle of SARS-CoV-2 spike and ACE2 by molecular dynamics simulation and free energy calculation. Chemrxiv. Org. 2020, 11877492, v1. [Google Scholar]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Singh, P.K.; Kulsum, U.; Rufai, S.B.; Mudliar, S.R.; Singh, S. Mutations in SARS-CoV-2 Leading to Antigenic Variations in Spike Protein: A Challenge in Vaccine Development. J. Lab. Phys. 2020, 12, 154. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.; ul Qamar, M.T.; Li, G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: Integrated computational approach. J. Mol. Liq. 2020, 320, 114493. [Google Scholar] [CrossRef]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Mutations in the SARS-CoV-2 spike RBD are responsible for stronger ACE2 binding and poor anti-SARS-CoV mAbs cross-neutralization. Comput. Struct. Biotechnol. J. 2020, 18, 3402–3414. [Google Scholar] [CrossRef]
- Zou, J.; Yin, J.; Fang, L.; Yang, M.; Wang, T.; Wu, W.; Bellucci, M.A.; Zhang, P. Computational Prediction of Mutational Effects on SARS-CoV-2 Binding by Relative Free Energy Calculations. J. Chem. Inf. Modeling 2020, 60, 5794–5802. [Google Scholar] [CrossRef]
- Mehalko, J.; Drew, M.; Snead, K.; Denson, J.-P.; Wall, V.; Taylor, T.; Sadtler, K.; Messing, S.; Gillette, W.; Esposito, D.J.; et al. Improved production of SARS-CoV-2 spike receptor-binding domain (RBD) for serology assays. Protein Expr. Purif. 2021, 179, 105802. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, Q.M.; Wilson, K.M.; Shen, M.; Itkin, Z.; Eastman, R.T.; Shinn, P.; Hall, M.D. Targeting ACE2–RBD Interaction as a Platform for COVID-19 Therapeutics: Development and Drug-Repurposing Screen of an AlphaLISA Proximity Assay. ACS Pharmacol. Transl. Sci. 2020, 3, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.C.; Lappala, A.; Fenimore, P.W.; Fischer, W.M.; McMahon, B.H.; Hengartner, N.W.; Sanbonmatsu, K.Y.; Tung, C.-S. Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail. Biomolecules 2021, 11, 124. [Google Scholar] [CrossRef]
- Kumari, A.; Rajput, V.S.; Nagpal, P.; Kukrety, H.; Grover, S.; Grover, A. Dual inhibition of SARS-CoV-2 spike and main protease through a repurposed drug, rutin. J. Biomol. Struct. Dyn. 2020, 1864476. [Google Scholar] [CrossRef]
- Ortuso, F.; Mercatelli, D.; Guzzi, P.H.; Giorgi, F.M. Structural Genetics of circulating variants affecting the SARS-CoV-2 Spike/human ACE2 complex. J. Biomol. Struct. Dyn. 2021, 1886175. [Google Scholar] [CrossRef]
- Verma, J.; Subbarao, N. Insilico study on the effect of SARS-CoV-2 RBD hotspot mutants’ interaction with ACE2 to understand the binding affinity and stability. Virology 2021, 561, 107–116. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Anshumali, M.; Vikash, V. Connections between biomechanics and higher infectivity: A tale of the D614G mutation in the SARS-CoV-2 spike protein. Signal Transduct. Target. Ther. 2021, 6, 11. [Google Scholar]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, G. The SARS-CoV-2 spike variant D614G favors an open conformational state. Biophys. J. 2021, 120, 298a. [Google Scholar] [CrossRef]
- Kim, J.-M.; Chung, Y.-S.; Jo, H.J.; Lee, N.-J.; Kim, M.S.; Woo, S.H.; Park, S.; Kim, J.W.; Kim, H.M.; Han, M.-G. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, A.K.; Kim, I.-H.; Kim, J.; Kim, J.-M.; Kim, H.M.; Young Lee, C.; Han, M.-G.; Rhie, G.-E.; Kwon, D.; Nam, J.-G. Genomic surveillance of SARS-CoV-2: Distribution of clades in the Republic of Korea in 2020. Osong Public Health Res. Perspect. 2021, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.-G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Mutant Type | A–B | A–C | B–C | SD (A–B) and (A–C) | SD (A–B) and (B–C) | SD (A–C) and (B–C) | SD (A–B–C) |
|---|---|---|---|---|---|---|---|---|
| 7A94 (1-open-complex form) | Wild | 3.00 | 3.01 | 0.25 | 0.01 | 1.95 | 1.95 | 1.59 |
| D614G | 3.05 | 2.99 | 0.27 | 0.04 | 1.97 | 1.93 | 1.59 | |
| D614A | 2.82 | 2.65 | 0.44 | 0.12 | 1.68 | 1.56 | 1.33 | |
| L455F | 3.13 | 3.23 | 0.25 | 0.07 | 2.04 | 2.11 | 1.69 | |
| F456L | 2.53 | 3.01 | 0.85 | 0.34 | 1.19 | 1.53 | 1.14 | |
| Q787H | 2.47 | 2.95 | 0.59 | 0.34 | 1.33 | 1.67 | 1.25 | |
| 7A97 (2-open-complex form) | Wild | 13.96 | 3.57 | 10.63 | 7.35 | 2.36 | 5.00 | 5.31 |
| D614G | 11.55 | 3.47 | 7.79 | 5.71 | 2.66 | 3.05 | 4.04 | |
| D614A | 11.65 | 3.72 | 7.72 | 5.61 | 2.78 | 2.83 | 3.97 | |
| L455F | 10.26 | 3.60 | 6.48 | 4.71 | 2.67 | 2.04 | 3.34 | |
| F456L | 10.30 | 3.79 | 6.20 | 4.60 | 2.90 | 1.70 | 3.29 | |
| Q787H | 12.55 | 4.03 | 8.38 | 6.03 | 2.95 | 3.08 | 4.26 | |
| 7A98 (3-open-complex form) | Wild | 8.23 | 10.32 | 9.75 | 1.48 | 1.08 | 0.40 | 1.08 |
| D614G | 9.61 | 8.81 | 9.14 | 0.56 | 0.33 | 0.23 | 0.40 | |
| D614A | 8.63 | 8.51 | 11.18 | 0.08 | 1.81 | 1.89 | 1.51 | |
| L455F | 9.33 | 8.65 | 10.05 | 0.48 | 0.51 | 0.99 | 0.70 | |
| F456L | 9.69 | 9.63 | 11.30 | 0.04 | 1.14 | 1.18 | 0.95 | |
| Q787H | 8.72 | 9.30 | 10.04 | 0.41 | 0.93 | 0.53 | 0.66 |
| PDB | Mutant Type | VdW | E | T |
|---|---|---|---|---|
| 7A94 | Wild | −1461.5 | −10,311.8 | −11,773.3 |
| D614G | −1129.2 | −11,082.0 | −12,211.2 | |
| D614A | −1186.4 | −10,417.5 | −11,603.9 | |
| L455F | −1185.4 | −9224.2 | −10,409.6 | |
| F456L | −1120.6 | −9704.0 | −10,824.6 | |
| Q787H | −1236.5 | −11,629.0 | −12,865.5 |
| PDB | Mutant Type | VdW | E | T |
|---|---|---|---|---|
| 7A97 A–D chain interaction | Wild | −1326.4 | −13,200.4 | −14,526.7 |
| D614G | −1290.0 | −13,844.8 | −15,134.7 | |
| D614A | −1307.6 | −12,053.5 | −13,361.1 | |
| L455F | −1237.6 | −11,097.9 | −12,335.5 | |
| F456L | −1081.2 | −10,342.6 | −11,423.8 | |
| Q787H | −948.2 | −12,017.3 | −12,965.5 | |
| 7A97 B–E chain interaction | Wild | −677.3 | −5553.0 | −6230.3 |
| D614G | −483.8 | −6481.6 | −6965.5 | |
| D614A | −674.9 | −6940.9 | −7615.8 | |
| L455F | −664.7 | −6134.4 | −6799.1 | |
| F456L | −646.3 | −5697.0 | −6343.3 | |
| Q787H | −693.7 | −5303.3 | −5997.0 | |
| 7A97 Total | Wild | −2003.7 | −18,753.4 | −20,757.0 |
| D614G | −1773.8 | −20,326.4 | −22,100.2 | |
| D614A | −1982.5 | −18,994.4 | −20,976.9 | |
| L455F | −1902.3 | −17,232.3 | −19,134.6 | |
| F456L | −1727.4 | −16,039.6 | −17,767.1 | |
| Q787H | −1641.9 | −17,320.6 | −18,962.5 |
| PDB | Type | VdW | E | T |
|---|---|---|---|---|
| 7A98 A–D chain interaction | Wild | −535.4 | −5278.5 | −5813.8 |
| D614G | −735.3 | −7198.1 | −7933.4 | |
| D614A | −1096.6 | −9024.3 | −10,120.8 | |
| L455F | −946.6 | −9730.1 | −10,676.7 | |
| F456L | −791.3 | −8938.8 | −9730.1 | |
| Q787H | −973.9 | −9216.2 | −10,190.1 | |
| 7A98 B–E chain interaction | Wild | −527.7 | −4872.3 | −5400.0 |
| D614G | −585.3 | −7306.0 | −7891.2 | |
| D614A | −651.4 | −6007.5 | −6658.8 | |
| L455F | −778.5 | −8190.2 | −8968.7 | |
| F456L | −492.9 | −6111.3 | −6604.2 | |
| Q787H | −759.2 | −7916.7 | −8675.9 | |
| 7A98 C–F chain interaction | Wild | −583.0 | −5587.4 | −6170.4 |
| D614G | −715.8 | −7689.6 | −8405.4 | |
| D614A | −638.3 | −8485.6 | −9124.0 | |
| L455F | −582.9 | −5819.0 | −6401.9 | |
| F456L | −1289.9 | −9654.7 | −10,944.6 | |
| Q787H | −434.2 | −5656.1 | −6090.3 | |
| 7A98 Total | Wild | −1646.1 | −15,738.2 | −17,384.2 |
| D614G | −2036.4 | −22,193.7 | −24,230.0 | |
| D614A | −2386.3 | −23,517.3 | −25,903.6 | |
| L455F | −2308.0 | −23,739.2 | −26,047.3 | |
| F456L | −2574.0 | −24,704.9 | −27,278.9 | |
| Q787H | −2167.3 | −22,789.0 | −24,956.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.-E.; Kim, J.-M.; Rhee, J.; Park, A.K.; Kim, E.-J.; Kang, N.S. Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 8714. https://doi.org/10.3390/ijms22168714
Choi K-E, Kim J-M, Rhee J, Park AK, Kim E-J, Kang NS. Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2. International Journal of Molecular Sciences. 2021; 22(16):8714. https://doi.org/10.3390/ijms22168714
Chicago/Turabian StyleChoi, Kwang-Eun, Jeong-Min Kim, JeeEun Rhee, Ae Kyung Park, Eun-Jin Kim, and Nam Sook Kang. 2021. "Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2" International Journal of Molecular Sciences 22, no. 16: 8714. https://doi.org/10.3390/ijms22168714
APA StyleChoi, K.-E., Kim, J.-M., Rhee, J., Park, A. K., Kim, E.-J., & Kang, N. S. (2021). Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2. International Journal of Molecular Sciences, 22(16), 8714. https://doi.org/10.3390/ijms22168714