
Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1


Abstract
:1. The Ubiquitin and SUMO System
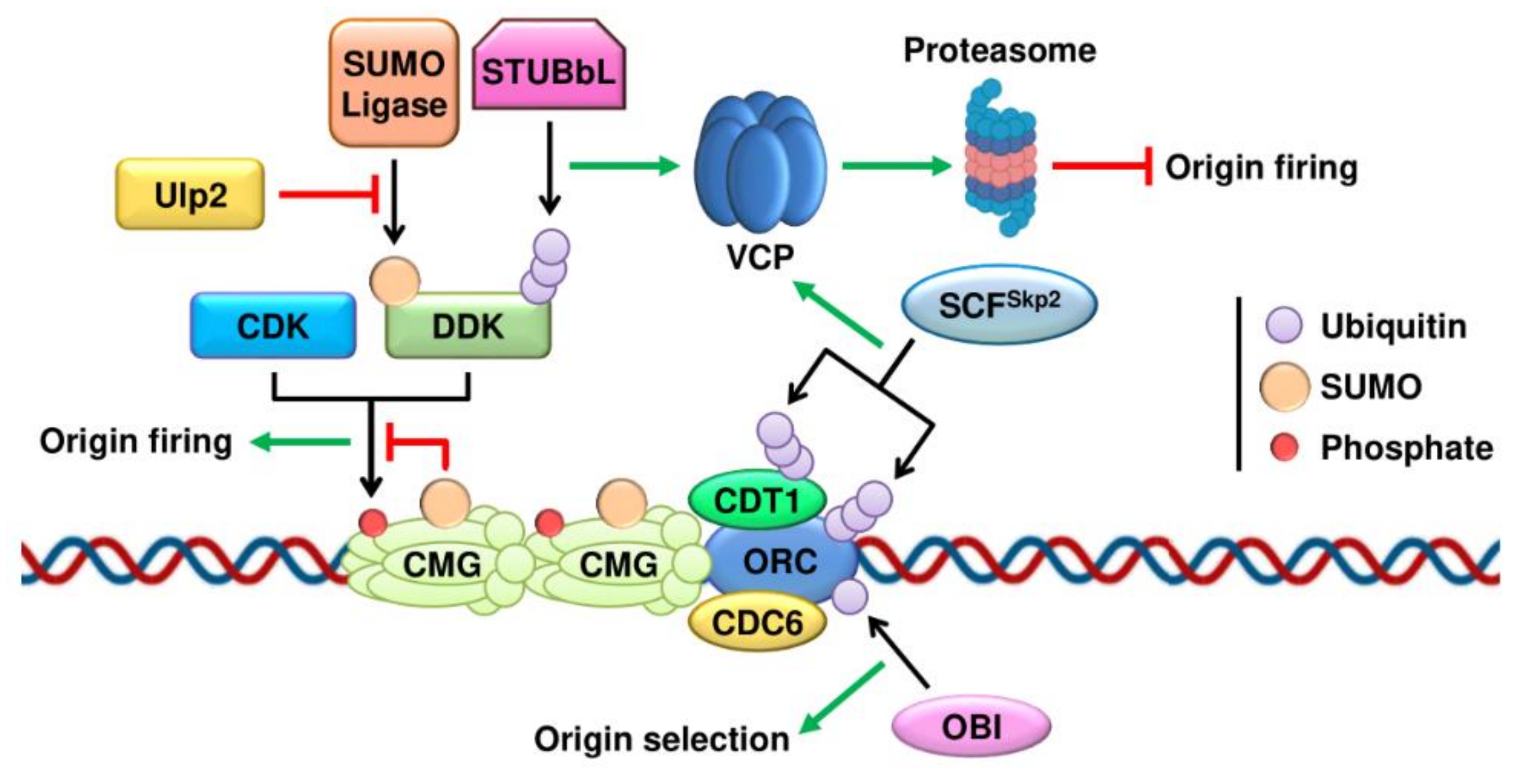
2. SUMO and Ubiquitin in DNA Replication
3. DNA Replication Termination
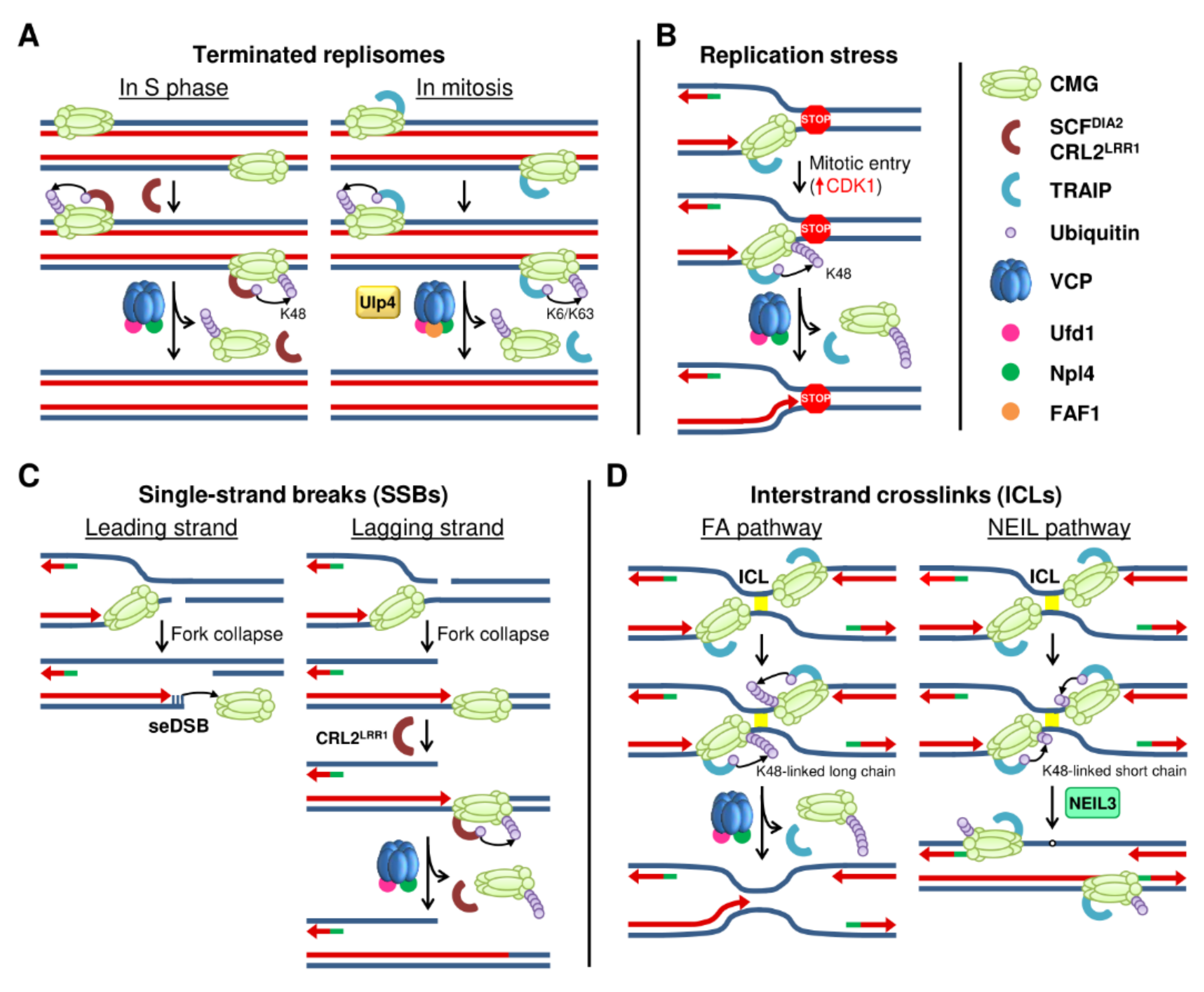
4. Replisome Disassembly at the End of DNA Replication
5. Replisome Disassembly after DNA Damage and outside the S Phase
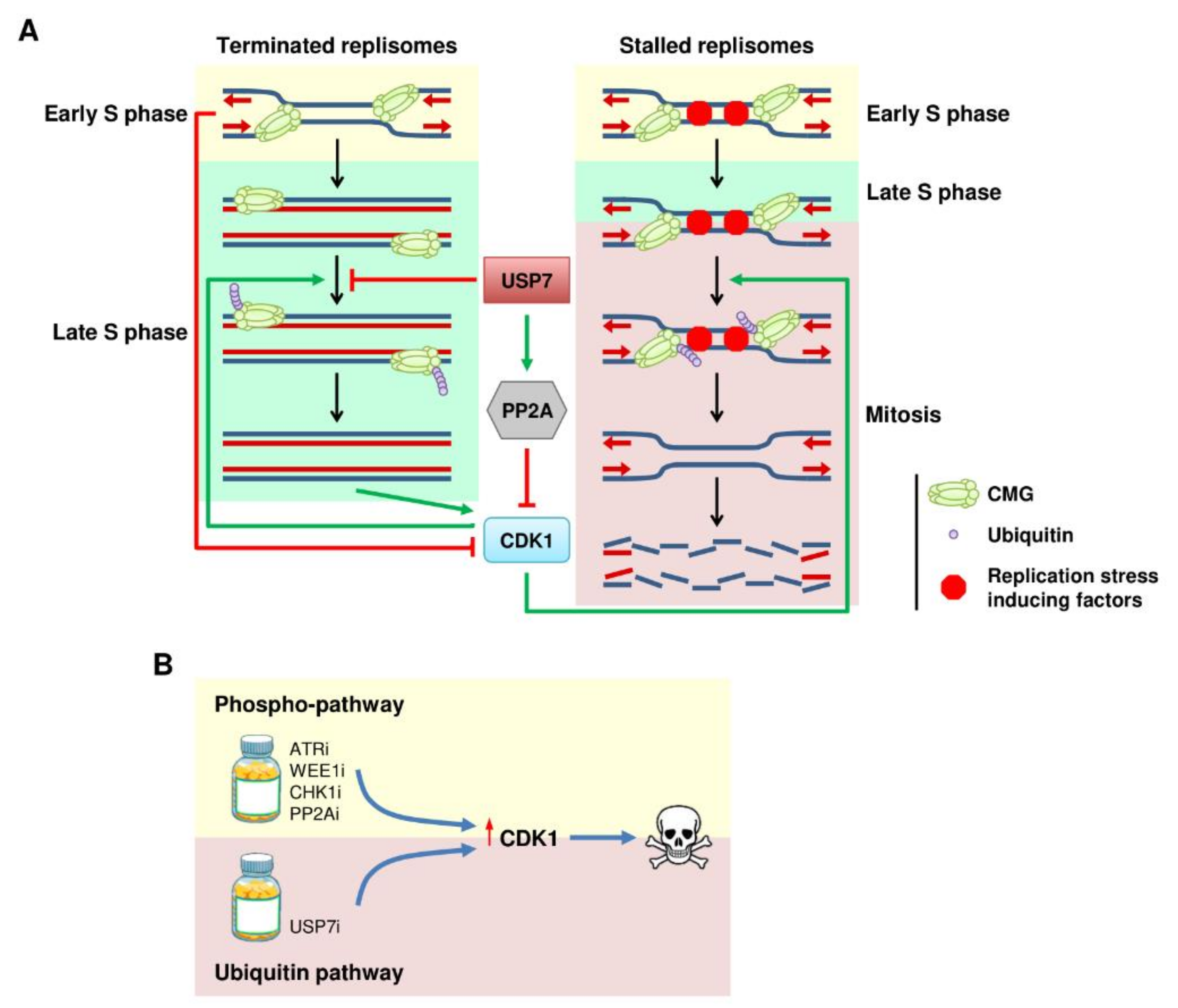
6. The Ubiquitin Connection between DNA Replication and CDK1 Activation
Author Contributions
Funding
Conflicts of Interest
References
- Damgaard, R.B. The ubiquitin system: From cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Meluh, P.B.; Koshland, D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol. Biol. Cell 1995, 6, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Seufert, W.; Futcher, B.; Jentsch, S. Role of a ubiquitin-conjugating enzyme in degradation of S- and M-phase cyclins. Nature 1995, 373, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.P.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric Chains of SUMO-2 and SUMO-3 are Conjugated to Protein Substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, A.; Ackermann, L.; Hoppe, T. Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front. Genet. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Vaz, B.; Halder, S.; Ramadan, K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y. Diverse functions with a common regulator: Ubiquitin takes command of an AAA ATPase. J. Struct. Biol. 2006, 156, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Dai, R.M.; Li, C.-C.H. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 2001, 3, 740–744. [Google Scholar] [CrossRef]
- Meyer, H.H.; Shorter, J.G.; Seemann, J.; Pappin, D.; Warren, G. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000, 19, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, C.; Yano, M.; DeMartino, G.N. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 2004, 117, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, W.; Verma, R.; Kleiger, G.; Oania, R.S.; Deshaies, R.J. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat. Struct. Mol. Biol. 2012, 19, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Bergink, S.; Ammon, T.; Kern, M.; Schermelleh, L.; Leonhardt, H.; Jentsch, S. Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat. Cell Biol. 2013, 15, 526–532. [Google Scholar] [CrossRef]
- Køhler, J.B.; Tammsalu, T.; Jørgensen, M.M.; Steen, N.; Hay, R.T.; Thon, G. Targeting of SUMO substrates to a Cdc48-Ufd1-Npl4 segregase and STUbL pathway in fission yeast. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Aslanian, A.; Prudden, J.; Heideker, J.; Vashisht, A.A.; Wohlschlegel, J.A.; Yates, J.R.; Boddy, M.N. Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem. 2012, 287, 29610–29619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.H.; Wang, Y.; Warren, G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002, 21, 5645–5652. [Google Scholar] [CrossRef]
- Elsasser, S.; Finley, D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 2005, 7, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.M.; Brasseur, C.; Meyer, H.H. The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Np14 and p47, through a common bipartite binding mechanism. J. Biol. Chem. 2004, 279, 49609–49616. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, P.; Buchberger, A.; Schindelin, H. Hierarchical binding of cofactors to the AAA ATPase p97. Structure 2011, 19, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- Kunz, K.; Piller, T.; Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Schunck, K.; Müller, S. SUMO Chains Rule on Chromatin Occupancy. Front. Cell Dev. Biol. 2020, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Rodriguez-Acebes, S.; Specks, J.; Lopez-Contreras, A.J.; Ruppen, I.; Murga, M.; Muñoz, J.; Mendez, J.; Fernandez-Capetillo, O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 2016, 23, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Schimmel, J.; Eifler, K.; Olsen, J.V.; Vertegaal, A.C.O. Ubiquitin-specific Protease 11 (USP11) Deubiquitinates Hybrid Small Ubiquitin-like Modifier (SUMO)-Ubiquitin Chains to Counteract RING Finger Protein 4 (RNF4). J. Biol. Chem. 2015, 290, 15526–15537. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Boddy, M.N. Cooperativity of the SUMO and ubiquitin pathways in genome stability. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Nassar, J.; Peiffer, I.; Stanojcic, S.; Sterkers, Y.; Delamarre, A.; Bocquet, S.; Méchali, M. The ORC ubiquitin ligase OBI1 promotes DNA replication origin firing. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Méndez, J.; Zou-Yang, X.H.; Kim, S.Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Q.; Liao, R.; Sun, P.; Wu, X. The SCF(Skp2) Ubiquitin Ligase Complex Interacts with the Human Replication Licensing Factor Cdt1 and Regulates Cdt1 Degradation*. J. Biol. Chem. 2003, 278, 30854–30858. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Pérez, S.; Cabrera, E.; Amoedo, H.; Rodríguez-Acebes, S.; Koundrioukoff, S.; Debatisse, M.; Méndez, J.; Freire, R. USP37 deubiquitinates Cdt1 and contributes to regulate DNA replication. Mol. Oncol. 2016, 10, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, H.; Sugimoto, N.; Roukos, V.; Nakanishi, Y.; Saijo, M.; Obuse, C.; Tsurimoto, T.; Nakayama, K.I.; Nakayama, K.; Fujita, M.; et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006, 25, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Blow, J.J. Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 2005, 24, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, N.; Kitabayashi, I.; Osano, S.; Tatsumi, Y.; Yugawa, T.; Narisawa-Saito, M.; Matsukage, A.; Kiyono, T.; Fujita, M. Identification of Novel Human Cdt1-binding Proteins by a Proteomics Approach: Proteolytic Regulation by APC/CCdh1. Mol. Biol. Cell 2008, 19, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2006, 8, 84–90. [Google Scholar] [CrossRef]
- Zhong, W.; Feng, H.; Santiago, F.E.; Kipreos, E.T. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 2003, 423, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Ralph, E.; Boye, E.; Kearsey, S.E. DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep. 2006, 7, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kipreos, E.T. The Caenorhabditis elegans Replication Licensing Factor CDT-1 Is Targeted for Degradation by the CUL-4/DDB-1 Complex. Mol. Cell. Biol. 2007, 27, 1394–1406. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009. [Google Scholar] [CrossRef]
- Senga, T.; Sivaprasad, U.; Zhu, W.; Jong, J.P.; Arias, E.E.; Walter, J.C.; Dutta, A. PCNA is a cofactor for CDT1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 2006, 281, 6246–6252. [Google Scholar] [CrossRef] [Green Version]
- Sansam, C.L.; Shepard, J.L.; Lai, K.; Ianari, A.; Danielian, P.S.; Amsterdam, A.; Hopkins, N.; Lees, J.A. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006, 20, 3117–3129. [Google Scholar] [CrossRef] [Green Version]
- Higa, L.A.; Banks, D.; Wu, M.; Kobayashi, R.; Sun, H.; Zhang, H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006, 5, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.O.; Wagener, C.; Marinoni, F.; Kramer, E.R.; Melixetian, M.; Denchi, E.L.; Gieffers, C.; Matteucci, C.; Peters, J.M.; Helin, K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000, 14, 2330–2343. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Diffley, J.F.X. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.; Hoffmann, S.; Komseli, E.S.; Rappsilber, J.; Gorgoulis, V.; Sørensen, C.S. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.; Calzada, A.; Bueno, A. The Cdc6 protein is ubiquitinated in vivo for proteolysis in Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 9092–9097. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.; Drury, L.S.; Diffley, J.F.X. Separate SCF(CDC4) recognition elements target Cdc6 for proteolysis in S phase and mitosis. EMBO J. 2001, 20, 4836–4845. [Google Scholar] [CrossRef] [Green Version]
- Drury, L.S.; Perkins, G.; Diffley, J.F.X. The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J. 1997, 16, 5966–5976. [Google Scholar] [CrossRef] [Green Version]
- Ballabeni, A.; Melixetian, M.; Zamponi, R.; Masiero, L.; Marinoni, F.; Helin, K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J. 2004, 23, 3122–3132. [Google Scholar] [CrossRef] [Green Version]
- McGarry, T.J.; Kirschner, M.W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Pérez, S.; Cabrera, E.; Salido, E.; Lim, M.; Reid, L.; Lakhani, S.R.; Khanna, K.K.; Saunus, J.M.; Freire, R. DUB3 and USP7 de-ubiquitinating enzymes control replication inhibitor Geminin: Molecular characterization and associations with breast cancer. Oncogene 2017, 36, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Charrasse, S.; Gharbi-Ayachi, A.; Burgess, A.; Vera, J.; Hached, K.; Raynaud, P.; Schwob, E.; Lorca, T.; Castro, A. Ensa controls S-phase length by modulating Treslin levels. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Pollok, S.; Grosse, F. Cdc45 degradation during differentiation and apoptosis. Biochem. Biophys. Res. Commun. 2007, 362, 910–915. [Google Scholar] [CrossRef]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 Axis Controls the G2 DNA-Damage-Response Checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Martín, Y.; Cabrera, E.; Amoedo, H.; Hernández-Pérez, S.; Domínguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058–1063. [Google Scholar] [CrossRef]
- McGarry, E.; Gaboriau, D.; Rainey, M.D.; Restuccia, U.; Bachi, A.; Santocanale, C. The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S-PhaseH. Cancer Res. 2016, 76, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of Claspin by SCFβTrCP Restrains Chk1 Activation and Facilitates Recovery from Genotoxic Stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCFβTrCP-Mediated Degradation of Claspin Regulates Recovery from the DNA Replication Checkpoint Response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like Kinase-1 Controls Proteasome-Dependent Degradation of Claspin during Checkpoint Recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustrup, H.; Bekker-Jensen, S.; Bartek, J.; Lukas, J.; Mailand, N. USP7 counteracts SCFβTrCP-but not APCCdh1-mediated proteolysis of Claspin. J. Cell Biol. 2009, 184, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Collyer, T.; Hardy, C.F. Cell cycle regulation of DNA replication Initiator Factor Dbf4p. Mol. Cell. Biol. 1999, 19, 4270–4278. [Google Scholar] [CrossRef] [Green Version]
- Godinho Ferreira, M.; Santocanale, C.; Drury, L.S.; Diffley, J.F.X. Dbf4p, an Essential S Phase-Promoting Factor, Is Targeted for Degradation by the Anaphase-Promoting Complex. Mol. Cell. Biol. 2000, 20, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Weinreich, M.; Stillman, B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 1999, 18, 5334–5346. [Google Scholar] [CrossRef]
- Psakhye, I.; Castellucci, F.; Branzei, D. SUMO-Chain-Regulated Proteasomal Degradation Timing Exemplified in DNA Replication Initiation. Mol. Cell 2019, 76, 632–645.e6. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wei, L.; Peng, X.P.; Zhao, X. Sumoylation of the DNA polymerase ε by the Smc5/6 complex contributes to DNA replication. PLoS Genet. 2019, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Winczura, A.; Appanah, R.; Tatham, M.H.; Hay, R.T.; De Piccoli, G. The S phase checkpoint promotes the Smc5/6 complex dependent SUMOylation of Pol2, the catalytic subunit of DNA polymerase ε. PLoS Genet. 2019, 15, e1008427. [Google Scholar] [CrossRef]
- Wei, L.; Zhao, X. A new MCM modification cycle regulates DNA replication initiation. Nat. Struct. Mol. Biol. 2016, 23, 209–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Albuquerque, C.P.; Liang, J.; Gaut, N.J.; Zhou, H. Molecular circuitry of the SUMO (Small Ubiquitin-like Modifier) pathway in controlling sumoylation homeostasis and suppressing genome rearrangements. J. Biol. Chem. 2016, 291, 8825–8835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346. [Google Scholar] [CrossRef] [Green Version]
- Villa, F.; Fujisawa, R.; Ainsworth, J.; Nishimura, K.; Lie-a-ling, M.; Lacaud, G.; Labib, K.P.M. CUL2LRR1, TRAIP and p97 control CMG helicase disassembly in the mammalian cell cycle. EMBO J. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Sonneville, R.; Moreno, S.P.; Knebel, A.; Johnson, C.; Hastie, C.J.; Gartner, A.; Gambus, A.; Labib, K. CUL-2LRR-1 and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol. 2017, 19, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Dewar, J.M.; Low, E.; Mann, M.; Räschle, M.; Walter, J.C. CRL2Lrr1 promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef] [Green Version]
- Vrtis, K.B.; Dewar, J.M.; Chistol, G.; Wu, R.A.; Graham, T.G.W.; Walter, J.C. Single-strand DNA breaks cause replisome disassembly. Mol. Cell 2021, 81, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Jones, R.M.; Poovathumkadavil, D.; Scaramuzza, S.; Gambus, A. Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci. Alliance 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 2019, 567, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diffley, J.F.X. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat. Cell Biol. 2002, 4, 198–207. [Google Scholar] [CrossRef]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Alabert, C.; Bukowski-Wills, J.C.; Lee, S.B.; Kustatscher, G.; Nakamura, K.; De Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. A SUMO and ubiquitin code coordinates protein traffic at replication factories. BioEssays 2016, 38, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Labib, K. Chromosome duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [Green Version]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Deegan, T.D.; Diffley, J.F.X. MCM: One ring to rule them all. Curr. Opin. Struct. Biol. 2016, 37, 145–151. [Google Scholar] [CrossRef]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.P.; Gambus, A. Mechanisms of eukaryotic replisome disassembly. Biochem. Soc. Trans. 2020, 48, 823–836. [Google Scholar] [CrossRef]
- Gambus, A. Termination of eukaryotic replication forks. Adv. Exp. Med. Biol. 2017, 1042, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The causes and consequences of topological stress during DNA replication. Genes 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Been, D.M.; Champoux, J.J. Breakage of single-stranded DNA by rat liver nicking-closing enzyme with the formation of a DNA-enzyme complex. Nucleic Acids Res. 1980, 8, 6129–6142. [Google Scholar] [CrossRef] [Green Version]
- Deegan, T.D.; Baxter, J.; Ortiz Bazán, M.Á.; Yeeles, J.T.P.; Labib, K.P.M. Pif1-Family Helicases Support Fork Convergence during DNA Replication Termination in Eukaryotes. Mol. Cell 2019, 74, 231–244.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espeli, O.; Levine, C.; Hassing, H.; Marians, K.J. Temporal regulation of topoisomerase IV activity in E. coli. Mol. Cell 2003, 11, 189–201. [Google Scholar] [CrossRef]
- Heintzman, D.R.; Campos, L.V.; Byl, J.A.W.; Osheroff, N.; Dewar, J.M. Topoisomerase II Is Crucial for Fork Convergence during Vertebrate Replication Termination. Cell Rep. 2019, 29, 422–436.e5. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, H.; Marians, K.J. Two distinct modes of strand unlinking during θ-type DNA replication. J. Biol. Chem. 1996, 271, 21529–21535. [Google Scholar] [CrossRef] [Green Version]
- Ishimi, Y.; Sugasawa, K.; Hanaoka, F.; Eki, T.; Hurwitz, J. Topoisomerase II plays an essential role as a swivelase in the late stage of SV40 chromosome replication in vitro. J. Biol. Chem. 1992, 267, 462–466. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.Y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Postow, L.; Crisona, N.J.; Peter, B.J.; Hardy, C.D.; Cozzarelli, N.R. Topological challenges to DNA replication: Conformations at the fork. Proc. Natl. Acad. Sci. USA 2001, 98, 8219–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Dewar, J.M.; Budzowska, M.; Walter, J.C. The mechanism of DNA replication termination in vertebrates. Nature 2015, 525, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Lucas, I.; Germe, T.; Chevrier-Miller, M.; Hyrien, O. Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J. 2001, 20, 6509–6519. [Google Scholar] [CrossRef] [Green Version]
- Zechiedrich, E.L.; Cozzarelli, N.R. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 11801–11805. [Google Scholar] [CrossRef] [Green Version]
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCMZ-7 Function Required for DNA Replication Fork Progression. Science 2000, 288, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Mukherjee, P.; Tatham, M.H.; Hay, R.; Labib, K. Ufd1-Npl4 Recruit Cdc48 for Disassembly of Ubiquitylated CMG Helicase at the End of Chromosome Replication. Cell Rep. 2017, 18, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, A.; Orth, M.; Pirson, P.A.; Sonneville, R.; Blow, J.J.; Gartner, A.; Stemmann, O.; Hoppe, T. CDC-48/p97 Coordinates CDT-1 Degradation with GINS Chromatin Dissociation to Ensure Faithful DNA Replication. Mol. Cell 2011, 44, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef]
- Low, E.; Chistol, G.; Zaher, M.S.; Kochenova, O.V.; Walter, J.C. The DNA replication fork suppresses CMG unloading from chromatin before termination. Genes Dev. 2020, 34, 1534–1545. [Google Scholar] [CrossRef]
- Deegan, T.D.; Mukherjee, P.P.; Fujisawa, R.; Rivera, C.P.; Labib, K. Cmg helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. Elife 2020, 9, 1–33. [Google Scholar] [CrossRef]
- Maculins, T.; Nkosi, P.J.; Nishikawa, H.; Labib, K. Tethering of SCFDia2 to the Replisome Promotes Efficient Ubiquitylation and Disassembly of the CMG Helicase. Curr. Biol. 2015, 25, 2254–2259. [Google Scholar] [CrossRef] [Green Version]
- Mimura, S.; Komata, M.; Kishi, T.; Shirahige, K.; Kamura, T. SCFDia2 regulates DNA replication forks during S-phase in budding yeast. EMBO J. 2009, 28, 3693–3705. [Google Scholar] [CrossRef] [Green Version]
- Morohashi, H.; Maculins, T.; Labib, K. The Amino-Terminal TPR Domain of Dia2 Tethers SCFDia2 to the Replisome Progression Complex. Curr. Biol. 2009, 19, 1943–1949. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.P.; Labib, K.P.M. In Vitro Reconstitution Defines the Minimal Requirements for Cdc48-Dependent Disassembly of the CMG Helicase in Budding Yeast. Cell Rep. 2019, 28, 2777–2783.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fujisawa, R.; Deegan, T.D.; Sonneville, R.; Labib, K.P.M. TIMELESS-TIPIN and UBXN-3 promote replisome disassembly during DNA replication termination in Caenorhabditis elegans. EMBO J. 2021, 1–18. [Google Scholar] [CrossRef]
- Jagannathan, M.; Nguyen, T.; Gallo, D.; Luthra, N.; Brown, G.W.; Saridakis, V.; Frappier, L. A Role for USP7 in DNA Replication. Mol. Cell. Biol. 2014, 34, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchberger, A.; Schindelin, H.; Hänzelmann, P. Control of p97 function by cofactor binding. FEBS Lett. 2015, 589, 2578–2589. [Google Scholar] [CrossRef] [Green Version]
- Fullbright, G.; Rycenga, H.B.; Gruber, J.D.; Long, D.T. p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol. Cell. Biol. 2016, 36, 2983–2994. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Smedegaard, S.; Nakamura, K.; Mortuza, G.B.; Räschle, M.; de Opakua, A.I.; Oka, Y.; Feng, Y.; Blanco, F.J.; Mann, M.; et al. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J. Cell Biol. 2016, 212, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Lalai, A.S.; Hoeijmakers, J.H.J. Fanconi anemia (cross)linked to DNA repair. Cell 2005, 123, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of Disease Susceptibility Pathways in Fanconi’s Anemia and Breast Cancer. N. Engl. J. Med. 2010, 362, 1909–1928. [Google Scholar] [CrossRef] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Long, D.T.; Joukov, V.; Budzowska, M.; Walter, J.C. BRCA1 promotes unloading of the CMG Helicase from a stalled DNA replication fork. Mol. Cell 2014, 56, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e14. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef]
- Akopyan, K.; Silva Cascales, H.; Hukasova, E.; Saurin, A.T.; Müllers, E.; Jaiswal, H.; Hollman, D.A.A.; Kops, G.J.P.L.; Medema, R.H.; Lindqvist, A. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 2014, 53, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128.e3. [Google Scholar] [CrossRef] [Green Version]
- Lemmens, B.; Lindqvist, A. DNA replication and mitotic entry: A brake model for cell cycle progression. J. Cell Biol. 2019, 218, 3892–3902. [Google Scholar] [CrossRef]
- Galarreta, A.; Valledor, P.; Ubieto-Capella, P.; Lafarga, V.; Zarzuela, E.; Muñoz, J.; Malumbres, M.; Lecona, E.; Fernandez-Capetillo, O. USP7 limits CDK1 activity throughout the cell cycle. EMBO J. 2021, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Target | Modification | E3 Ligase | Protease | Organism | Role | Cellular Context | References |
|---|---|---|---|---|---|---|---|
| ORC3/ORC5 | Ubiquitin | OBI1 | Human | Origin selection for firing | Replication initiation, G1/S | [27] | |
| ORC1 | Ubiquitin | SCFSkp2 | Human | Prevent re-licensing | Replication initiation, G1/S | [28] | |
| CDT1 | Ubiquitin | SCFSkp2 | USP37 | Human | Prevent re-licensing | Replication initiation, G1/S | [29,30,31] |
| APC/C-CDH1 | Human, Xenopus | Regulate licensing. Prevent licensing in quiescence. | M/G1 | [32,33] | |||
| CRL4-DDB1CDT2 | Human, yeast, Xenopus, C. elegans, zebrafish | Prevent re-licensing. Prevent licensing upon DNA damage | Replication initiation, G1/S | [31,34,35,36,37,38,39,40,41] | |||
| CDC6 (Cdc18 in yeast) | Ubiquitin | APC/C-CDH1 | Human | Prevent re-licensing. | Early G1 | [42] | |
| Prevent pre-RC assembly | Quiescence | [43] | |||||
| SCFCyclinF | Prevent re-licensing | G2 | [44] | ||||
| SCFCdc4 | Yeast | G1/S | [45,46,47] | ||||
| Geminin | Ubiquitin | APC/C-CDH1 | Dub3, USP7 | Xenopus | Promote origin licensing | M/G1 | [48,49,50] |
| Treslin | Ubiquitin | CRLs | Human | Prevent origin firing | G1/S | [51] | |
| CDC45 | Ubiquitin | APC/C-CDH1 | Human | Prevent origin firing | M/G1 | [52] | |
| Claspin | Ubiquitin | APC/C-CDH1 | USP28, USP29, USP9X | Human | Prevent origin firing. Recovery from G2 checkpoint response. | M/G1 | [53,54,55] |
| SCFβTrCP | USP7 | Human | Recovery from checkpoint response | G2/M | [56,57,58,59] | ||
| DBF4 | Ubiquitin | APC/C-Cdc20 | Yeast | Prevent re-replication. Prevent new pre-RC firing | M/G1 | [60,61,62] | |
| DDK | SUMO | Siz1, Siz2 | Ulp2 | Yeast | Prevent origin firing | Replication initiation | [63] |
| Ubiquitin | Slx5/Slx8 | Yeast | [63] | ||||
| Polymerase ε (Pol2 subunit) | SUMO | Smc5/6 complex (Mms21 subunit) | Yeast | Promote fork progression under replication stress | Elongation, S phase | [64,65] | |
| MCM7 | SUMO | Mms21, Siz1, Siz2 | Ulp2 | Yeast | Prevent origin firing | Replication initiation, G1 | [66,67] |
| Ubiquitin | SCFDia2 | Yeast | Trigger replisome disassembly | Replication termination | [68] | ||
| CRL2LRR1 | C. elegans, Xenopus, mouse embryonic stem cells | [69,70,71] | |||||
| Xenopus | Under lagging strand SSBs | [72] | |||||
| TRAIP | C. elegans, Xenopus, mouse embryonic stem cells | Trigger replisome disassembly | Mitosis | [69,73,74] | |||
| C. elegans, Xenopus | Trigger replisome disassembly | Stalled replisomes upon RS | [73] | ||||
| Xenopus | Trigger replisome disassembly | ICL repair by FA pathway | [75] | ||||
| NEIL3 recruitment | ICL repair by NEIL3 glycosylase pathway | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galarreta, A.; Valledor, P.; Fernandez-Capetillo, O.; Lecona, E. Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1. Int. J. Mol. Sci. 2021, 22, 8796. https://doi.org/10.3390/ijms22168796
Galarreta A, Valledor P, Fernandez-Capetillo O, Lecona E. Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1. International Journal of Molecular Sciences. 2021; 22(16):8796. https://doi.org/10.3390/ijms22168796
Chicago/Turabian StyleGalarreta, Antonio, Pablo Valledor, Oscar Fernandez-Capetillo, and Emilio Lecona. 2021. "Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1" International Journal of Molecular Sciences 22, no. 16: 8796. https://doi.org/10.3390/ijms22168796
APA StyleGalarreta, A., Valledor, P., Fernandez-Capetillo, O., & Lecona, E. (2021). Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1. International Journal of Molecular Sciences, 22(16), 8796. https://doi.org/10.3390/ijms22168796