
Pathological Role of Phosphoglycerate Kinase 1 in Balloon Angioplasty-Induced Neointima Formation

 , and
, and 
Abstract
:1. Introduction
2. Results
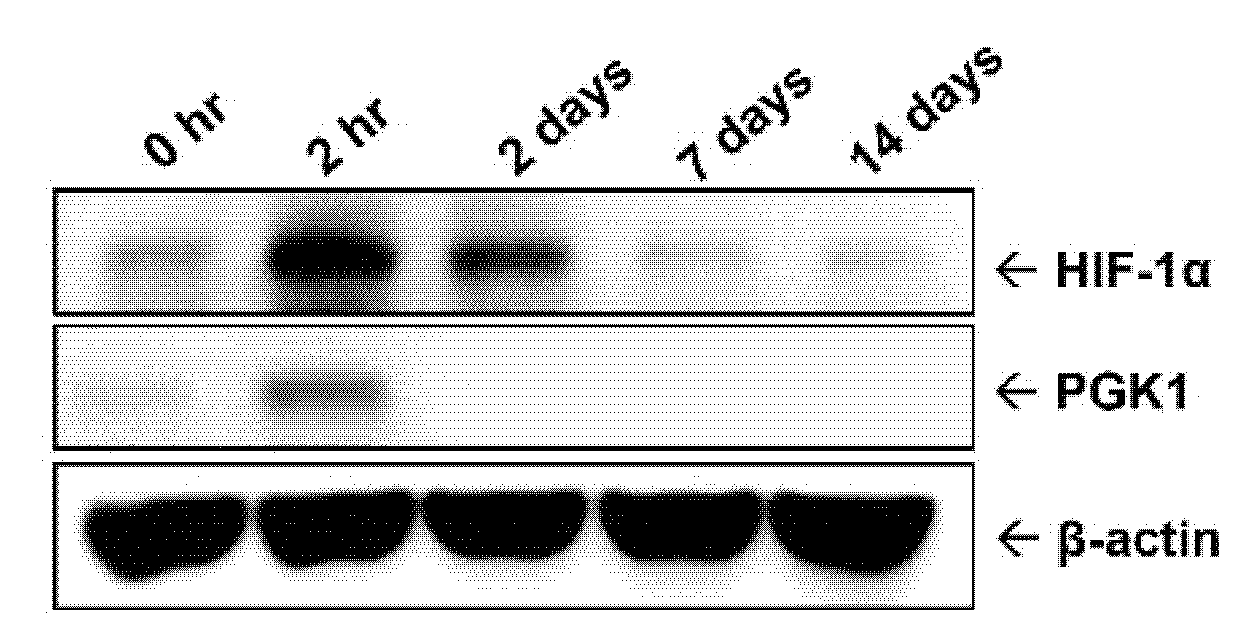
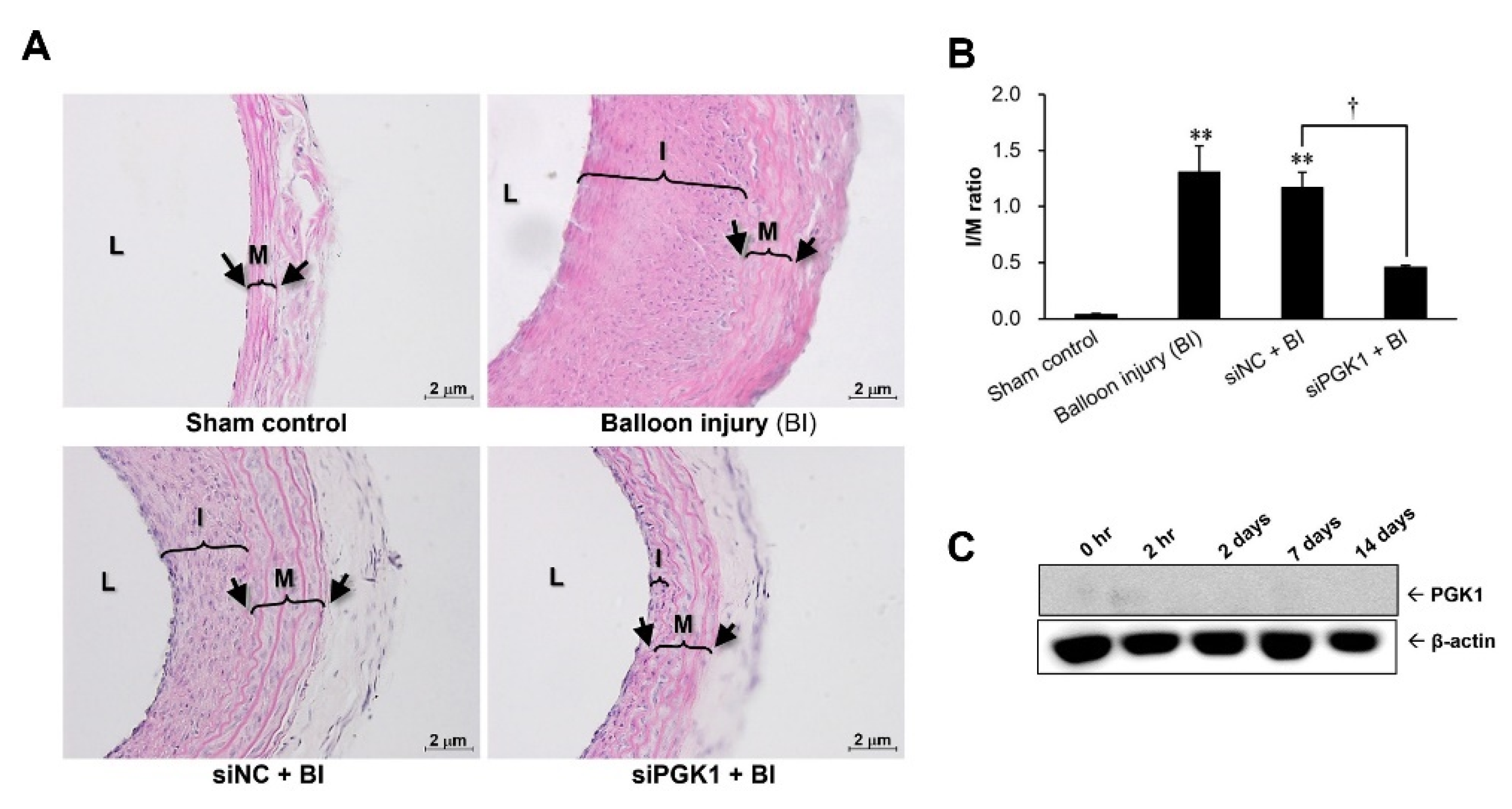
2.1. Protein Level of PGK1 Was Rapidly Elevated in Arteries after Balloon Angioplasty
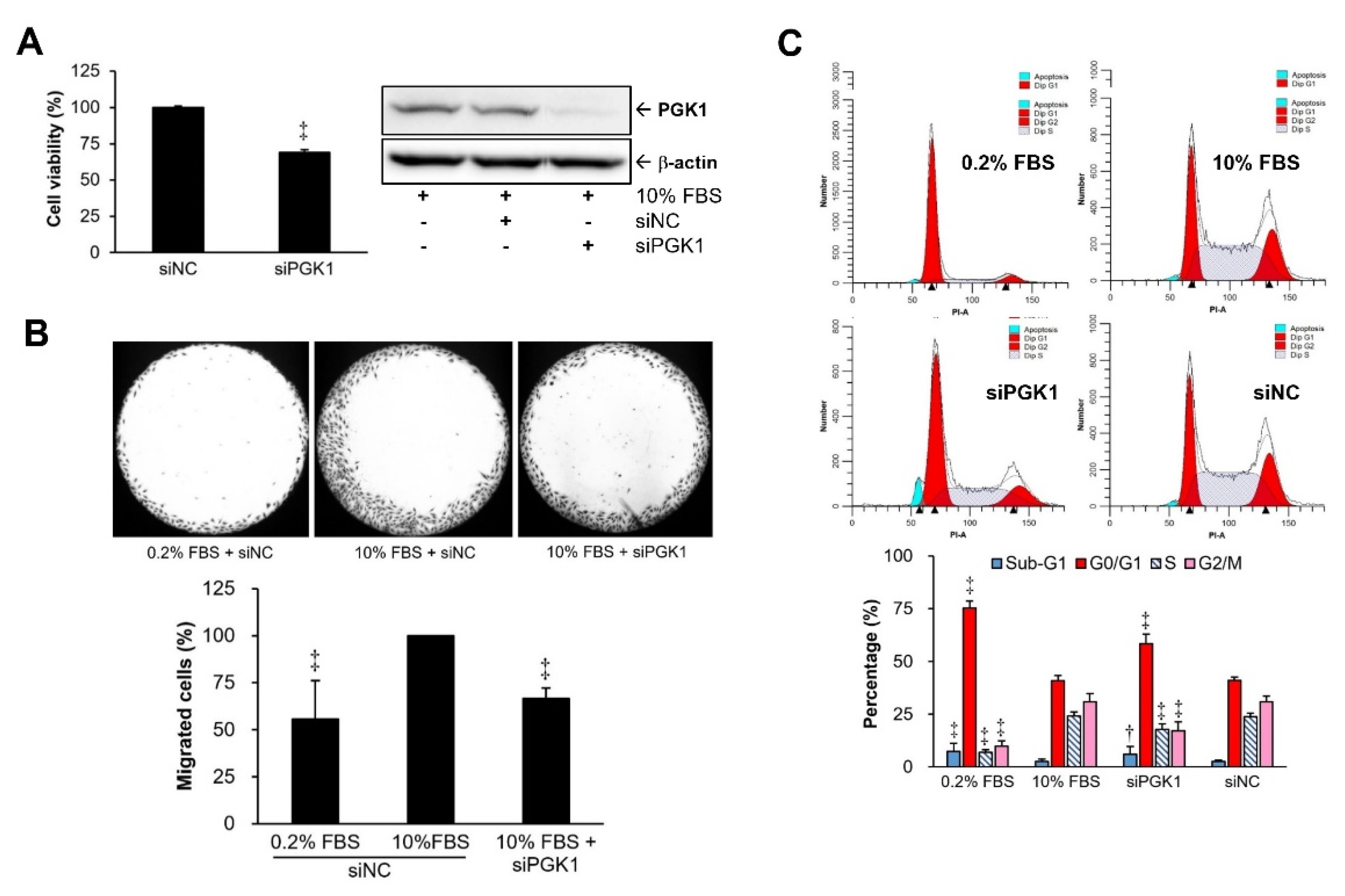
2.2. PGK1 Knockdown Resulted in Decreases in VSMC Proliferation and Migration
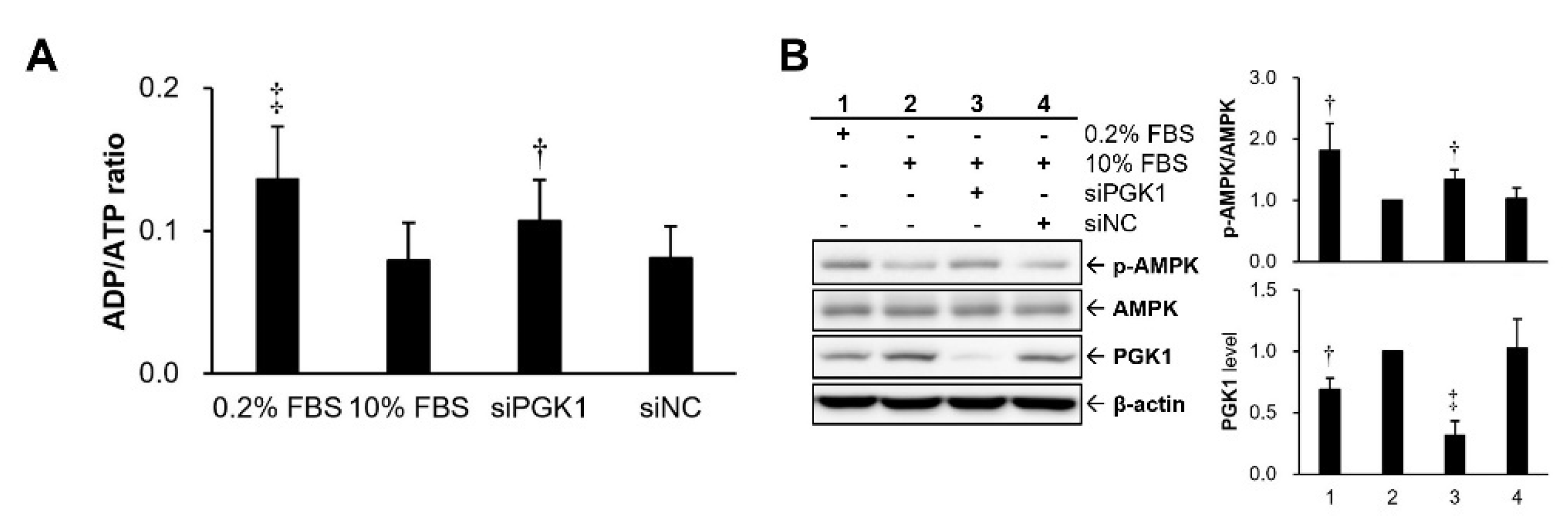
2.3. Silencing PGK1 Increased Intracellular Adenosine Diphosphate (ADP)/ATP Ratio to Trigger Activation of Adenosine 5’-Monophosphate (AMP)-Activated Protein Kinase (AMPK)
2.4. Suppressing PGK1 Can Improve Balloon Injury-Induced Neointimal Formation
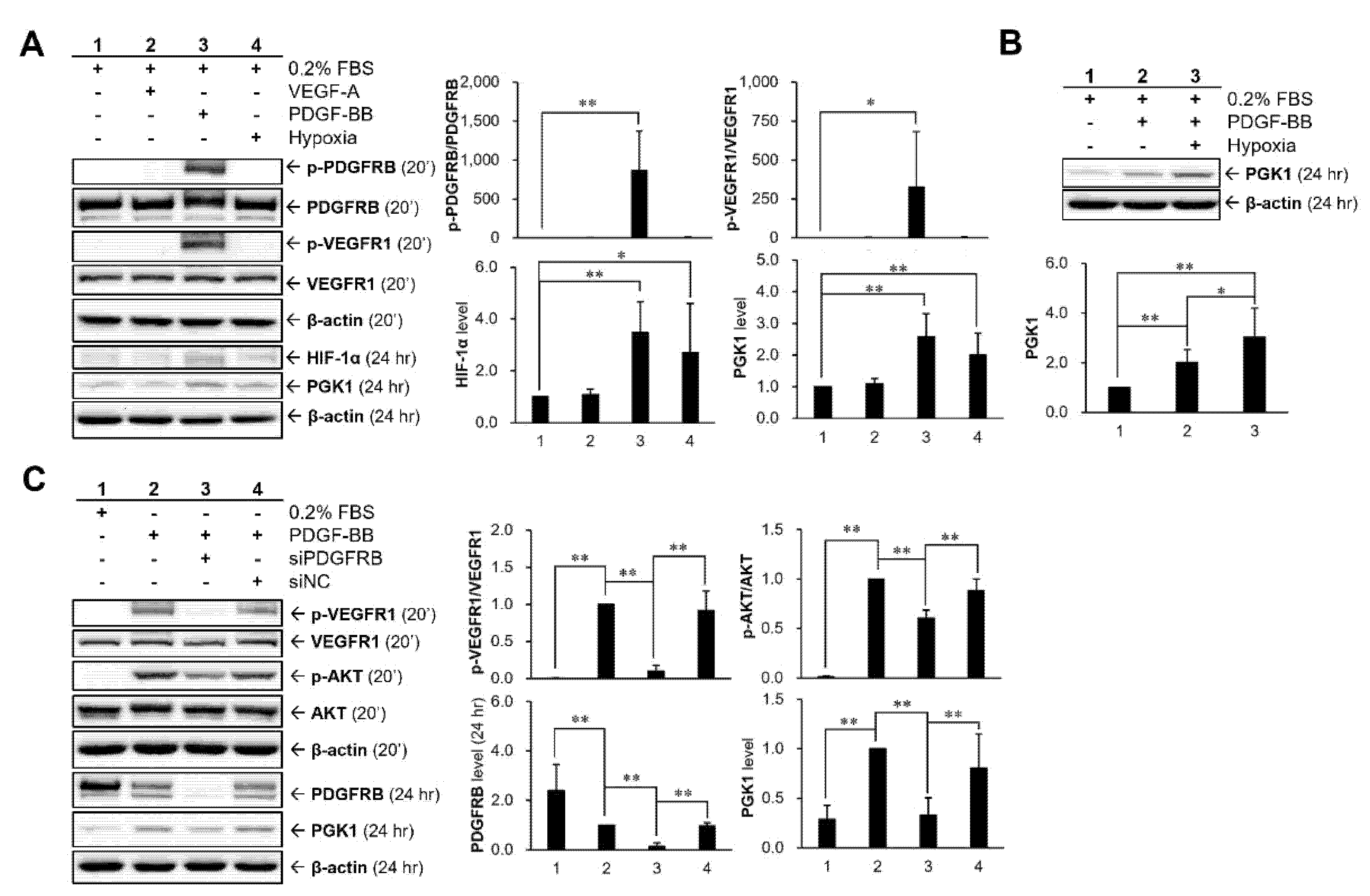
2.5. Hypoxia and PDGF-BB Can Be Served as Stimulatory Factors for PGK1 Expression in VSMCs
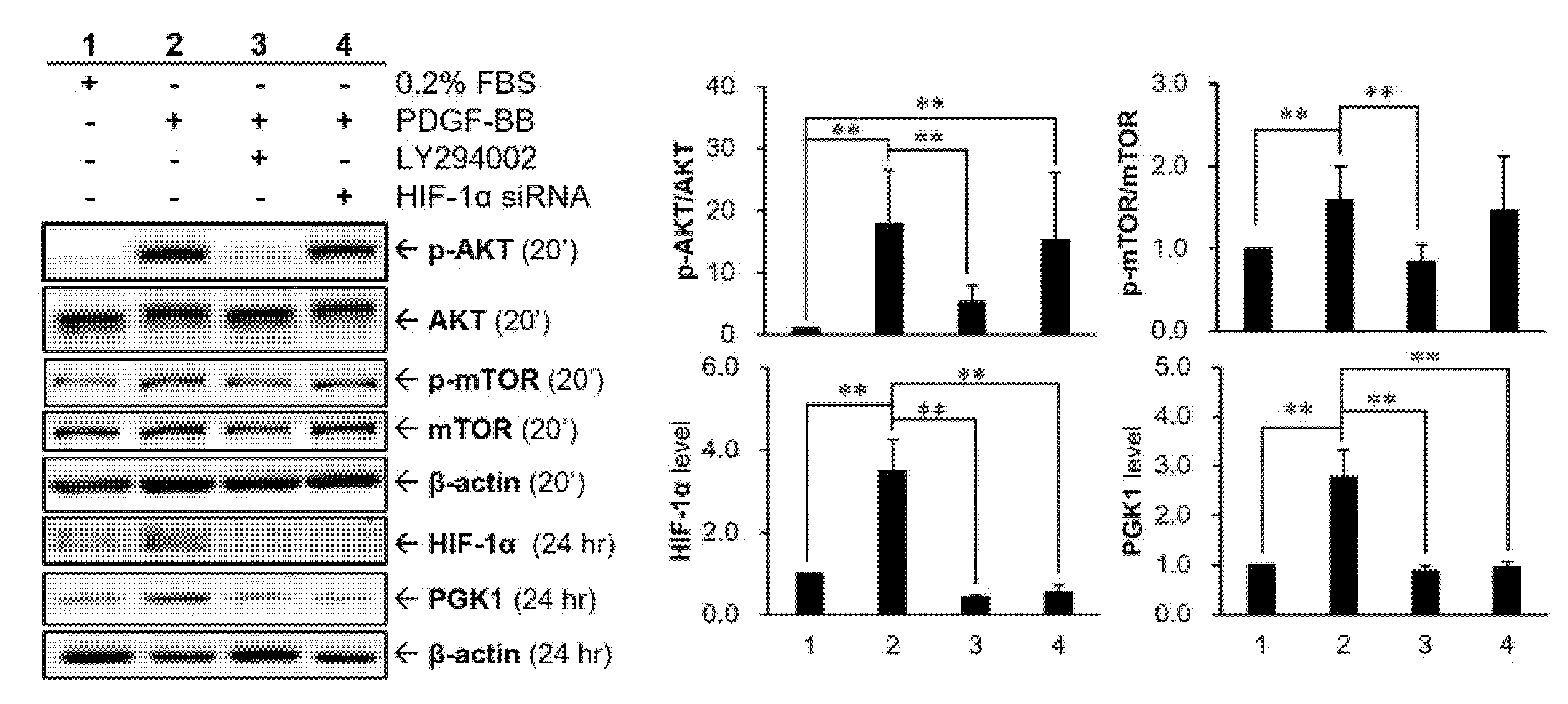
2.6. HIF-1α Participated in PDGF-BB-Mediated PGK1 Expression in VSMCs
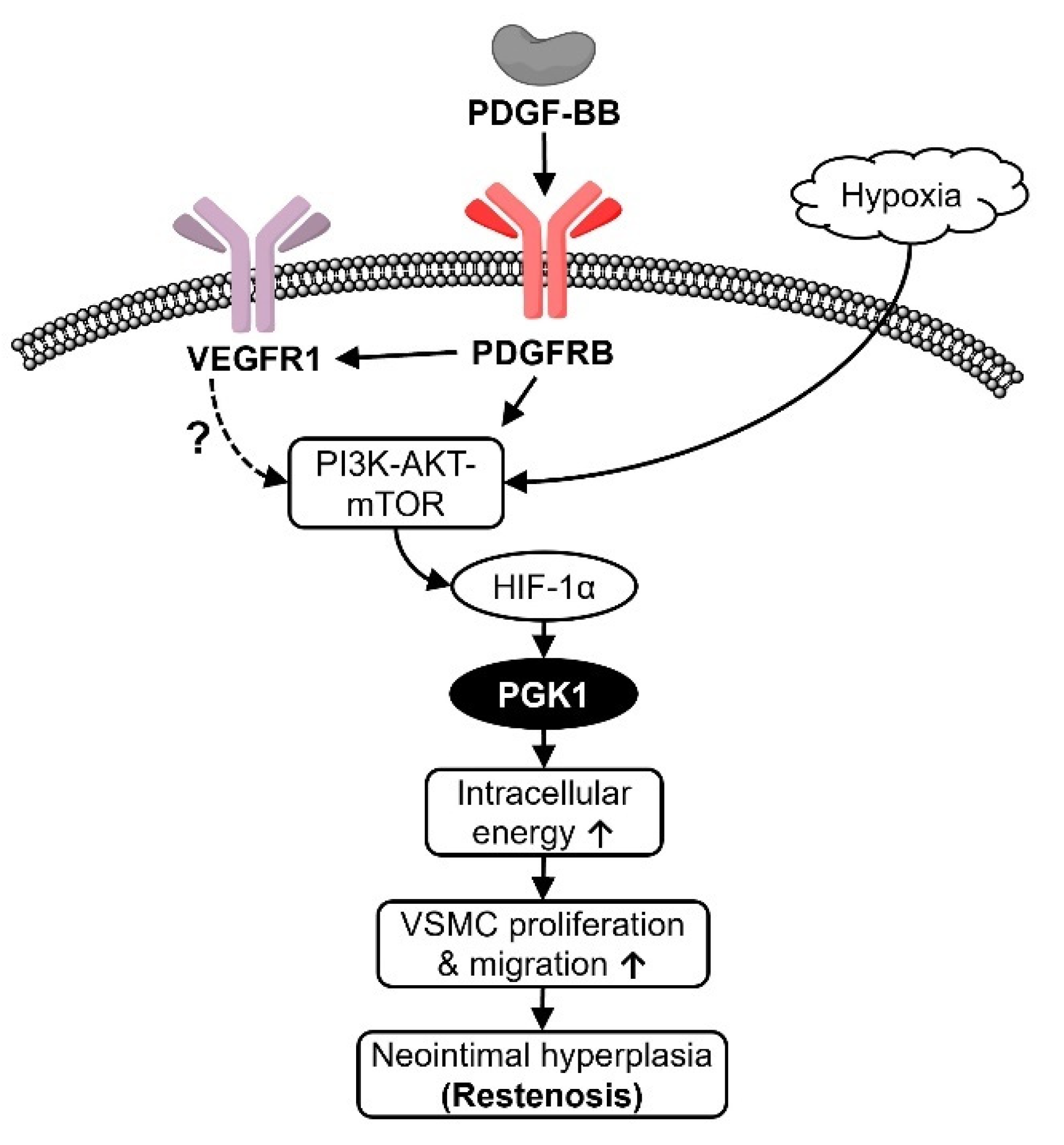
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Transfection of Specific siRNA
4.4. Cell Viability Assay (MTT Assay)
4.5. Cell Migration Assay
4.6. Cell Cycle Analysis
4.7. Measurement of Cellular Energy Status
4.8. Induction of Hypoxic Condition (Anaerobic Cultivation)
4.9. Rat Carotid Artery Balloon Angioplasty
4.10. Protein Extraction and Sample Preparation
4.11. Immunoblot
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pakala, R.; Willerson, J.T.; Benedict, C.R. Effect of serotonin, thromboxane A2, and specific receptor antagonists on vascular smooth muscle cell proliferation. Circulation 1997, 96, 2280–2286. [Google Scholar] [CrossRef]
- Atkinson, S.; Fox, S.B. Vascular endothelial growth factor (VEGF)-A and platelet-derived growth factor (PDGF) play a central role in the pathogenesis of digital clubbing. J. Pathol. 2004, 203, 721–728. [Google Scholar] [CrossRef]
- Parenti, A.; Brogelli, L.; Filippi, S.; Donnini, S.; Ledda, F. Effect of hypoxia and endothelial loss on vascular smooth muscle cell responsiveness to VEGF-A: Role of flt-1/VEGF-receptor-1. Cardiovasc. Res. 2002, 55, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Clowes, A.W.; Reidy, M.A.; Clowes, M.M. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab. Investig. J. Tech. Methods Pathol. 1983, 49, 327–333. [Google Scholar]
- Ross, R.; Glomset, J.; Kariya, B.; Harker, L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc. Natl. Acad. Sci. USA 1974, 71, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyberg, J. Phenotypic modulation of smooth muscle cells during formation of neointimal thickenings following vascular injury. Histol. Histopathol. 1998, 13, 871–891. [Google Scholar]
- Misra, S.; Fu, A.A.; Rajan, D.K.; Juncos, L.A.; McKusick, M.A.; Bjarnason, H.; Mukhopadhyay, D. Expression of hypoxia inducible factor-1 alpha, macrophage migration inhibition factor, matrix metalloproteinase-2 and -9, and their inhibitors in hemodialysis grafts and arteriovenous fistulas. J. Vasc. Interv. Radiol. 2008, 19, 252–259. [Google Scholar] [CrossRef]
- Karshovska, E.; Zernecke, A.; Sevilmis, G.; Millet, A.; Hristov, M.; Cohen, C.D.; Schmid, H.; Krotz, F.; Sohn, H.Y.; Klauss, V.; et al. Expression of HIF-1alpha in injured arteries controls SDF-1alpha mediated neointima formation in apolipoprotein E deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Christoph, M.; Ibrahim, K.; Hesse, K.; Augstein, A.; Schmeisser, A.; Braun-Dullaeus, R.C.; Simonis, G.; Wunderlich, C.; Quick, S.; Strasser, R.H.; et al. Local inhibition of hypoxia-inducible factor reduces neointima formation after arterial injury in ApoE-/- mice. Atherosclerosis 2014, 233, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Fu, D.; He, C.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. PGK1 is a potential survival biomarker and invasion promoter by regulating the HIF-1alpha-mediated epithelial-mesenchymal transition process in breast cancer. Cell Physiol. Biochem. 2018, 51, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, H.; Liao, Y.; Zhu, Y.; Wang, F.; Hou, J. Activation of PGK1 under hypoxic conditions promotes glycolysis and increases stem celllike properties and the epithelialmesenchymal transition in oral squamous cell carcinoma cells via the AKT signalling pathway. Int. J. Oncol. 2020, 57, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Senaratne, S.G.; Pirianov, G.; Mansi, J.L.; Arnett, T.R.; Colston, K.W. Bisphosphonates induce apoptosis in human breast cancer cell lines. Br. J. Cancer 2000, 82, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clézardin, P.; Ebetino, F.H.; Fournier, P.G. Bisphosphonates and cancer-induced bone disease: Beyond their antiresorptive activity. Cancer Res. 2005, 65, 4971–4974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, A. Emerging role of bisphosphonates in the clinic—Antitumor activity and prevention of metastasis to bone. Cancer Treat. Rev. 2008, 34, S25–S30. [Google Scholar] [CrossRef]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J. Biol. Chem. 1995, 270, 13333–13340. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, D.; Kim, K.; Noha, M.; Teramoto, A. Hypoxia inducible factor 1-alpha regulates of platelet derived growth factor-B in human glioblastoma cells. J. Neurooncol. 2006, 76, 13–21. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Roy, H.; Heikura, T.; Yla-Herttuala, S. VEGF-A, VEGF-D and VEGF-D(DeltaNDeltaC) induced intimal hyperplasia in carotid arteries. Eur. J. Clin. Investig. 2005, 35, 669–676. [Google Scholar] [CrossRef]
- Bilder, G.; Wentz, T.; Leadley, R.; Amin, D.; Byan, L.; O’Conner, B.; Needle, S.; Galczenski, H.; Bostwick, J.; Kasiewski, C.; et al. Restenosis following angioplasty in the swine coronary artery is inhibited by an orally active PDGF-receptor tyrosine kinase inhibitor, RPR101511A. Circulation 1999, 99, 3292–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.M.; Zhang, X.; Nelson, P.R.; Odgren, P.R.; Nelson, J.D.; Vasiliu, C.; Park, J.; Morris, M.; Lian, J.; Cutler, B.S.; et al. Temporal evolution of gene expression in rat carotid artery following balloon angioplasty. J. Cell. Biochem. 2007, 101, 399–410. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.C.; Villafranca, J.E.; et al. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a009092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamer, S.B.; Chen, S.; Weddell, J.C.; Palasz, A.; Wittenkeller, A.; Kumar, M.; Imoukhuede, P.I. Discovery of high-affinity PDGF-VEGFR interactions: Redefining RTK dynamics. Sci. Rep. 2017, 7, 16439. [Google Scholar] [CrossRef] [Green Version]
- Ball, S.G.; Shuttleworth, C.A.; Kielty, C.M. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J. Cell Biol. 2007, 177, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Weddell, J.C.; Chen, S.; Imoukhuede, P.I. VEGFR1 promotes cell migration and proliferation through PLCgamma and PI3K pathways. NPJ Syst. Biol. Appl. 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, D.; Greenberg, M.A.; Menegus, M.A.; Breitbart, S. Should all patients undergoing cardiac catheterization or percutaneous transluminal coronary angioplasty receive oxygen? Chest 1994, 105, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: Similarities with the erythropoietin 3’ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef] [Green Version]
- Zieker, D.; Konigsrainer, I.; Tritschler, I.; Loffler, M.; Beckert, S.; Traub, F.; Nieselt, K.; Buhler, S.; Weller, M.; Gaedcke, J.; et al. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int. J. Cancer 2010, 126, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ying, G.; Jung, Y.; Lu, J.; Zhu, J.; Pienta, K.J.; Taichman, R.S. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer Res. 2010, 70, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jindal, H.K.; Vishwanatha, J.K. Functional identity of a primer recognition protein as phosphoglycerate kinase. J. Biol. Chem. 1990, 265, 6540–6543. [Google Scholar] [CrossRef]
- Schito, L.; Rey, S.; Tafani, M.; Zhang, H.; Wong, C.C.; Russo, A.; Russo, M.A.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2707–E2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mermis, J.; Gu, H.; Xue, B.; Li, F.; Tawfik, O.; Buch, S.; Bartolome, S.; O’Brien-Ladner, A.; Dhillon, N.K. Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir. Res. 2011, 12, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, S.; Zhang, S.; Driss, A.; Liu, Z.R.; Kim, H.R.; Wang, Y.; Ritenour, C.; Zhau, H.E.; Kucuk, O.; Chung, L.W.; et al. PDGF upregulates Mcl-1 through activation of beta-catenin and HIF-1alpha-dependent signaling in human prostate cancer cells. PLoS ONE 2012, 7, e30764. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.; Fanburg, B.L.; Beasley, D. Hypoxia and hypoxia-inducible factor-1alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2528–H2534. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.K.; Yang, Z.F.; Ho, D.W.; Ng, M.N.; Yeoh, G.C.; Poon, R.T.; Fan, S.T. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin. Cancer Res. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [Green Version]
- Gowans, G.J.; Hardie, D.G. AMPK: A cellular energy sensor primarily regulated by AMP. Biochem. Soc. Trans. 2014, 42, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B. The Energy Sensor AMPK: Adaptations to exercise, nutritional and hormonal signals. In Hormones, Metabolism and the Benefits of Exercise; Spiegelman, B., Ed.; Springer: Cham, Switzerland, 2017; pp. 13–24. [Google Scholar]
- Kim, S.M.; Yun, M.R.; Hong, Y.K.; Solca, F.; Kim, J.H.; Kim, H.J.; Cho, B.C. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer Ther. 2013, 12, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology 2006, 21, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Zhang, Y.W.; Lee, J.H.; Zeng, S.X.; Wang, Y.V.; Luo, Z.; Dong, X.C.; Viollet, B.; Wahl, G.M.; Lu, H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell. Biol. 2014, 34, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Kudo, F.A.; Muto, A.; Maloney, S.P.; Pimiento, J.M.; Bergaya, S.; Fitzgerald, T.N.; Westvik, T.S.; Frattini, J.C.; Breuer, C.K.; Cha, C.H.; et al. Venous identity is lost but arterial identity is not gained during vein graft adaptation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1562–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gou, R.; Hu, Y.; Liu, O.; Dong, H.; Gao, L.; Wang, S.; Zheng, M.; Li, X.; Lin, B. PGK1 is a key target for anti-glycolytic therapy of ovarian cancer: Based on the comprehensive analysis of glycolysis-related genes. Front. Oncol. 2021, 11, 682461. [Google Scholar] [CrossRef]
- Wang, W.L.; Jiang, Z.R.; Hu, C.; Chen, C.; Hu, Z.Q.; Wang, A.L.; Wang, L.; Liu, J.; Wang, W.C.; Liu, Q.S. Pharmacologically inhibiting phosphoglycerate kinase 1 for glioma with NG52. Acta Pharmacol. Sin. 2021, 42, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhu, J.L.; Zhang, Y.; Zhang, H.; Yang, Y.; Tang, D.R.; Sun, J. PGK1 inhibitor CBR-470-1 protects neuronal cells from MPP+. Aging 2020, 12, 13388–13399. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes (Accession No.) | siRNA Sequences (5′-3′) |
|---|---|
| HIF-1α (NM_024359) | GGAAAGAGAGUCAUAGAAATT (sense) UUUCUAUGACUCUCUUUCCTT (antisense) |
| PDGFRB (NM_031525) | GGUGGUGUUUGAGGCUUAUTT (sense) AUAAGCCUCAAACACCACCTT (antisense) |
| PGK1 (NM_053291) | GUACUGAGAGCAGUAAGAATT (sense) UUCUUACUGCUCUCAGUACTT (antisense) |
| Negative control | UUCUCCGAACGUGUCACGUTT (sense) ACGUGACACGUUCGGAGAATT (antisense) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, C.-H.; Chien, Y.-C.; Sung, M.-S.; Huang, H.-Y.; Sheu, M.-J.; Wu, C.-H. Pathological Role of Phosphoglycerate Kinase 1 in Balloon Angioplasty-Induced Neointima Formation. Int. J. Mol. Sci. 2021, 22, 8822. https://doi.org/10.3390/ijms22168822
Pan C-H, Chien Y-C, Sung M-S, Huang H-Y, Sheu M-J, Wu C-H. Pathological Role of Phosphoglycerate Kinase 1 in Balloon Angioplasty-Induced Neointima Formation. International Journal of Molecular Sciences. 2021; 22(16):8822. https://doi.org/10.3390/ijms22168822
Chicago/Turabian StylePan, Chun-Hsu, Yi-Chung Chien, Min-Shan Sung, Hui-Yu Huang, Ming-Jyh Sheu, and Chieh-Hsi Wu. 2021. "Pathological Role of Phosphoglycerate Kinase 1 in Balloon Angioplasty-Induced Neointima Formation" International Journal of Molecular Sciences 22, no. 16: 8822. https://doi.org/10.3390/ijms22168822
APA StylePan, C.-H., Chien, Y.-C., Sung, M.-S., Huang, H.-Y., Sheu, M.-J., & Wu, C.-H. (2021). Pathological Role of Phosphoglycerate Kinase 1 in Balloon Angioplasty-Induced Neointima Formation. International Journal of Molecular Sciences, 22(16), 8822. https://doi.org/10.3390/ijms22168822