1. Introduction
Diatoms are an abundant group of eukaryotic photosynthetic microalgae, which belong to the pennates (
Bacillariophyceae) class. They occur mainly in marine and freshwater environments. Among diatoms,
Phaeodactylum tricornutum was the first with a completely sequenced genome [
1].
P. tricornutum has the ability to produce significant amounts of omega-3 very long-chain polyunsaturated fatty acids (VLC-PUFAs), especially eicosapentaenoic acid (EPA; 20:5
Δ5,8,11,14,17) constituting up to 30% of total fatty acids [
2].
Omega-3 VLC-PUFAs such as EPA and docosahexaenoic acid (DHA; 22:6
∆4,7,10,13,16,19) are essential dietary nutrients with numerous beneficial effects on human health. Consumption of these fatty acids helps in preventing the onset of cardiovascular diseases, age-associated declines in cognition and promotes visual system development [
3]. The main source of EPA and DHA in the human diet is marine fish. Accelerating overexploitation of marine stocks and contamination of marine environments call for finding new sources of omega-3 VLC-PUFAs [
4]. The marine diatoms (as the primary producers of omega-3 VLC-PUFAs) have become the main candidate for the investigation of genes encoding the enzymes (desaturases and elongases) taking part in the biosynthesis of these high-value polyunsaturated fatty acids [
5].
The biosynthesis of EPA and DHA can occur via two different pathways called “conventional” Δ6-pathway and “alternative” Δ8-pathway. In
P. tricornutum and other marine microorganisms, the “conventional” Δ6-pathway is the most active [
4].
The 18:1 (oleic acid) desaturation was established as the starting point of VLC-PUFAs biosynthesis. Subsequent reactions rely on a series of alternating desaturation and elongation steps. Enzymes responsible for the production of VLC-PUFAs, fatty acids composed of more than 18 carbon atoms, are located in the endoplasmic reticulum [
1]. The EPA biosynthetic pathway was reconstituted in yeast using genes coding Δ5-desaturase, Δ6-desaturase from
P. tricornutum, and Δ6-elongase from
Physcomitrella patens. Further investigations of the substrate selectivity of Δ5- and Δ6- desaturases revealed that they convert omega-3 and omega-6 substrates with similar efficiencies [
6]. Using pulse-chased experiments with [
14C] fatty acids, Arao and Yamada [
7] proposed the predominant route of EPA biosynthesis in
P. tricornutum. The first step in this pathway is Δ6-desaturation of linoleic acid (LA; 18:2
Δ9,12) to γ-linolenic acid (GLA; 18:3
Δ6,9,12), which is then Δ15-desaturated to produce stearidonic acid (SDA; 18:4
Δ6,9,12,15). SDA is elongated by two carbons and desaturated to yield eicosapentaenoic acid (EPA; 20:5
Δ5,8,11,14,17). In diatoms that can synthesize DHA, created EPA can undergo the second step of elongation to docosapentaenoic acid (DPA; 22:5
Δ7,10,13,16,19) by the action of Δ5-elongase. Subsequently, DPA can be desaturated to DHA by Δ4-desaturase. However,
P. tricornutum produces DHA in trace levels only [
6]. After biosynthesis in the endoplasmic reticulum, EPA can be transported to the plastid and incorporated into galactolipids, especially MGDG and DGDG [
8,
9]. The mechanism of trafficking EPA between the endoplasmic reticulum and plastid is unclear and requires further investigations [
1,
9]. It is speculated that direct contact of the outer plastidial membrane and ER membrane can facilitate EPA translocation [
1].
The family of enzymes called acyl-CoA:lysophospholipids acyltransferase (LPLAT) occurs commonly in animals, plants, and microorganisms. LPLATs can be divided into groups named, e.g., LPAAT, LPEAT, LPCAT. These groups of enzymes differ from each other in substrate specificity toward fatty acid acceptors. LPAAT preferentially uses LPA and LPEAT—LPE as fatty acid acceptors. LPCAT group of enzymes is characterized by their highest specificity for lysophosphatidylcholine [
10,
11]. LPLATs in forward reaction use lysophospholipids (LPLs) and acyl-CoA to produce phospholipids (PLs). In reverse reaction, they produce LPL and acyl-CoA from PL and coenzyme A. LPCATs play the most important role in supplying the cytosolic acyl-CoA pool with acyl groups from PL. They catalyze preferentially the turnover of acyls between PC (the main membrane lipids and place of fatty acids modification) and acyl-CoA pools [
10,
11,
12,
13]. The genes coding LPCAT enzymes were cloned, and LPCATs from various plants were characterized, e.g.,
Arabidopsis thaliana [
14],
Brassica napus [
15],
Nicotiana benthamiana [
16],
Ricinus communis [
7],
Hiptage benghalensis,
Lesquerella fendleri,
Carthamus tinctorius [
12], and
Helianthus annuus [
17].
Synthesis of EPA in
P. tricornutum is highly efficient, which implies that this diatom has developed a high ability to channel intermediates of its biochemical pathway between different lipid pools [
8]. Desaturases from
P. tricornutum are phospholipid-dependent, as opposed to elongases, which are acyl-CoA-dependent [
18]. Therefore, it is predicted that this organism has a highly efficient mechanism of acyl exchange between PC pool and acyl-CoA pool catalyzed by the action of LPCATs, similar to the one present in plants [
8].
Transgenic oilseed plants with introduced EPA and DHA biosynthetic pathways derived from marine diatoms or other microorganisms synthesizing such fatty acids could become alternative sources of omega-3 VLC-PUFA. Many attempts have been made to produce such transgenic plants. However, low product accumulation obtained so far necessitates further improvement to allow for profitable industrial-scale production of such fatty acids [
19]. That low efficiency indicates some metabolic bottlenecks in these transgenic plants. One of these bottlenecks could be the substrate dichotomy between desaturases and elongases derived from diatoms. It is suggested that LPCAT type of enzymes from marine organisms might overcome substrate dichotomy by switching intermediates between acyl-CoA pool and PC. The action of these enzymes may lead to increased yields of EPA in transgenic plants due to potentially different characteristics of LPCATs from marine diatoms compared to those find in plants [
20].
LPCATs from microalgae have not been characterized yet, so in this study, we investigate the biochemical properties, positional specificity, and substrate specificity of PtLPCAT1 cloned by us. Additionally, we investigated substrate specificity of LPCATs of a microsomal fraction of developing seeds of Camelina sativa toward different acyl-CoA with polyunsaturated fatty acids. Based on the results of this study and literature data, we discuss the similarities and differences between LPCATs from diatoms and plants.
3. Discussion
Until now, biochemical characterization of LPLAT enzymes from diatoms has not been performed. Nevertheless, putative genes encoding LPLAT enzymes have been identified in the
P. tricornutum genome by homology in a genome database [
21]. In our studies, we have cloned six such genes, which, after introduction to yeast
ale1 mutant, passed the lyso-PAF sensitivity test. From these six genes, only “Phatr3_J20460” produced in the yeast system a very active enzyme with LPCAT activity. In the presented work, we have characterized the biochemical properties and substrate specificities of this
PtLPCAT1 using in vitro assays applying different radioactive and non-radioactive substrates and microsomal fractions from yeast
ale1 mutant harboring the “Phatr3_J20460” gene as an enzyme source.
In contrast to diatom, the plant LPCAT enzymes have been much better characterized [
11,
12,
17]. Currently, it is considered that they are, at least partially, responsible for controlling of acyl composition of membrane phospholipids (particularly phosphatidylcholine) and cytosolic acyl-CoA pool via forward and backward reactions. It is assumed that they are one of the most important players in supplying cytosolic acyl-CoA pool with polyunsaturated fatty acids used further in phospholipids acyl editing process, storage lipid biosynthesis, or for elongation [
11,
12]. Mühlroth et al. [
22] speculated that in diatoms, LPCAT type of enzymes could also play an important role in polyunsaturated fatty acids flux between PC and cytosolic acyl-CoA pool during the very long-chain polyunsaturated fatty acid biosynthesis process. They assumed, however, that most likely, these enzymes have different substrate specificities in comparison to the plant LPCATs.
In the presented studies, we have shown that
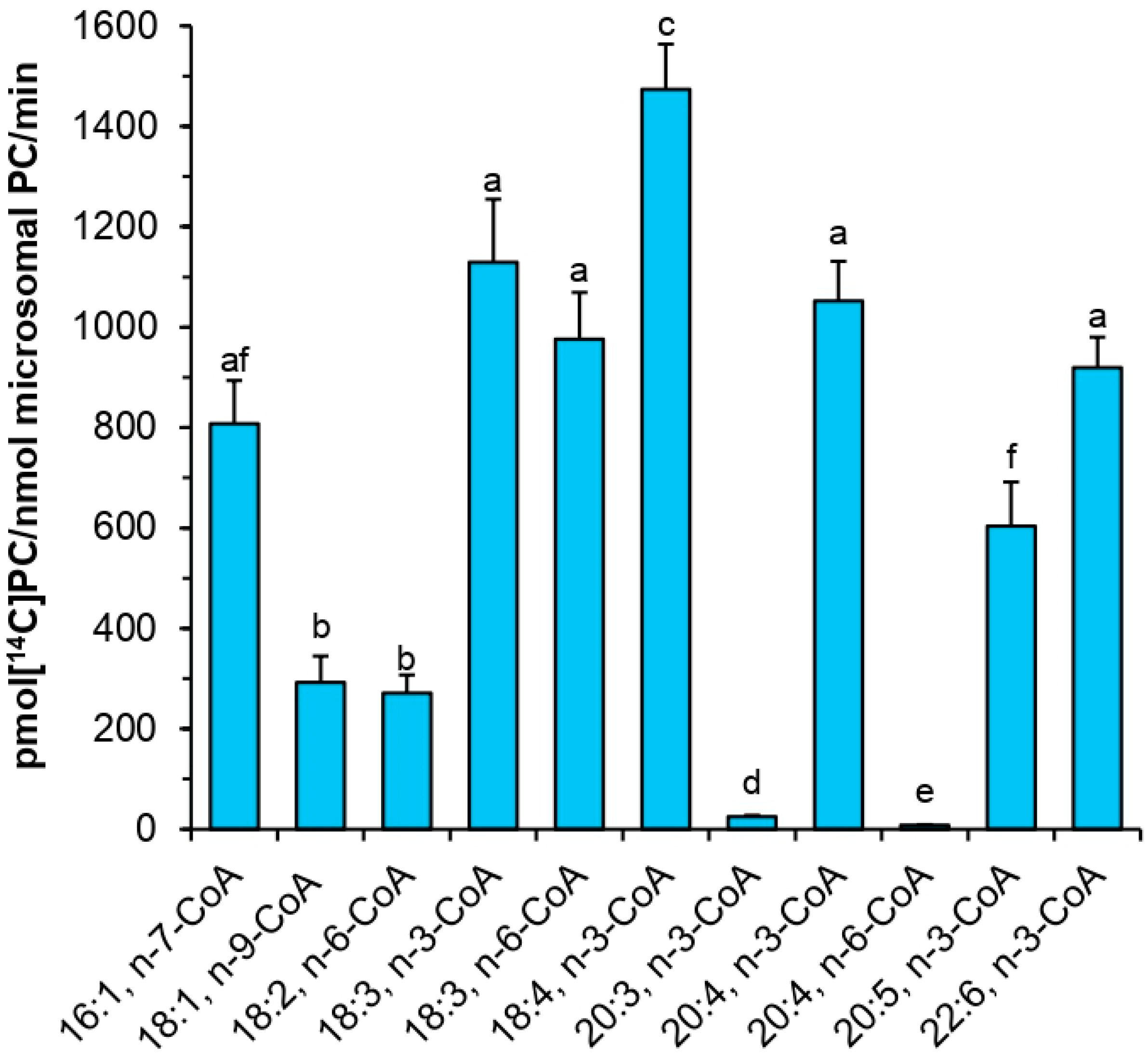
PtLPCAT1 was active toward various acyl-CoA with unsaturated fatty acids. The highest activity was observed toward 18:4-CoA n-3, and still high toward 18:3-CoA n-3 > 20:4-CoA n-3 > 18:3-CoA n-6 > 22:6-CoA n-3 > 16:1-CoA n-7 > 20:5-CoA n-3 > 18:1-CoA n-9 > 18:2-CoA n-6. Contrary to that, acyl-CoA with saturated fatty acids was almost not accepted by the enzyme. Surprisingly, its activity toward 20:3-CoA n-3 and toward 20:4-CoA n-6 was also very low. Thus, it seems that for the tested
PtLPCAT1 activity, not only the presence of double bonds is important, but also their place in the acyl moieties. When the first double bond is located further than at Δ9 position, such as in the mentioned 20:3-CoA (first double bond is at Δ11 position) or the last double bond is located closer than at n-3 in fatty acids longer than 18C (such as in 20:4-CoA n-6) it seems that acyl-CoA with such fatty acids cannot be accepted (or are used very badly) by the enzyme. The tested
PtLPCAT1 activity toward acyl-CoA with unsaturated C18 acyl groups was similar to the substrate specificity of plant LPCATs [
11,
12]. However, some differences were noted in the case of saturated fatty acids. Plant LPCATs seem to be able to better use such acyl-CoA. It was shown that
B. napus and
C. sativa LPCATs exhibited activity toward 16:0-CoA and 18:0-CoA [
11,
16] higher than the tested
PtLPCAT1. The use of acyl-CoA with VLC-PUFA by plant LPCATs was not tested so far. Klińska et al. [
11] showed that
C. sativa LPCATs cannot use 20:1-CoA and 22:1-CoA; however, fatty acids in these acyl-CoA have the first double bond in a position further than at Δ9; thus, the situation can be similar such as with 20:3-CoA (n-3) tested in this study. The data obtained in the presented studies revealed, however, that at least LPCATs of microsomal fractions of
C. sativa developing seeds cannot use not only 20:3-CoA (n-3) and 20:4-CoA (n-6) such as
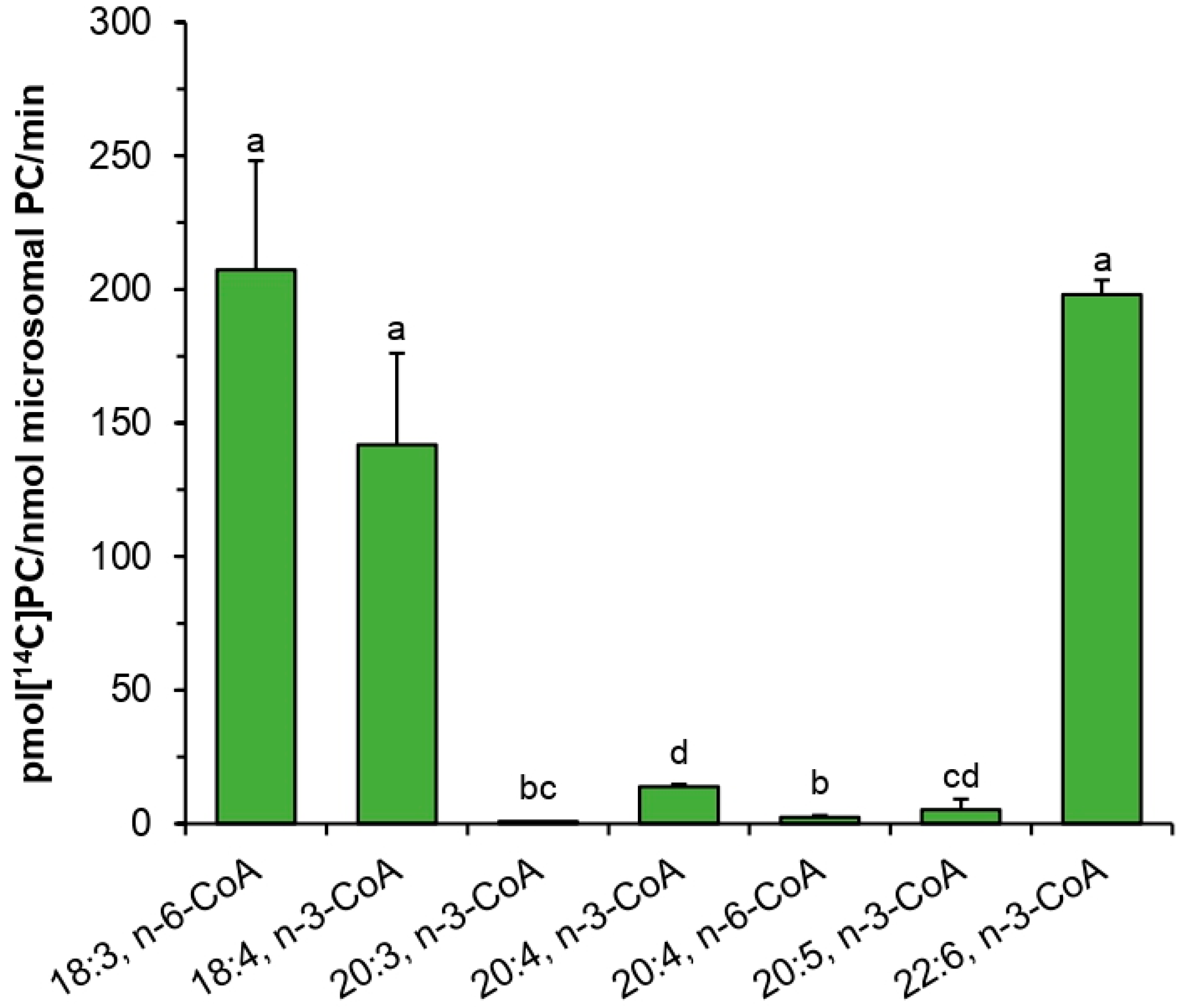
PtLPCAT1 but also 20:4-CoA (n-3) and 20:5-CoA (n-3).
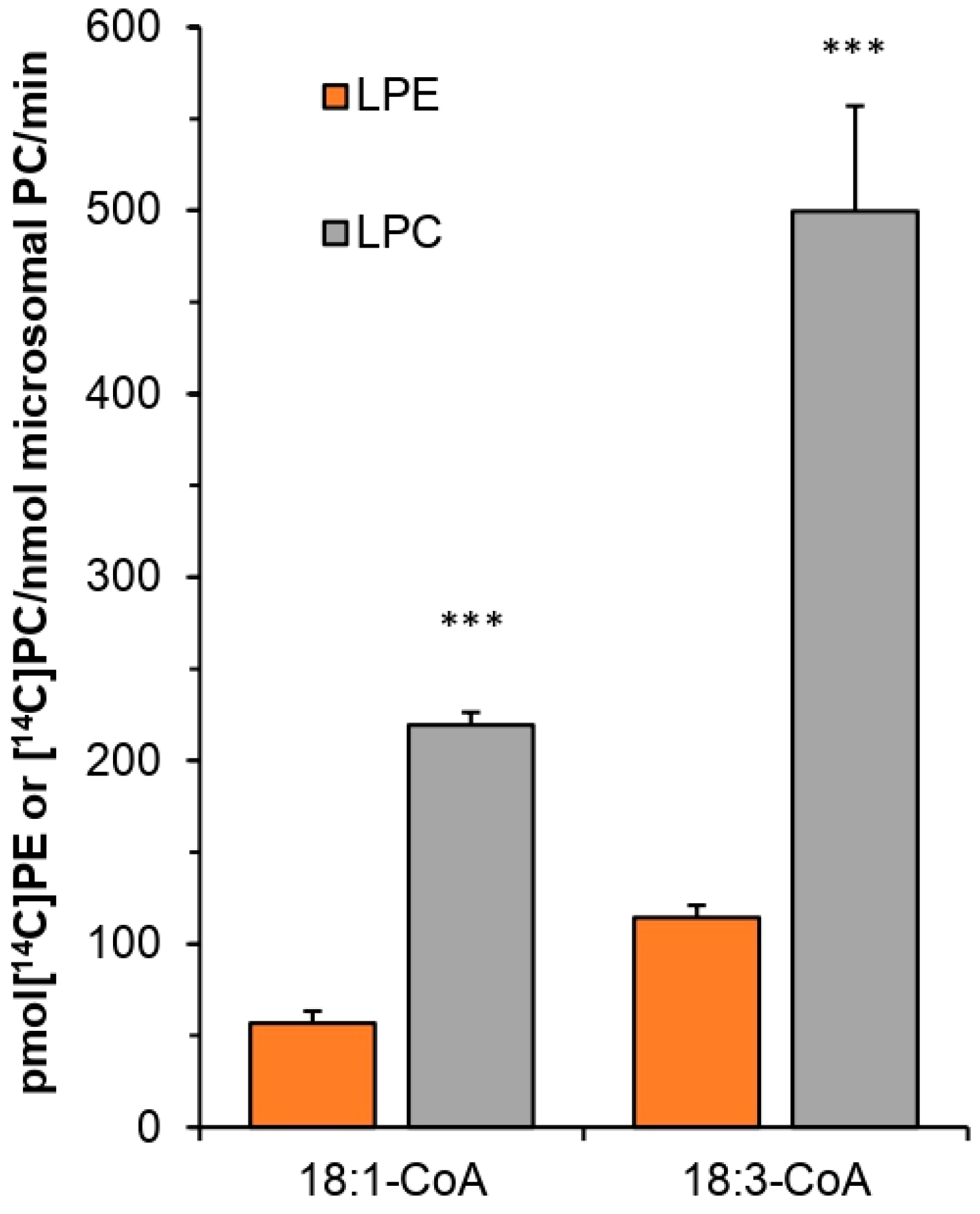
Obtained results are confirmation of earlier assumptions that
PtLPCATs may play an important role during the VLC-PUFAs biosynthetic pathway. Tested
PtLPCAT1 showed high substrate specificity toward acyl-CoA derivatives of 20:4 (ETA, n-3). This is in agreement with the proposed biosynthesis pathway of EPA and DHA in
P. tricornutum through 20:4 (n-3), but not by 20:4 (n-6) [
1]. Taking into consideration that the Δ5 desaturation of 20:4 (ETA) to 20:5 (EPA) takes place in PC, and it is the last step of the EPA biosynthesis pathway, the preceding action of
PtLPCAT1 can play the major role in the production of the high level of EPA in
P. tricornutum. Moreover, ETA fatty acid constitutes only approximately 2.2% of the fatty acid profile of
P. tricornutum cultivated under normal growth conditions [
1]; therefore, ETA fatty acids are probably quickly introduced to PC for desaturation by the action of
PtLPCAT1. These findings could suggest that the tested
PtLPCAT1 can potentially eliminate the acyltransferase bottlenecks in transgenic oilseed plants producing EPA and DHA fatty acids [
23], as at least LPCATs of
C. sativa seeds cannot use 20:4-CoA (n-3), which is the key step in the biosynthetic pathway of EPA and in consequence also in DHA fatty acids.
In the biosynthesis of VLC-PUFAs, not only the introduction of proper acyl-CoA to the PC pool for the desaturation process is important, but also the transfer of some intermediates after modification in PC to cytosol for elongation. The tested PtLPCAT1 can also be involved in this process. It has been proven that these types of enzymes in the backward reaction can transfer fatty acids from PC to the acyl-CoA pool (see Introduction). We have not tested this, however, in this study. It will be one of the subjects of our future research. It will require some modifications of microsomal membranes used in the studies with PC species containing different very long-chain polyunsaturated fatty acids. We are currently developing the precise protocol for performing these experiments.
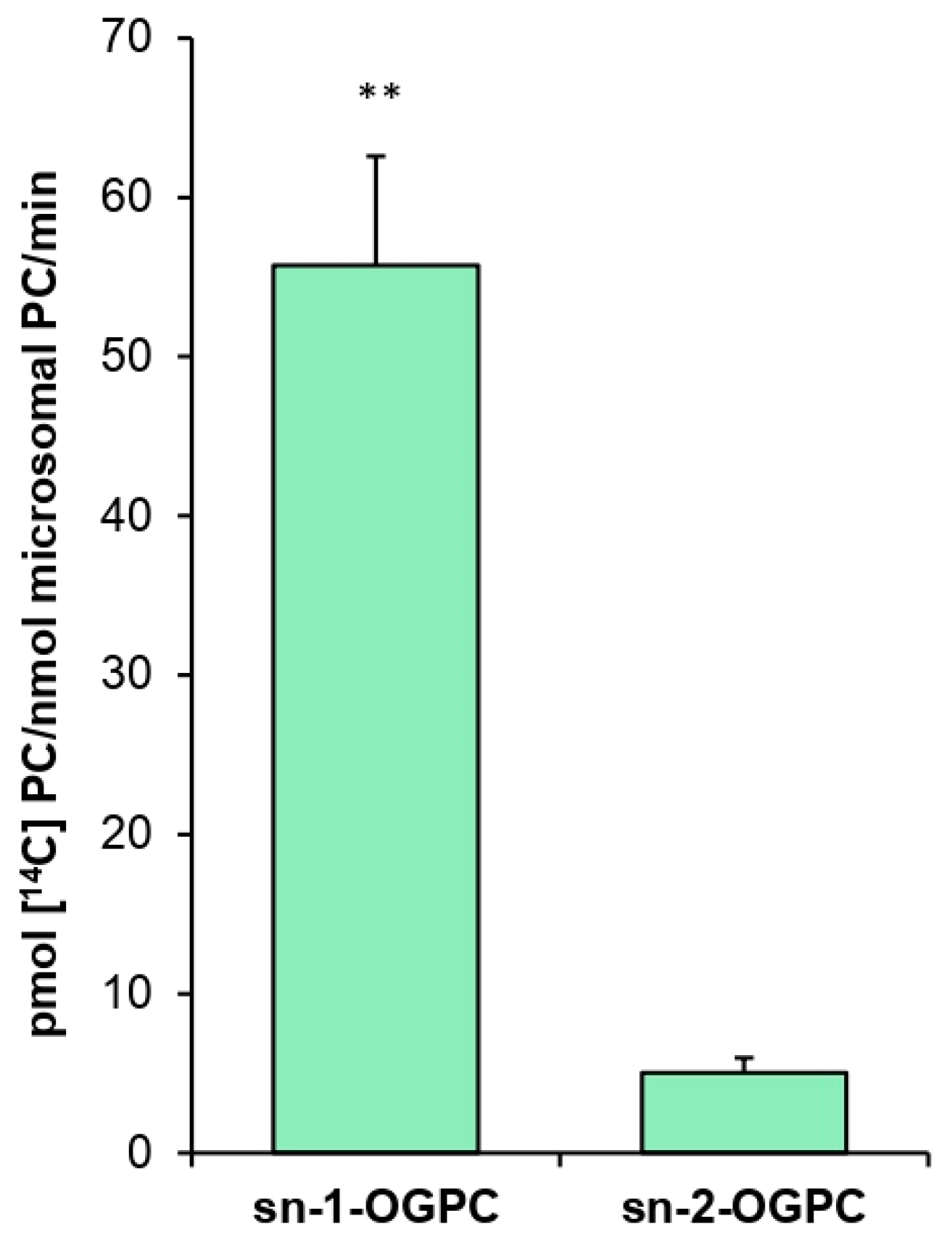
The positional specificity of
PtLPCAT1 proved to be 11 times higher toward the
sn-2 position than toward the
sn-1 position of LPC when 18:1-CoA was used as a fatty acid donor. A similar result was obtained, for example, in an experiment conducted by Lager et al. [
12], who demonstrated that LPCAT1 and LPCAT2 from
A. thaliana are 7–8 times more efficient toward the
sn-2 position than toward the
sn-1 position of LPC with 18:1-CoA as acyl donor. Klińska et al. [
11] arrived at the same conclusion in the case of
C. sativa, which LPCATs were approximately eight times more active toward
the sn-2 position. These data indicate that the positional specificity of the tested
PtLPCAT1 does not differ significantly from that of plant LPCATs.
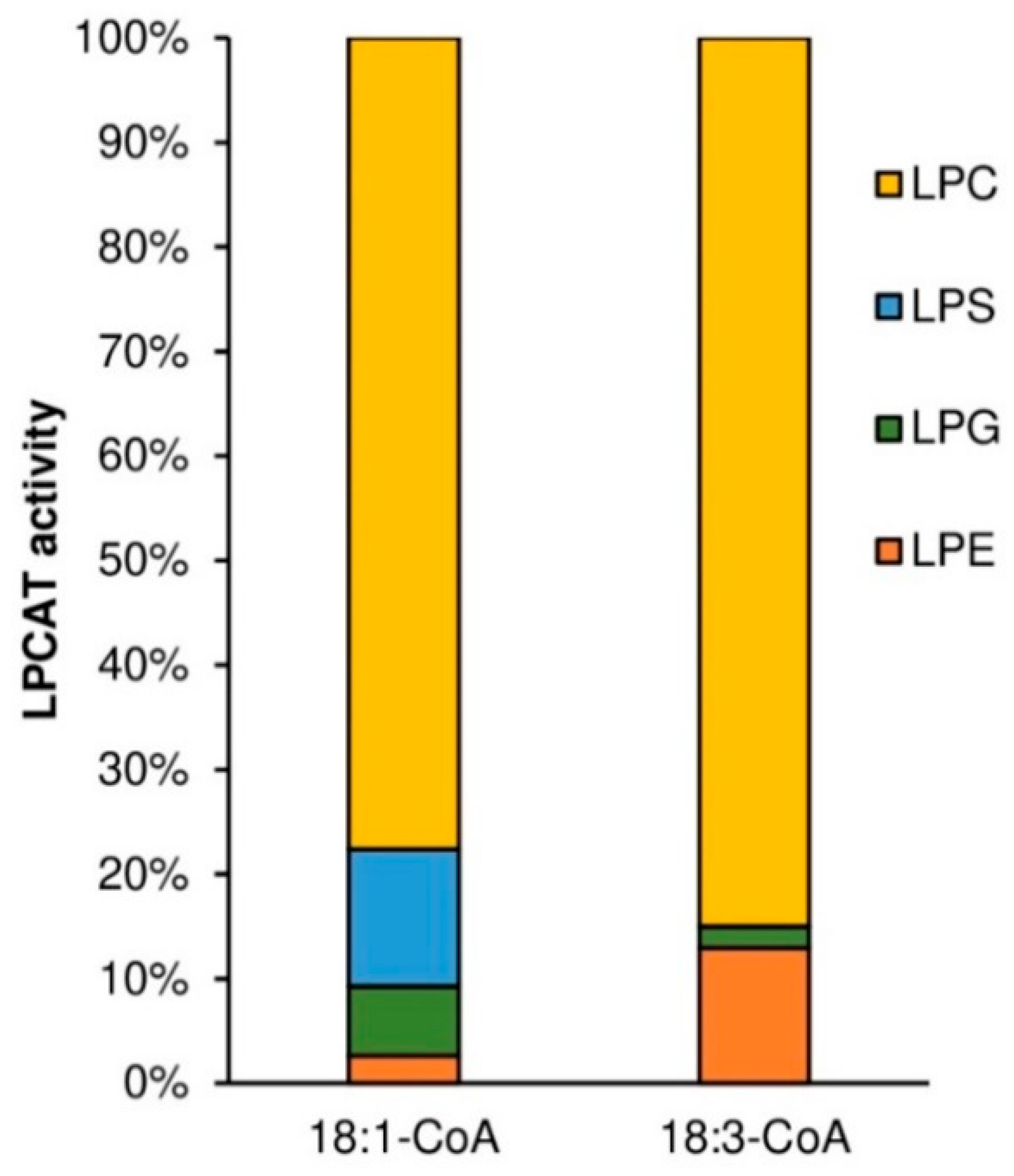
Similar to plant and yeast LPCATs, the tested
PtLPCAT1 also showed specificity toward different lysophospholipids [
10,
14]. Similar to the previously mentioned LPCATs, the highest activity was toward LPC. However, the differences in specificity toward, e.g., LPC and LPE were smaller than presented by plant LPCATs and much more similar to specificity presented by yeast ALE1 in single LPL assays (the competence tests with a mixture of different LPLs were not performed for other LPCATs). From among different LPC used in our assays, the tested
PtLPCAT1 preferentially used
sn-1-16:0-LPC and with somewhat lower intensity
sn-1-18:1-LPC and
sn-1-18:0-LPC and was almost not active toward
sn-1-20:0-LPC. The previous research concerning plant LPCATs was almost exclusively based on
sn-1-18:1-LPC [
10,
11,
14]. Lager et al. [
12] usage of
sn-1-ric-LPC (LPC with ricinoleic acid) resulted in a lower affinity of tested plant LPCATs than toward
sn-1-18:1-LPC.
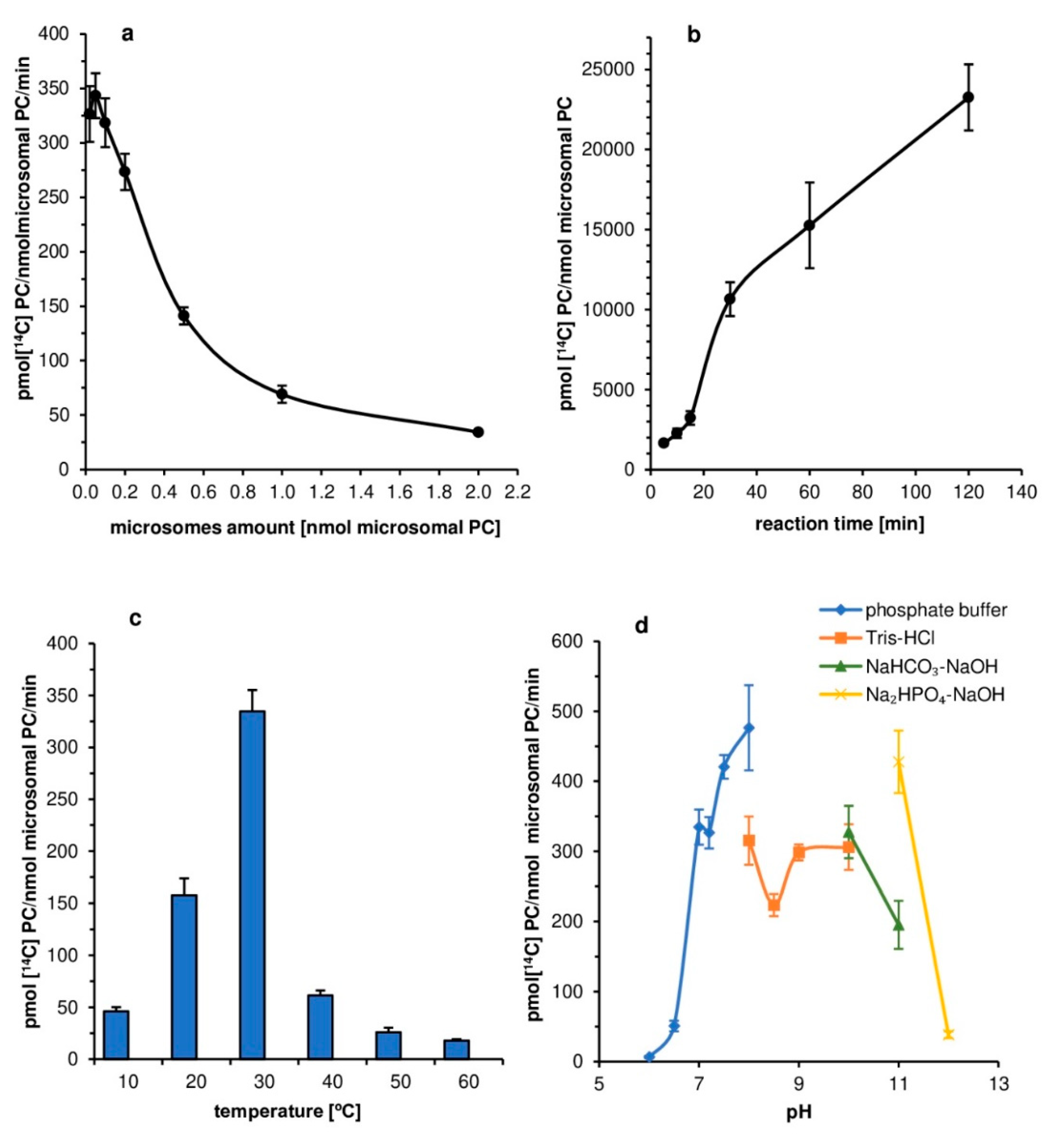
The tested
PtLPCAT1 was the most active at 30 °C, and its activity sharply decreased at either higher or lower temperatures. The 30 °C was also the optimal temperature for
C. sativa LPCATs; however, in that case, the temperatures below and higher than optimal caused less drastic changes [
11]. The physiological significance of this high-temperature dependency of
PtLPCAT1 is unknown. However, for
P. tricornutum the temperatures below 15 °C and above 21 °C are regarded as stress conditions [
24]. Yongmanitchai and Ward [
25] reported that the EPA production is most effective in
P. tricornutum in a temperature range of 21.5–23 °C; however, the optimal temperature for growth is estimated as 15 to 20 °C. The observed doubling of activity of
PtLPCAT1 between 20 and 30 °C could be in some way connected with this stress increase in EPA production.
The pH-level dependency of the analyzed diatom LPCAT activity was somewhat similar to that expressed by LPCATs of
C. sativa seeds’ microsomal fraction [
11]. Both types of LPCAT were most active in alkaline pH (pH 8–11); however, some differences in the dependency curves were noted. The maximum LPCAT activity in alkaline pH was also reported in the case of
B. napus [
26,
27] and mice [
28]. Thus, it seems to be a common feature of LPCAT type of enzymes, with, however, yet unknown physiological significance. When
P. tricornutum is cultivated in high alkaline pH, cells are mobilized to produce more lipids than during the cultivation in neutral pH [
29,
30]. However, so far, there is no evidence to connect this with the high activity of LPCAT at such pH.
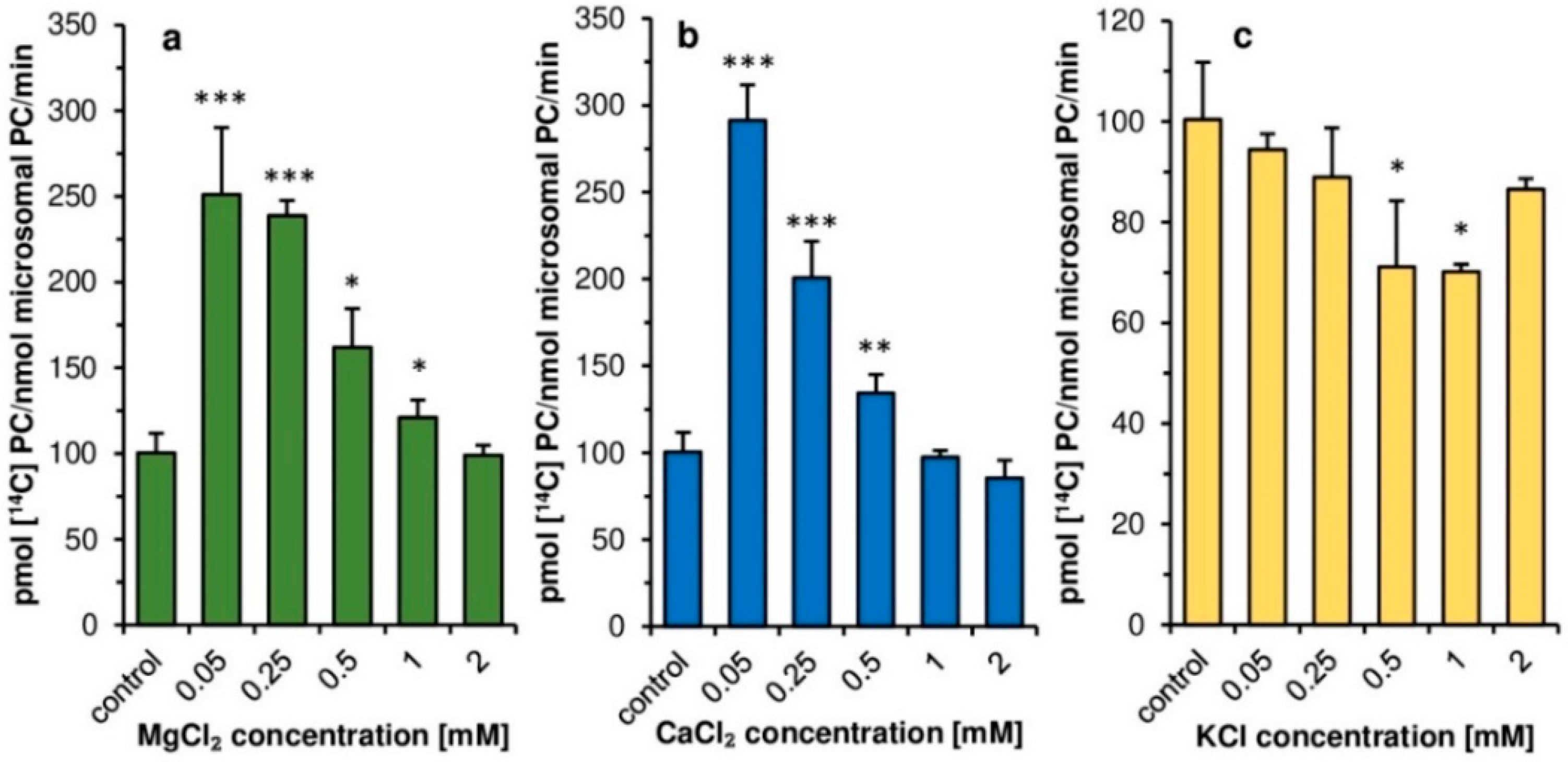
Contrary to plant LPCATs [
11], the addition of magnesium and calcium ions at concentrations 0.05–0.5 mM had a clear stimulatory effect on the activity of
PtLPCAT1. Such calcium and magnesium concentrations, however, significantly inhibit LPCATs activity of developing seeds of
C. sativa. Thus, in this aspect, plant and diatom LPCATs behaving completely differently. Similar to plant LPCATs, the potassium ions had some inhibitory effect on
PtLPCAT1 activity. However, the inhibitory effect was rather small in both cases, and depending on potassium ions, concentration ranged from a few to max 30% compared to assays without these ions. The higher stimulation of
PtLPCAT1 activity observed in lower magnesium or calcium ions concentrations compared to that observed at higher concentrations (≥0.5 mM) could be connected with the impeding of acyl-CoA solubility by high concentrations of these ions [
31]
As the tested ions concentrations in the diatom cells (or different compartments of the cells) could be different from these used in our assays, we could not directly transfer the results obtained in vitro to the situation in vivo and rather treat these findings as features of the characterized enzyme. In the case of calcium ions, we expected that intracellular concentration could be close to the lowest concentrations used in our assays. In diatoms, calcium ions are actively transported from the cell by Ca
2+-ATPase located in the cell membrane to maintain low intracellular concentration as opposed to their extracellular concentration [
32]. In the case of magnesium and potassium ions, their intracellular concentration might be higher than used in our assays. Dickson and Kirst [
33] showed, for instance, that the intracellular concentration of K
+ and Mg
2+ in
P. tricornutum was about 200 and 32 mM, respectively, when cells were cultured in media with salinity corresponding to that existing in the natural habitat. Similarly, a high concentration of K
+, accounting for about 140 mM were observed by Overnell [
34] in the cells of
P. tricornutum grew in medium with potassium concentration at 6.7 mM. The concentrations of tested ions could be, however, different in direct environment of the enzyme and could be more similar to that used in our assays. Nevertheless, we should have in mind that calcium, magnesium and potassium ions could affect
PtLPCAT1 activity.
To sum up, we can state that the substrate specificity of the tested PtLPCAT1 indicated that it can completely supply PC with all fatty acids connected with DHA biosynthetic pathway for the desaturation process. However, the biosynthetic process of VLC-PUFA in diatoms also requires the transfer of fatty acids from PC to cytosolic acyl-CoA pool for the elongation process. The involvement of the tested PtLPCAT1 in this process needs to be elucidated in further studies. We can also mention that biochemical properties of the tested PtLPCAT1 are in some cases (such as the dependency of its activity on pH value) similar, differ moderately (such as in response to temperature changes), or express completely different properties (such as in reaction to calcium and magnesium ions or toward some acyl-CoA with 20C polyunsaturated fatty acids) compared to plant LPCATs.
4. Materials and Methods
4.1. Chemicals
[
14C]-labeled fatty acids were purchased from PerkinElmer Life Science (Waltham, MA, USA).
sn-1-18:1-
sn-2-[
14C]18:2-phosphatidic acid,
sn-1-18:1-
sn-2-[
14C]18:2-phosphatidylethanolamine and
sn-1-18:1-
sn-2-[
14C]18:2-phosphatidylcholine used as TLC standard were synthesized biochemically with use of a microsomal fraction of yeast overexpressing ALE1 and
sn-1-18:1-LPA,
sn-1-18:1-LPE or
sn-1-18:1-LPC together with [
14C]18:2-CoA as substrates in assays system described below. Synthesized [
14C]-phospholipids were separated on TLC and eluted to chloroform as described below for separation of [
14C]-LPC.
sn-1-[1-
14C]18:1-LPC was prepared by phospholipase A
2 (Sigma-Aldrich, St. Louis, MO, USA) treatment of
sn-1-
sn-2-[1-
14C]18:1-phosphatidylcholine purchased from American Radiolabeled Chemicals (Saint Louis, MO, USA). Reaction products were separated on TLC plates in polar solvent system consisting of chloroform:methanol:acetic acid:water (90:17,5:10:3,5;
v:
v:
v:
v). The radioactive LPC was eluted from silica gel with methanol:chloroform (2:1;
v:
v) by sonication and was subsequently extracted according to the method of Bligh and Dyer [
35]. Non-radioactive fatty acids and lysophospholipids were obtained from Larodan Fine Chemicals (Malmö, Sweden). The [
14C]acyl-CoAs and non-radioactive acyl-CoAs were synthesized by the method described by Sánchez et al. [
36] with modifications. Other chemicals were obtained from Sigma-Aldrich or Merck (Darmstadt, Germany).
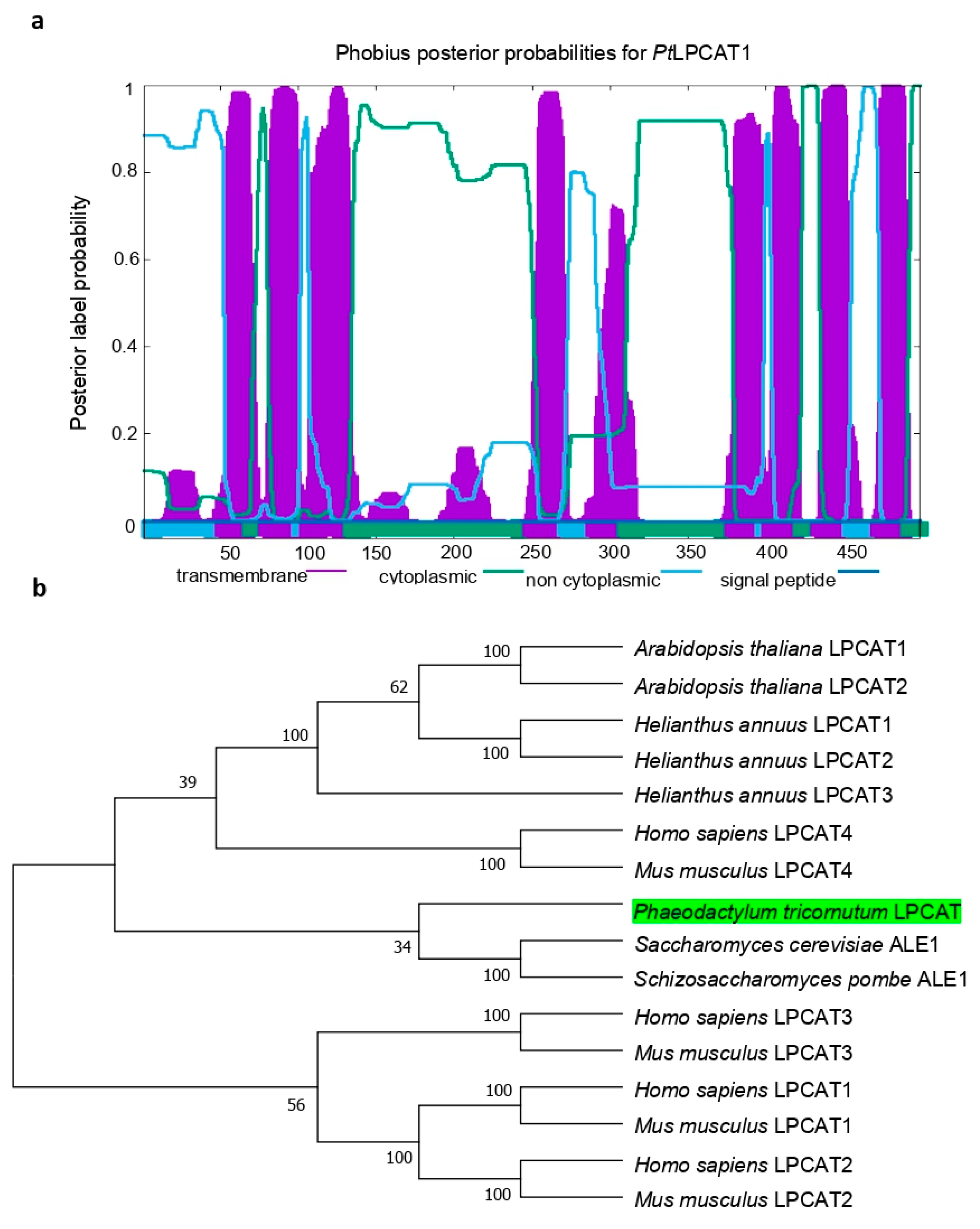
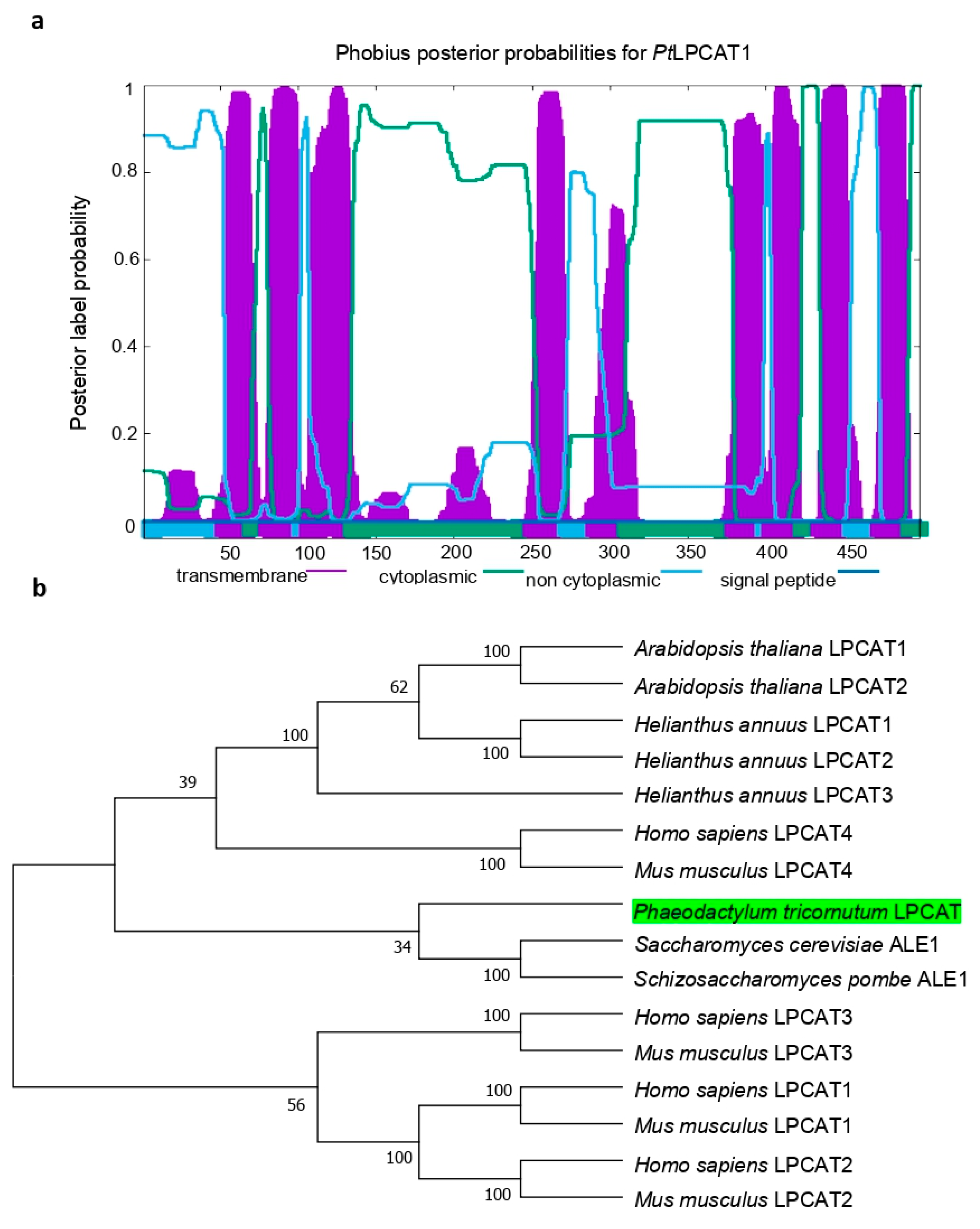
4.2. Gene Cloning and Sequence Analysis
The coding sequence of the LPCAT candidate gene (
PtLPCAT1, Phatr3_J20460) was retrieved from the
P. tricornutum genome database (
http://protists.ensembl.org/Phaeodactylum_tricornutum/Info/Index/; accessed on 11 July 2018), and amplified by reverse-transcription PCR (RT-PCR) using total RNA as a template according to the supplier’s recommendations with gene-specific sense primer (5′-
AAGCTTATGAGTCTCCCCGAAGCC-3′) and antisense primer (5′
TCTAGATTCTTTCTTTTCTTTCTTGGGCG-3′), which were equipped with terminal restriction sites of
HindIII and
XbaI, respectively. The ~1.5 kb DNA fragment produced by RT-PCR was ligated into the yeast expression vector pYES2/CT (Invitrogen; Waltham, MA, USA), sequenced, and introduced for heterologous expression into the
Saccharomyces cerevisiae mutant Y02431 (Δ
lca/Δ
ale1) disrupted in endogenous LPCAT enzyme activity. To search for the orthologs from other organisms for the construction of a phylogenetic tree, we performed Basic Local Alignment Search Tool (BLAST) analysis using the amino acid sequence of
PtLPCAT1 as a query. Analysis of the presence of conserved acyltransferase domains was conducted using hmmer searches [
37]. Prediction of the potential transmembrane domains within the primary sequence of
PtLPCAT1 protein was performed using Phobius (
https://phobius.sbc.su.se/; accessed on 18 August 2021) [
38]. The protein sequence was also analyzed with PredSL (
http://aias.biol.uoa.gr/PredSL/input.html; accessed on 10 July 2021) and PSORTII (
http://psort.hgc.jp/form2.html; accessed on 18 August 2021) to search for the presence of putative N-terminal targeting sequences. The theoretical molecular weight and isoelectric point (
pI) of
PtLPCAT1 were computed by the Compute pI/Mw server (
http://web.expasy.org/compute_pi/; accessed on 14 July 2021). For the construction of a phylogenetic tree of LPCATs, a total of 16 amino acid sequences were aligned using ClustalW [
39] in the Alignment Explorer, MEGA 4.0 [
40]. Gaps and ambiguously aligned regions were omitted from further analysis. Phylogenetic and molecular evolutionary analyses were performed using MEGA 4.0 version by the neighbor-joining method [
41]. The bootstrap value of 1000 replicas was selected together with the JTT model. Amino acid sequences selected from GenBank were:
Phaeodactylum tricornutum LPCAT, Phatr3_J20460 (The ID number for the diatom LPCAT is according to the
P. tricornutum genome database);
Arabidopsis thaliana LPCAT1, NP_172724;
Arabidopsis thaliana LPCAT2, NP_176493;
Homo sapiens LPCAT1, NP_079106;
Homo sapiens LPCAT2, NP_060309;
Homo sapiens LPCAT3, NP_005759;
Homo sapiens LPCAT4, NP_620154;
Mus musculus LPCAT1, BAE94687;
Mus musculus LPCAT2, BAF47695;
Mus musculus LPCAT3, BAG12120;
Mus musculus LPCAT4, BAG12122;
Saccharomyces cerevisiae ALE1, NP_014818;
Schizosaccharomyces pombe ALE1, NP_596779;
Helianthus annuus LPCAT1, AER57988;
Helianthus annuus LPCAT2, AER57989; and
Helianthus annuus LPCAT3, ARQ87991.
4.3. Microsomal Preparation and Enzymes Assays
The yeast Δ
ale1 cells, with introduced plasmids pYES2/CT or plasmids pYES2/CT harboring
PtLPCAT1 gene (Phatr3_J20460), were cultured with uracil dropout medium for 24 h. After that time, the yeast cultures were rejuvenated to an OD
600 = 0.2 and grew for another 24 h. On the third day, galactose was added to the cultures in an amount equivalent to 2% of the final concentration. The yeast cells were grown for at least 24 h to an OD
600 = 3–4 and harvested by centrifugation at 1500×
g for 10 min. The pellets were resuspended in 20 mL of Tris buffer (25 mM Tris-HCl pH 7.9) in order to wash cells from the residue of the medium. Then yeast cells were collected once again and resuspended in 1.5 mL of glass bead disruption buffer (20 mM Tris-HCl, pH 7.9, 10 mM MgCl
2, 1 mM EDTA, 5% glycerol, 0.3 M ammonium sulfate) with protease inhibitors (Complete Tablets, Roche). Glass disruption beads were added to the resuspended yeast cells, which were subsequently homogenized in Mini Bead Beater (BioSpec Products, Bartlesville, OK, USA). The homogenization process was carried out for 5 × 30 s, with 30 s breaks in between. After a 5 min break, the above process was repeated once again. The glass disruption beads and not crashed cells were separated from the homogenates by centrifugation for 10 min at 1500×
g. Supernatants were filtered through Miracloth to ultra-centrifuge tubes and centrifugated at 100,000×
g for 2 h. The pellets were washed with 0.1 M potassium phosphate buffer pH 7.2 (without vortexing) and homogenized with the addition of a new, small portion of potassium phosphate buffer in glass homogenizers. Obtained homogenates of microsomes were divided on aliquots and stored at −80 °C. To estimate microsomes “concentrations”, the content of microsomal endogenous phosphatidylcholine (PC) was evaluated with the use of TLC and gas chromatography analysis [
11].
In addition to the yeast microsomal fractions, the microsomal fraction of
C. sativa developing seeds was prepared according to the method described by Klińska et al. [
11].
First assays aimed at optimization of the conditions of the in vitro reaction catalyzed by tested
PtLPCAT1 enzyme. The influence of factors such as the amount of microsomal fraction, reaction time, temperature, buffer pH, and effect of ions (K
+, Ca
2+, Mg
2+) on the
PtLPCAT1 activity were investigated, as enzyme substrates 5 nmol of [
14C]18:1-CoA (acyl donor) and 5 nmol of
sn-1-18:1-lysophosphatidylcholine (acyl acceptor) were used in these assays. The reactions were carried out in Eppendorf tubes with 0.1 M potassium phosphate buffer (100 µL), with two exceptions. The first concerned assays aimed at estimation of the effects of pH on enzyme activity (used buffers are presented in the legend of
Figure 2d). The second concerned assays measured the effect of selected ions on enzyme activity, where HEPES buffer (pH 7.2) was used, due to the danger of forming insoluble salts of Mg
2+ and Ca
2+ ions in potassium phosphate buffer.
Further studies concerned the specific activity of PtLPCAT1 toward various substrates. Positional specificity was tested in assays containing 5 nmol of [14C]18:1-CoA and 5 nmol of one of the ether analogs: sn-1-18:1-lyso-PC (sn-1-O-GPC) or sn-2-18:1-lyso-PC (sn-2-O-GPC).
Substrate specificity of tested PtLPCAT1 enzyme toward LPA, LPE, and LPC, was studied with two acyl donors [14C]18:1-CoA and [14C]18:3-CoA. Substrate selectivity assays were performed with LPA, LPE, LPC, and LPG added to the reaction in equimolar concentration, together with acyl donors: [14C]18:1-CoA or [14C]18:3-CoA. Activity toward four different LPC: sn-1-16:0-LPC, sn-1-18:0-LPC, sn-1-18:1-LPC, sn-1-20:0-LPC was measured with seven different [14C]acyl-CoA: palmitoyl-CoA ([14C]16:0-CoA), stearoyl-CoA ([14C]18:0-CoA), oleoyl-CoA ([14C]18:1-CoA), linoleoyl-CoA ([14C]18:2-CoA), linolenoyl-CoA ([14C]18:3-CoA), arachidoyl-CoA ([14C]20:0-CoA) and erucoyl-CoA ([14C]22:1-CoA). In these assays, 5 nmol of respective lysophospholipid and 5 nmol of respective acyl-CoA were added together to the reaction mixtures, with the exception of assays measuring substrate selectivity to which 1 nmol of each lysophospholipid was added.
Assays measuring the substrate specificity toward acyl donors were performed with twenty one non-radioactive acyl-CoAs: myristoyl-CoA (14:0-CoA), palmitoyl-CoA (16:0-CoA), palmitoleoyl-CoA (16:1Δ9-CoA), stearoyl-CoA (18:0-CoA), oleoyl-CoA (18:1Δ9-CoA), linoleoyl-CoA (18:2Δ9,12-CoA), α-linolenoyl-CoA (18:3Δ9,12,15-CoA), γ-linolenoyl-CoA (18:3Δ6,9,12-CoA), eicosatetraenoyl-CoA (18:4Δ6,9,12,15-CoA), arachidoyl-CoA (20:0-CoA), eicosenoyl-CoA (20:1Δ11-CoA), eicosatrienoyl-CoA (20:3Δ11,14,17-CoA), arachidonoyl-CoA (20:4Δ5,8,11,14-CoA), eicosatetraenoyl-CoA (20:4Δ8,11,14,17-CoA), eicosapentaenoyl-CoA (20:5Δ5,8,11,14,17-CoA), docosanoyl-CoA (22:0-CoA), erucoyl-CoA (22:1Δ9-CoA), docosapentaenoyl-CoA (22:6Δ4,7,10,13,16,19-CoA), tetracosanoyl-CoA (24:0-CoA), nervonoyl-CoA (24:1Δ9-CoA), and hexacosanoyl-CoA (26:0-CoA). The single reaction contained 5 nmol of respective acyl-CoA and 5 nmol of sn-1-[14C]18:1-LPC. The assays were carried out with 0.1 M potassium phosphate buffer (pH 7.2; 100 µL). Microsomal fractions (source of the tested enzyme) were added in amount equivalent to 0.05 nmol of microsomal PC (approximately 0.22 µg of microsomal proteins). Assays were incubated for 30 min at 30 °C in Eppendorf Thermomixer Compact with continuous shaking at 1250 rpm. Enzymatic reactions were terminated by addition of 375 µL chloroform:methanol (1:2; v:v), 5 µL of glacial acetic acid, 125 µL of chloroform and 125 µL of water. The samples were mixed vigorously and centrifuged to separate bottom chloroform layer from upper methanol-water layer. Chloroform fractions containing lipids were collected and separated by thin-layer chromatography method on silica gel 60 plates from Merck using polar solvent system consisting of chloroform:methanol:acetic acid:water (90:15:10:2.5 or 90:17.5:10:3.5; v:v:v:v). [14C]PC products were visualized and measured by electronic autoradiography (Instant Imager, Packard Instrument Co., Downers Grove, IL, USA).
Additionally to the assays measuring substrate specificity toward acyl donors of the tested
PtLPCAT1, we performed assays to evaluate the corresponding substrate specificity of LPCATs of the microsomal fraction of developing
C. sativa seeds (24 days after flowering). We used
sn-1-[
14C]18:1-LPC as acyl acceptor and several acyl-CoA with long and very long-chain polyunsaturated fatty acids as acyl donors. The assays conditions were as described above for yeast microsomal fraction with aliquots of microsomes containing 0.2 nmol microsomal PC/assay (for additional information, see Klińska et al. [
11]).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}