
Simultaneous Monitoring of Monoclonal Antibody Variants by Strong Cation-Exchange Chromatography Hyphenated to Mass Spectrometry to Assess Quality Attributes of Rituximab-Based Biotherapeutics

 , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
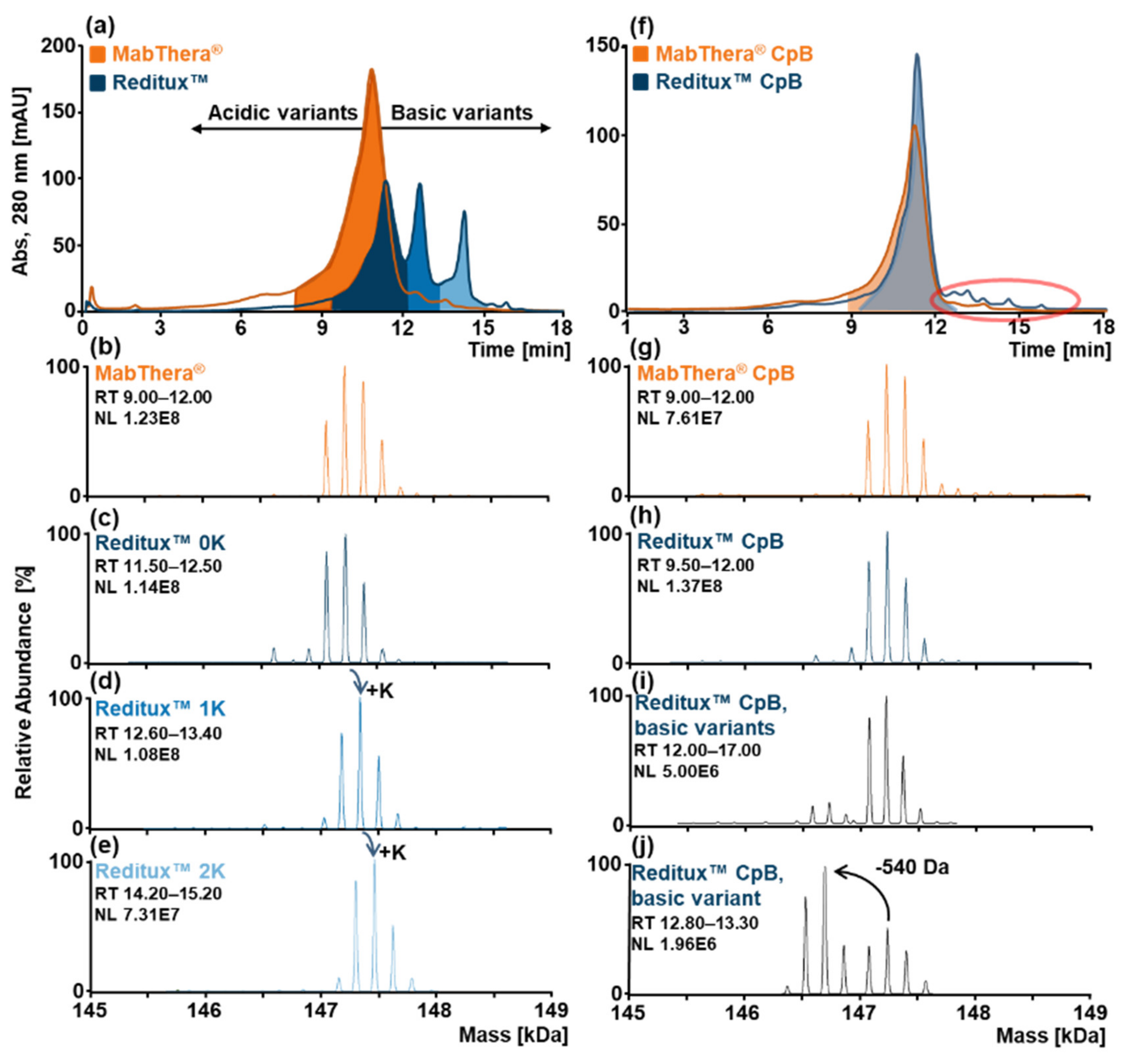
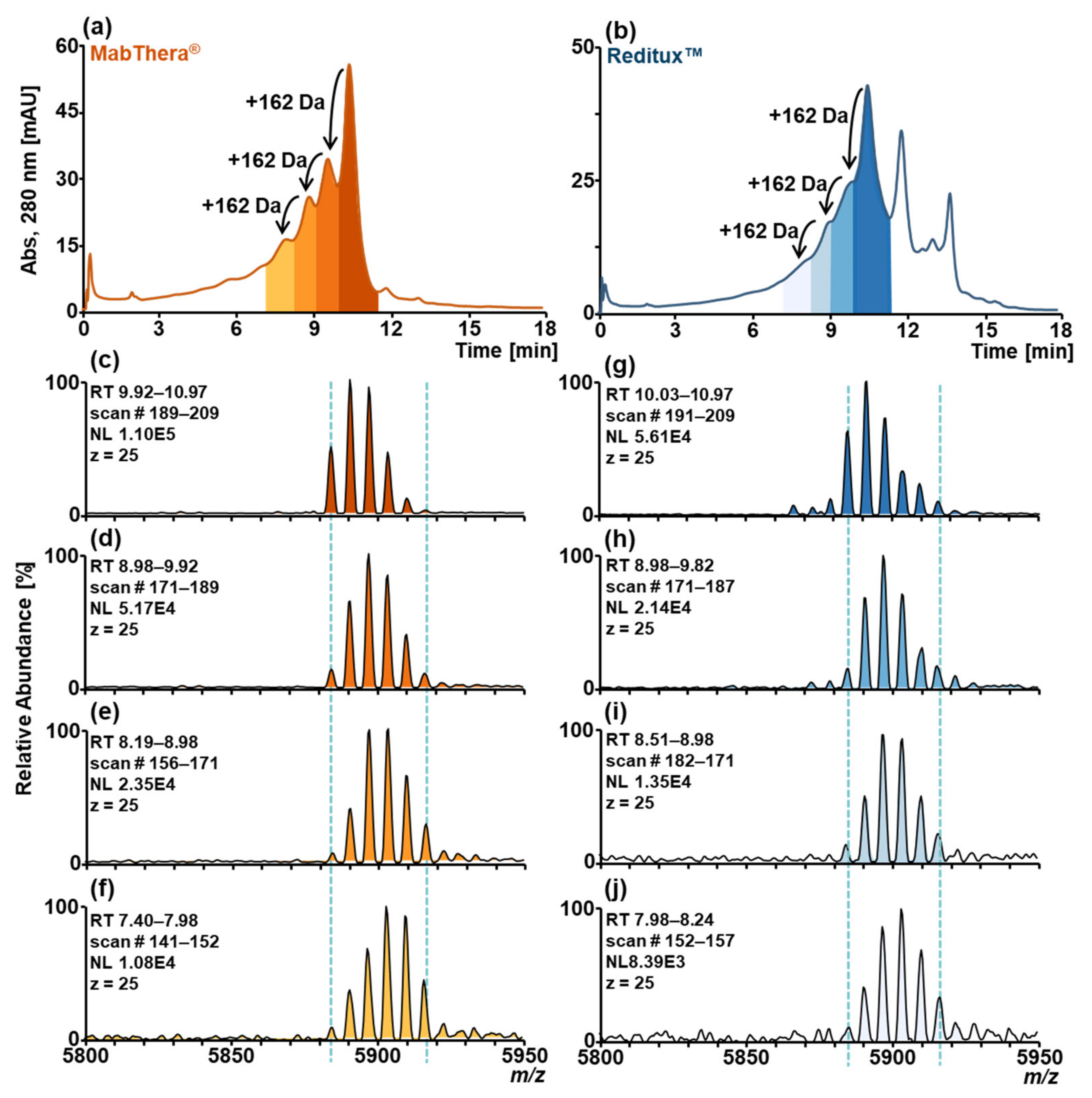
2.1. SCX-HPLC-MS Charge Patterns of MabThera® and Reditux™
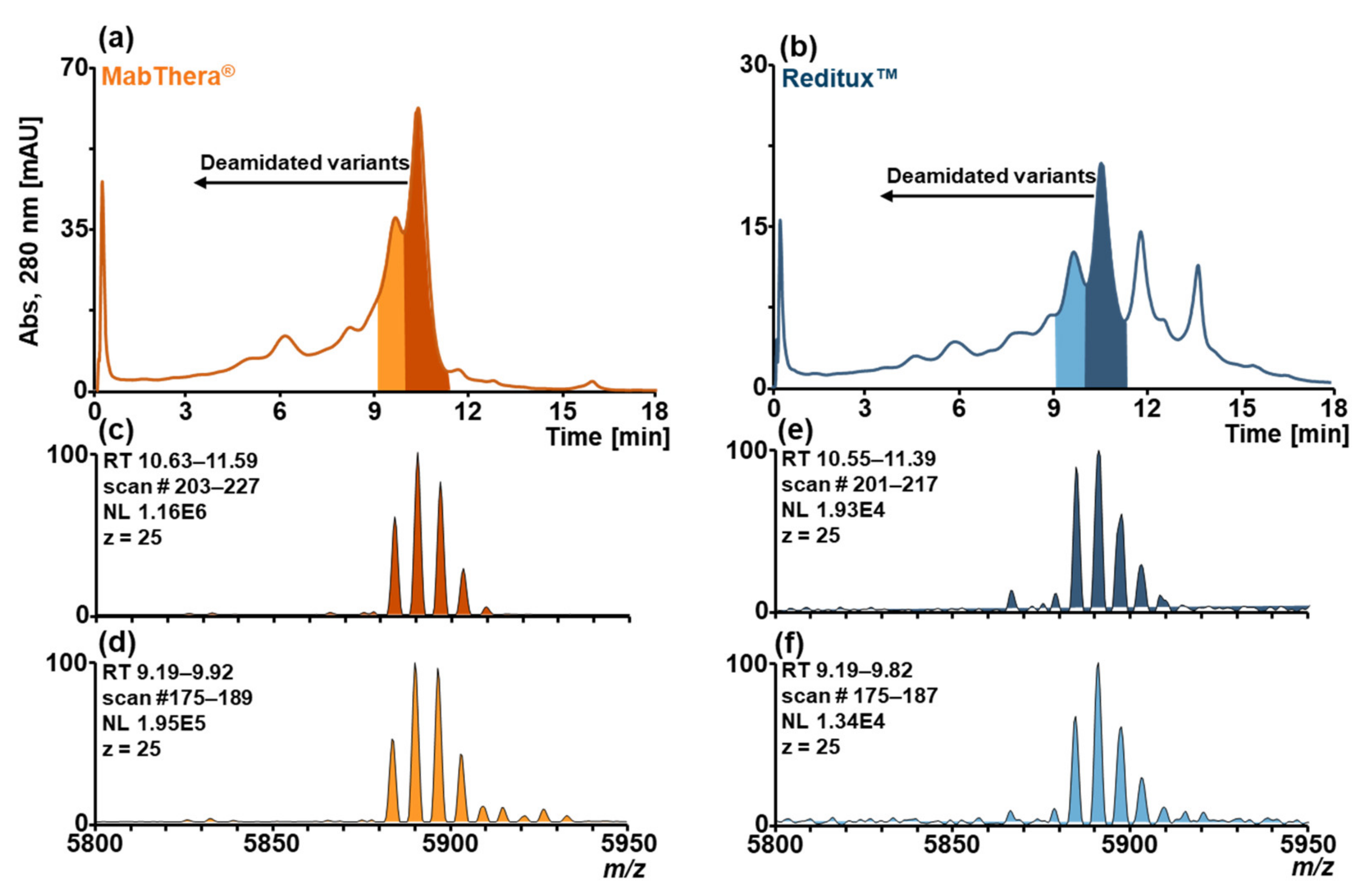
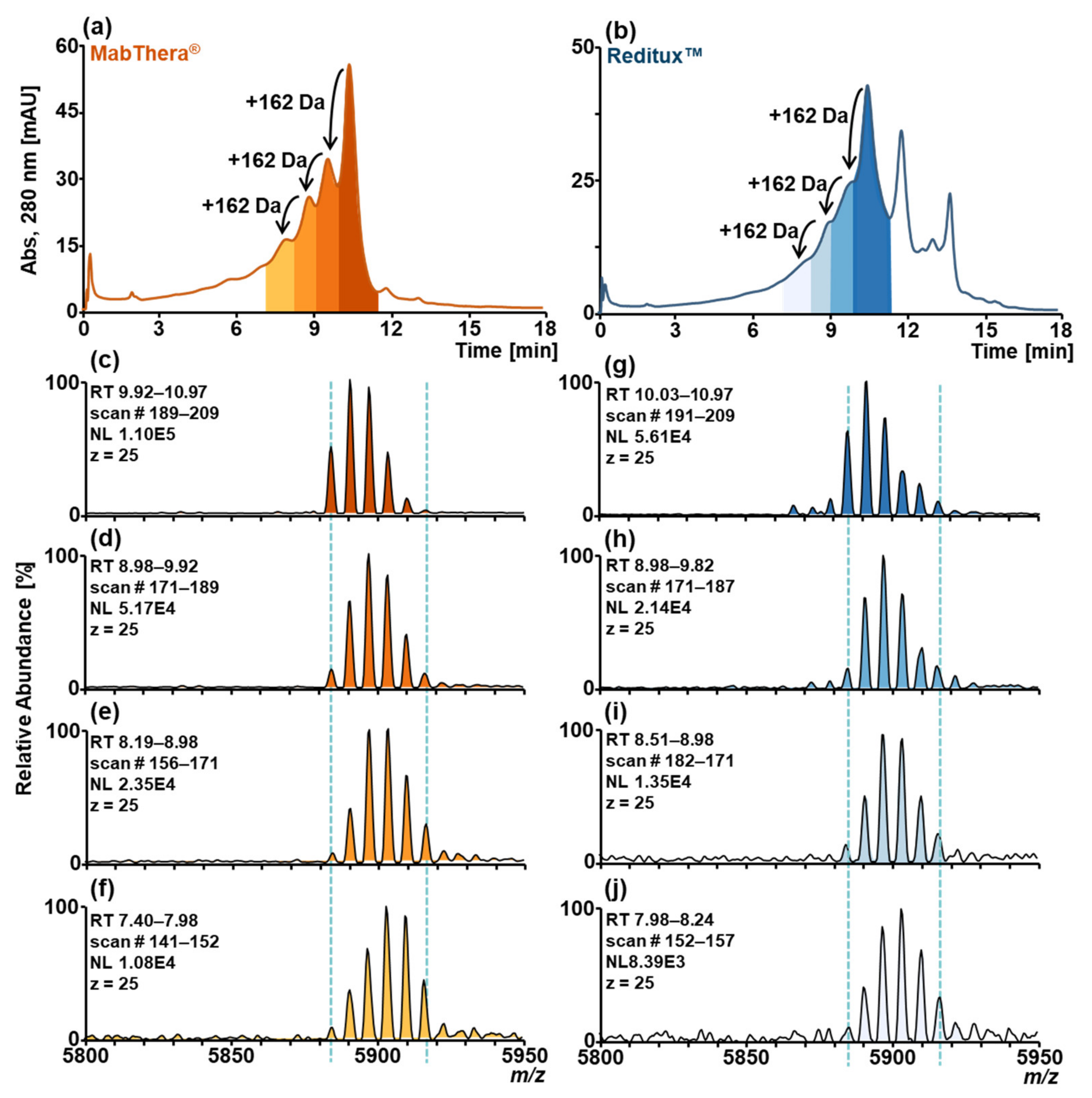
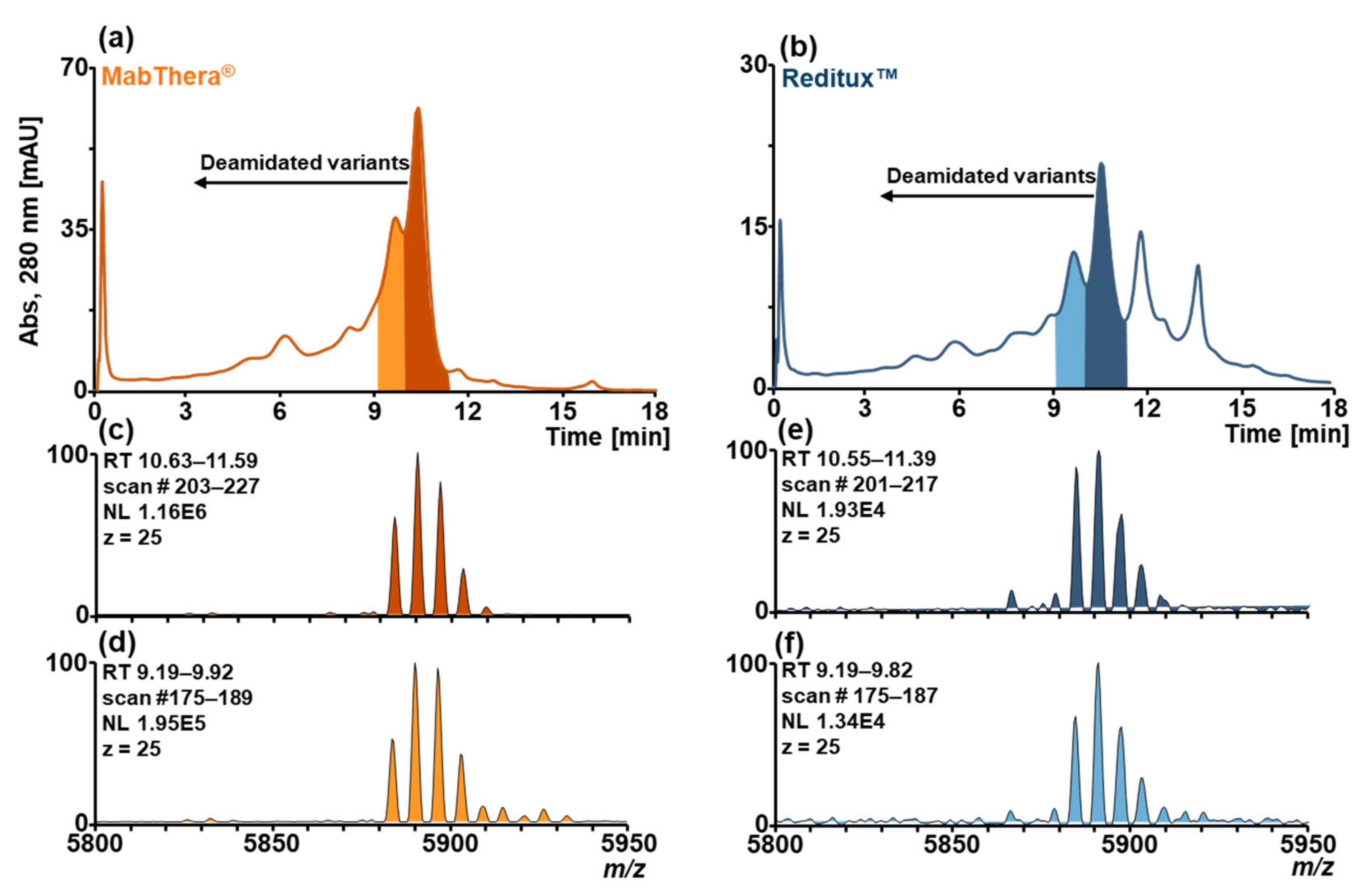
2.2. Forced Deamidation of MabThera® and Reditux™
2.3. Forced Glycation of MabThera® and Reditux™
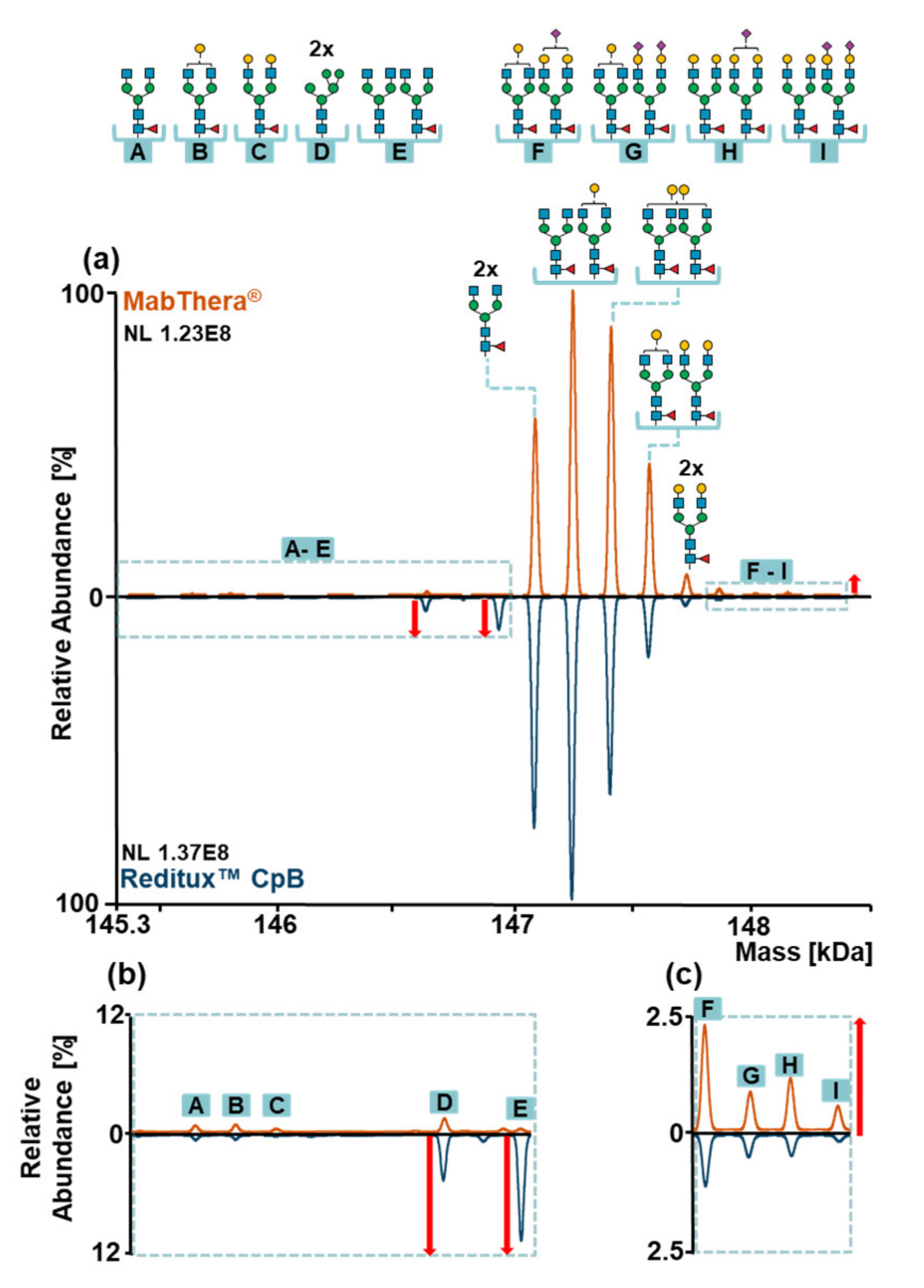
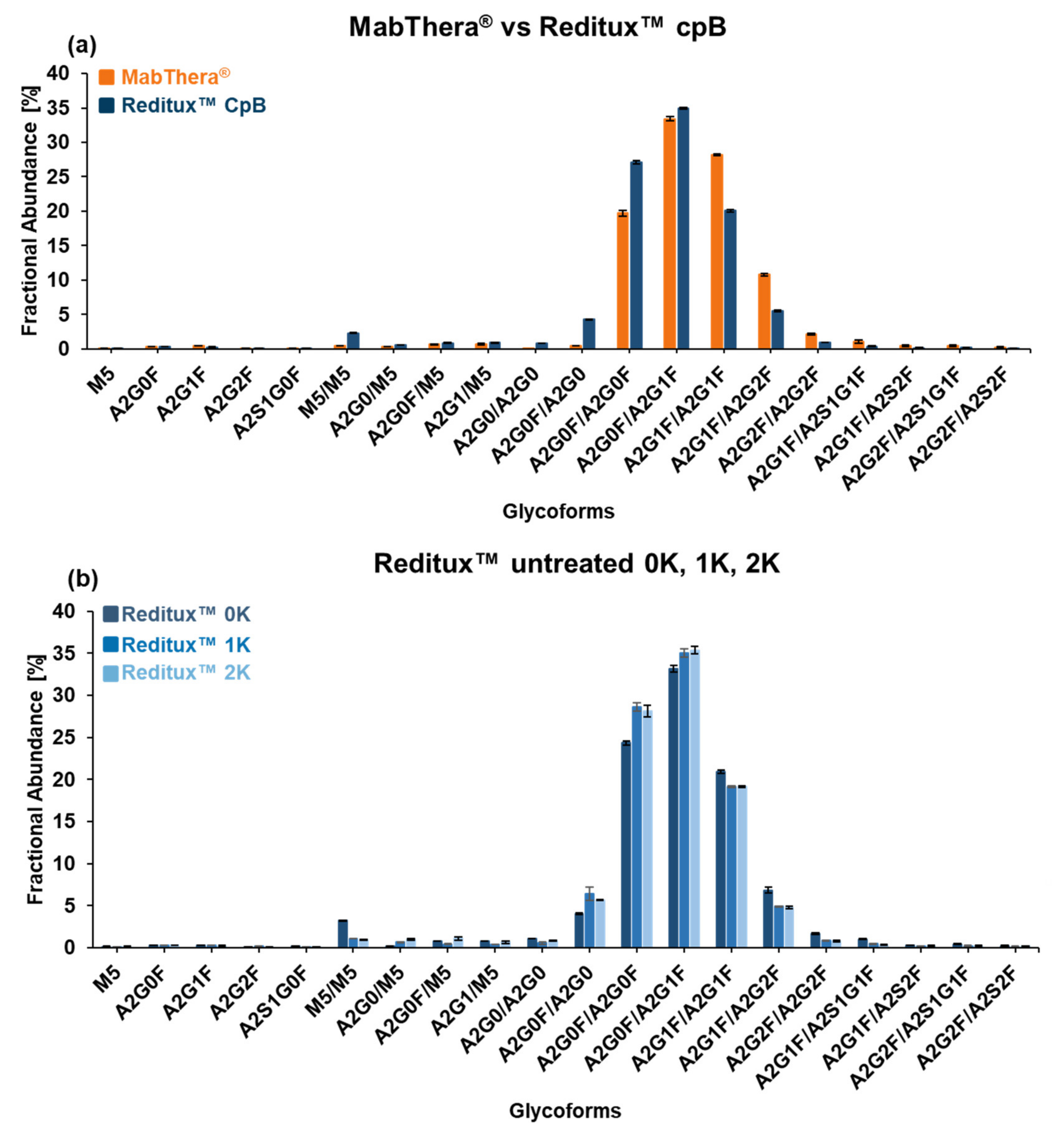
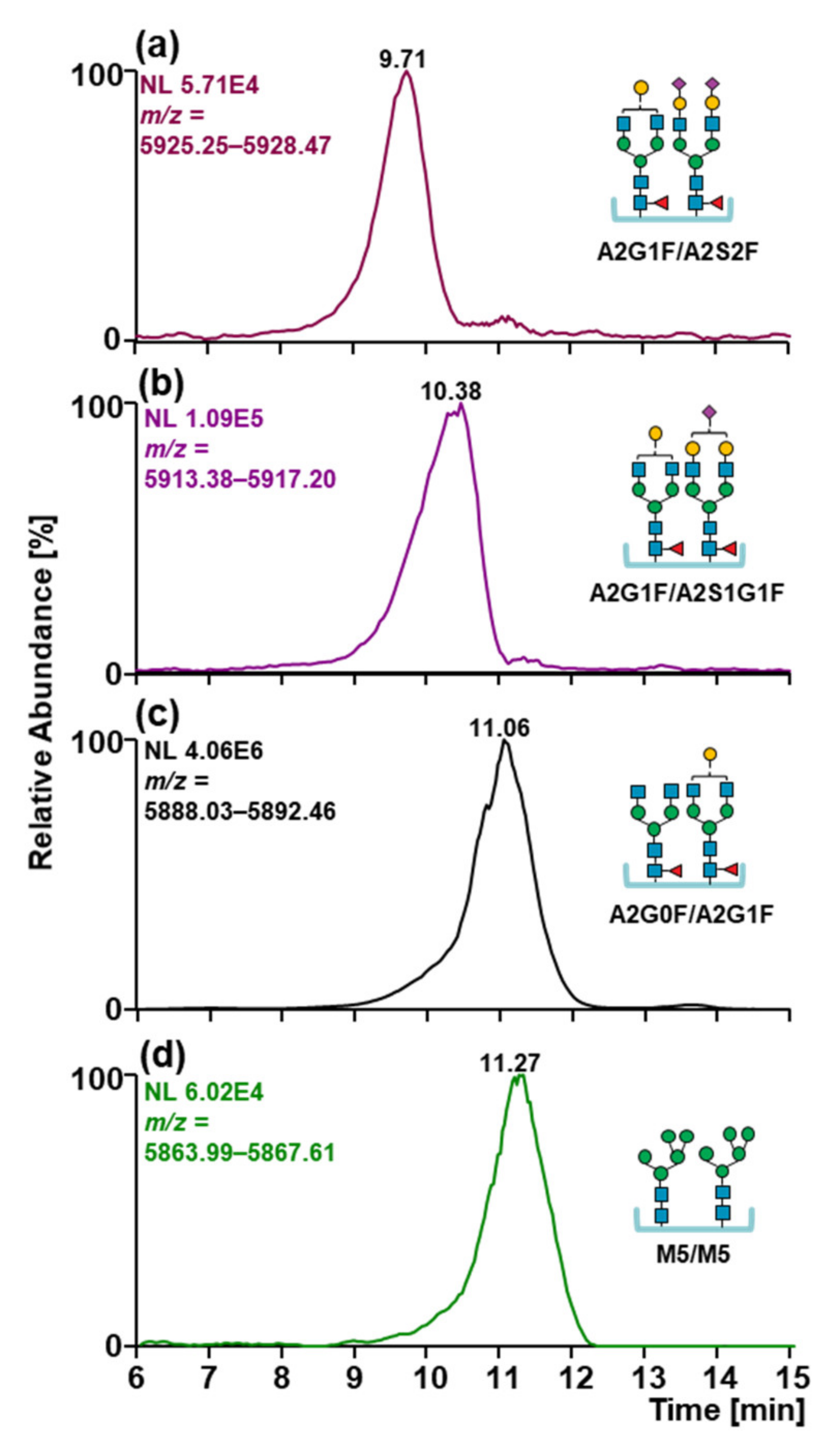
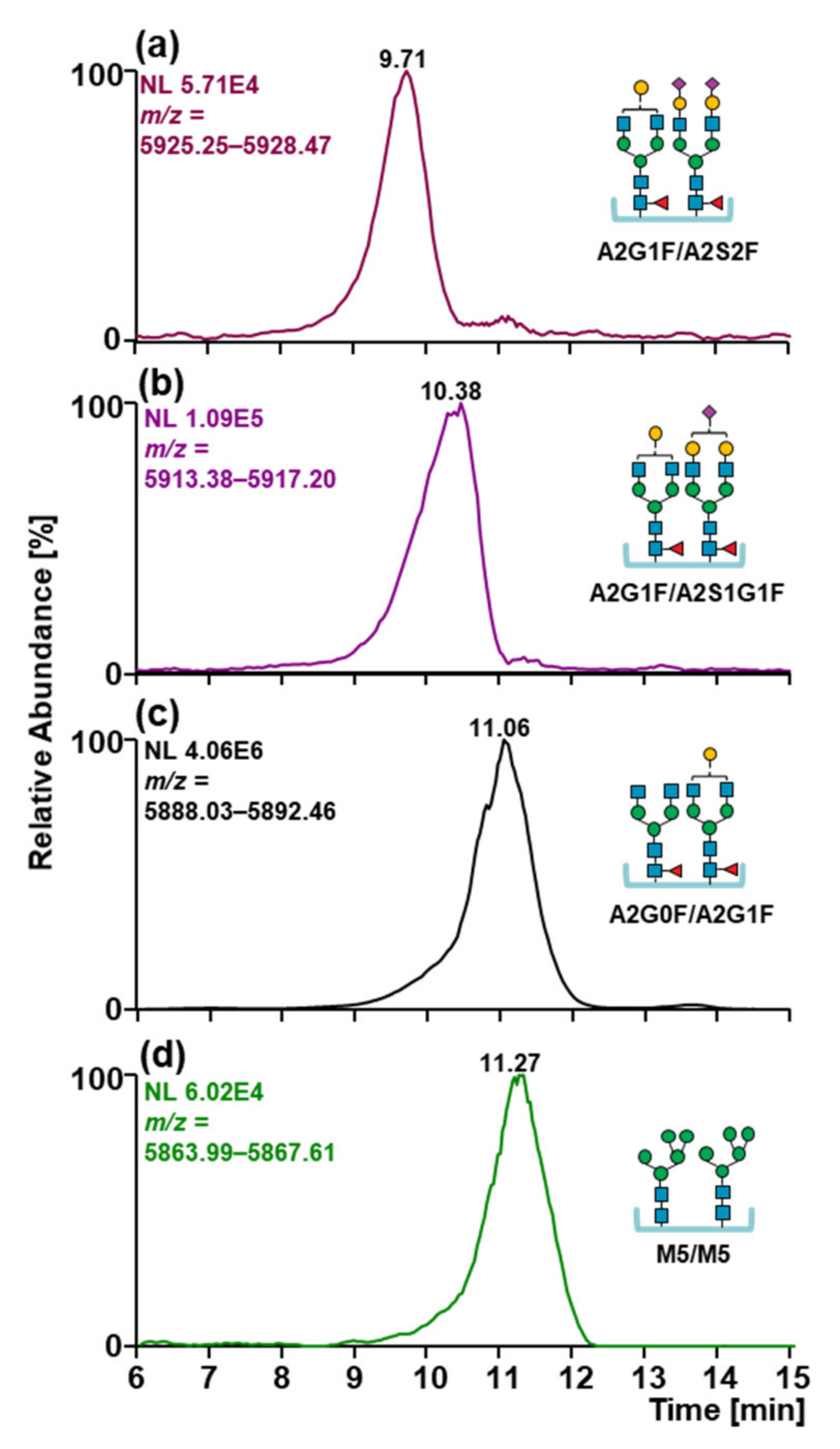
2.4. Glycosylation Patterns of MabThera® and Reditux™
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. SCX-HPLC-MS Analyses of MabThera® and Reditux™
4.4. Peptide Mapping of Untreated and Stressed MabThera® and Reditux™ by Nano RP-HPLC-MS/MS
4.5. Glycoprofiling of Released N-Glycans of MabThera® and Reditux™ by xCGE-LIF
4.6. Data Evaluation
4.7. Data and Code Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, M.; Bielsky, M.-C.; De Smet, K.; Ehmann, F.; Ekman, N.; Narayanan, G.; Heim, H.-K.; Heinonen, E.; Ho, K.; Thorpe, R.; et al. Biosimilars—why terminology matters. Nat. Biotechnol. 2011, 29, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-N.; Thorpe, R.; Knezevic, I.; Blades, C.D.R.Z.; Levano, M.C.; Chew, J.Y.; Chilufya, M.B.; Chirachanakul, P.; Chua, H.M.; Farahani, A.V.; et al. The regulatory landscape of biosimilars: WHO efforts and progress made from 2009 to 2019. Biologicals 2020, 65, 1–9. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Limper, C.; Richards, K.L. Past, present, and future of rituximab—The World’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Qureshi, Z.P.; Singh, S.; Magwood, J.S. Rituximab and biosimilars—equivalence and reciprocity. Biosimilars 2013, 3, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczorek, M. Biosimilar mit hoher und zunehmender Akzeptanz. InFo Onkol. 2018, 21, 65. [Google Scholar] [CrossRef]
- Smith, L.M.; The Consortium for top down proteomics; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Wagner-Rousset, E.; Ayoub, D.; Van Dorsselaer, A.; Cianferani, S. Characterization of therapeutic antibodies and related products. Anal. Chem. 2012, 85, 715–736. [Google Scholar] [CrossRef]
- Beck, A.; Diemer, H.; Ayoub, D.; Debaene, F.; Wagner-Rousset, E.; Carapito, C.; Van Dorsselaer, A.; Sanglier-Cianférani, S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. TrAC Trends Anal. Chem. 2013, 48, 81–95. [Google Scholar] [CrossRef]
- Beck, A.; Sanglier-Cianferani, S.; Van Dorsselaer, A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal. Chem. 2012, 84, 4637–4646. [Google Scholar] [CrossRef]
- Beck, A.; Debaene, F.; Diemer, H.; Wagner-Rousset, E.; Colas, O.; Van Dorsselaer, A.; Cianférani, S. Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J. Mass Spectrom. 2015, 50, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Liu, H. Macro and micro-heterogeneity of natural and recombinant IgG antibodies. Antibodies 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In Vitro and In Vivo modifications of recombinant and human IgG antibodies. mAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Garza, C.E.E.-; Perdomo-Abúndez, F.C.; Padilla-Calderón, J.; Uribe-Wiechers, J.M.; Perez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Analysis of recombinant monoclonal antibodies by capillary zone electrophoresis. Electrophoresis 2013, 34, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Beck, A.; Veuthey, J.-L.; Guillarme, D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2015, 113, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Beck, A.; Fekete, J.; Guillarme, D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part II: pH gradient approach. J. Pharm. Biomed. Anal. 2015, 102, 282–289. [Google Scholar] [CrossRef]
- Fekete, S.; Beck, A.; Fekete, J.; Guillarme, D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part I: Salt gradient approach. J. Pharm. Biomed. Anal. 2015, 102, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Goyon, A.; Excoffier, M.; Janin-Bussat, M.-C.; Bobaly, B.; Fekete, S.; Guillarme, D.; Beck, A. Determination of isoelectric points and relative charge variants of 23 therapeutic monoclonal antibodies. J. Chromatogr. B 2017, 1065–1066, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Trappe, A.; Füssl, F.; Carillo, S.; Zaborowska, I.; Meleady, P.; Bones, J. Rapid charge variant analysis of monoclonal antibodies to support lead candidate biopharmaceutical development. J. Chromatogr. B 2018, 1095, 166–176. [Google Scholar] [CrossRef]
- Van Schaick, G.; Gstöttner, C.; Büttner, A.; Reusch, D.; Wuhrer, M.; Domínguez-Vega, E. Anion exchange chromatography—Mass spectrometry for monitoring multiple quality attributes of erythropoietin biopharmaceuticals. Anal. Chim. Acta 2021, 1143, 166–172. [Google Scholar] [CrossRef]
- Talebi, M.; Nordborg, A.; Gaspar, A.; Lacher, N.A.; Wang, Q.; He, X.Z.; Haddad, P.R.; Hilder, E. Charge heterogeneity profiling of monoclonal antibodies using low ionic strength ion-exchange chromatography and well-controlled pH gradients on monolithic columns. J. Chromatogr. A 2013, 1317, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Raoufi, F.; Bailly, M.A.; Fayadat-Dilman, L.; Tomazela, D. Hyphenation of strong cation exchange chromatography to native mass spectrometry for high throughput online characterization of charge heterogeneity of therapeutic monoclonal antibodies. mAbs 2020, 12, 1763762. [Google Scholar] [CrossRef] [PubMed]
- Füssl, F.; Cook, K.; Scheffler, K.; Farrell, A.; Mittermayr, S.; Bones, J. Charge variant analysis of monoclonal antibodies using direct coupled ph gradient cation exchange chromatography to high-resolution native mass spectrometry. Anal. Chem. 2018, 90, 4669–4676. [Google Scholar] [CrossRef] [PubMed]
- Füssl, F.; Trappe, A.; Cook, K.; Scheffler, K.; FitzGerald, O.; Bones, J. Comprehensive characterisation of the heterogeneity of adalimumab via charge variant analysis hyphenated on-line to native high resolution Orbitrap mass spectrometry. mAbs 2019, 11, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Fussl, F.; Trappe, A.; Carillo, S.; Jakes, C.; Bones, J. Comparative elucidation of cetuximab heterogeneity on the intact protein level by cation exchange chromatography and capillary electrophoresis coupled to mass spectrometry. Anal. Chem. 2020, 92, 5431–5438. [Google Scholar] [CrossRef] [PubMed]
- Millán-Martín, S.; Carillo, S.; Füssl, F.; Sutton, J.; Gazis, P.; Cook, K.; Scheffler, K.; Bones, J. Optimisation of the use of sliding window deconvolution for comprehensive characterisation of trastuzumab and adalimumab charge variants by native high resolution mass spectrometry. Eur. J. Pharm. Biopharm. 2021, 158, 83–95. [Google Scholar] [CrossRef]
- Shi, R.L.; Xiao, G.; Dillon, T.M.; Ricci, M.S.; Bondarenko, P.V. Characterization of therapeutic proteins by cation exchange chromatography-mass spectrometry and top-down analysis. mAbs 2020, 12, 1739825. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Roy, P.S.; John, S.; Karankal, S.; Kannan, S.; Pawaskar, P.; Gawande, J.; Bagal, B.; Khattry, N.; Sengar, M.; et al. Comparison of the efficacy and safety of Rituximab (Mabthera™) and its biosimilar (Reditux™) in diffuse large B-cell lymphoma patients treated with chemo-immunotherapy: A retrospective analysis. Indian J. Med Paediatr. Oncol. 2013, 34, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Bankar, A.; Korula, A.; Abraham, A.; Viswabandya, A.; George, B.; Srivastava, A.; Mathews, V. Comparison of the efficacy of innovator rituximab and its biosimilars in diffuse large B cell lymphoma patients: A retrospective analysis. Indian J. Hematol. Blood Transfus. 2020, 36, 71–77. [Google Scholar] [CrossRef]
- Flores-Ortiz, L.F.; Campos-García, V.R.; Perdomo-Abúndez, F.C.; Perez, N.O.; Medina-Rivero, E. Physicochemical properties of rituximab. J. Liq. Chromatogr. Relat. Technol. 2014, 37, 1438–1452. [Google Scholar] [CrossRef]
- Wang, B.; Gucinski, A.C.; Keire, D.; Buhse, L.F.; Ii, M.T.B. Structural comparison of two anti-CD20 monoclonal antibody drug products using middle-down mass spectrometry. Analyst 2013, 138, 3058–3065. [Google Scholar] [CrossRef]
- Zhang, Z.; Perrault, R.; Zhao, Y.; Ding, J. SpeB proteolysis with imaged capillary isoelectric focusing for the characterization of domain-specific charge heterogeneities of reference and biosimilar Rituximab. J. Chromatogr. B 2016, 1020, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Nupur, N.; Chhabra, N.; Dash, R.; Rathore, A.S. Assessment of structural and functional similarity of biosimilar products: Rituximab as a case study. mAbs 2017, 10, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Wohlschlager, T.; Scheffler, K.; Forstenlehner, I.C.; Esser-Skala, W.; Senn, S.; Damoc, E.; Holzmann, J.; Huber, C.G. Native mass spectrometry combined with enzymatic dissection unravels glycoform heterogeneity of biopharmaceuticals. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, C.; Cheung, J.K.; Dellatore, S.M.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. mAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelius, D.; Rehder, A.D.S.; Bondarenko, P.V. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal. Chem. 2005, 77, 6004–6011. [Google Scholar] [CrossRef] [PubMed]
- Antes, B.; Amon, S.; Rizzi, A.; Wiederkum, S.; Kainer, M.; Szolar, O.; Fido, M.; Kircheis, R.; Nechansky, A. Analysis of lysine clipping of a humanized Lewis-Y specific IgG antibody and its relation to Fc-mediated effector function. J. Chromatogr. B 2007, 852, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Faid, V.; Leblanc, Y.; Berger, M.; Seifert, A.; Bihoreau, N.; Chevreux, G. C-terminal lysine clipping of IgG1: Impact on binding to human FcγRIIIa and neonatal Fc receptors. Eur. J. Pharm. Sci. 2021, 159, 105730. [Google Scholar] [CrossRef]
- Bremer, E.T.V.D.; Beurskens, F.J.; Voorhorst, M.; Engelberts, P.J.; De Jong, R.N.; Van der Boom, B.G.; Cook, E.M.; Lindorfer, M.A.; Taylor, R.P.; Van Berkel, P.H.; et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. mAbs 2015, 7, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Regl, C.; Wohlschlager, T.; Esser-Skala, W.; Wagner, I.; Samonig, M.; Holzmann, J.; Huber, C.G. Dilute-and-shoot analysis of therapeutic monoclonal antibody variants in fermentation broth: A method capability study. mAbs 2019, 11, 569–582. [Google Scholar] [CrossRef]
- Kaschak, T.; Boyd, D.; Lu, F.; Derfus, G.; Kluck, B.; Nogal, B.; Emery, C.; Summers, C.; Zheng, K.; Bayer, R.; et al. Characterization of the basic charge variants of a human IgG1. mAbs 2011, 3, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Walsh, A.; Ehrick, R.; Xu, W.; May, K.; Liu, H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. mAbs 2012, 4, 578–585. [Google Scholar] [CrossRef]
- Ayoub, D.; Bertaccini, D.; Diemer, H.; Wagner-Rousset, E.; Colas, O.; Cianférani, S.; Van Dorsselaer, A.; Beck, A.; Schaeffer-Reiss, C. Characterization of the N-Terminal heterogeneities of monoclonal antibodies using In-Gel charge derivatization of α-Amines and LC-MS/MS. Anal. Chem. 2015, 87, 3784–3790. [Google Scholar] [CrossRef]
- Laptoš, T.; Omersel, J. The importance of handling high-value biologicals: Physico-chemical instability and immunogenicity of monoclonal antibodies (Review). Exp. Ther. Med. 2018, 15, 3161–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Alcala, E.; Petkovska, I.; Matthews, D.; Canova-Davis, E.; Taticek, R.; Ma, S. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: Where it is, how it got there, and how it affects charge-based behavior. Anal. Biochem. 2008, 373, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Seidl, A.; Sörgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Kim, S.Y.; Vallejo, D.; Hageman, T.S.; White, D.R.; Benet, A.; Coghlan, J.; Sen, K.I.; Ford, M.; Saveliev, S.; et al. Multifaceted assessment of rituximab biosimilarity: The impact of glycan microheterogeneity on Fc function. Eur. J. Pharm. Biopharm. 2019, 146, 111–124. [Google Scholar] [CrossRef]
- Wei, B.; Gao, X.; Cadang, L.; Izadi, S.; Liu, P.; Zhang, H.-M.; Hecht, E.; Shim, J.; Magill, G.; Pabon, J.R.; et al. Fc galactosylation follows consecutive reaction kinetics and enhances immunoglobulin G hexamerization for complement activation. mAbs 2021, 13, 1893427. [Google Scholar] [CrossRef]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. mAbs 2018, 10, 693–711. [Google Scholar] [CrossRef]
- Yu, M.; Brown, D.; Reed, C.; Chung, S.; Lutman, J.; Stefanich, E.; Wong, A.; Stephan, J.-P.; Bayer, R. Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. mAbs 2012, 4, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scallon, B.J.; Tam, S.H.; McCarthy, S.G.; Cai, A.N.; Raju, T.S. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol. Immunol. 2007, 44, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Ai, J.; Bao, Y.; Yuan, Z.; Qin, Y.; Xie, Y.-W.; Tao, D.; Fu, D.; Peng, Y. Investigation of the correlation between charge and glycosylation of IgG1 variants by liquid chromatography–mass spectrometry. Anal. Biochem. 2014, 448, 82–91. [Google Scholar] [CrossRef]
- Strasser, L.; Farrell, A.; Ho, J.T.C.; Scheffler, K.; Cook, K.; Pankert, P.; Mowlds, P.; Viner, R.; Karger, B.L.; Bones, J. Proteomic profiling of IgG1 Producing CHO Cells using LC/LC-SPS-MS3: The effects of bioprocessing conditions on productivity and product quality. Front. Bioeng. Biotechnol. 2021, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Torkashvand, F.; Mahboudi, F.; Vossoughi, M.; Fatemi, E.; Basri, S.M.M.; Vaziri, B. Ouantitative proteomic analysis of cellular responses to a designed amino acid feed in a monoclonal antibody producing chinese hamster ovary cell line. Iran. Biomed. J. 2018, 22, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Ou, J.; Si, Y.; Goh, K.Y.; Flanigan, D.D.; Han, X.; Yang, Y.; Yang, S.-T.; Zhou, L.; Liu, X. (Margaret) Proteomics insight into the production of monoclonal antibody. Biochem. Eng. J. 2019, 145, 177–185. [Google Scholar] [CrossRef]
- Cajic, S.; Hennig, R.; Burock, R.; Rapp, E. Chapter 3: Capillary (gel) electrophoresis-based methods for immunoglobulin (G) glycosylation analysis. Chapter accepted. In Antibody Glycosylation. Experientia Supplementum; Pezer, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2021; Volume 112, ISBN 978-3-030-76912-3. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Marco, F.; Berger, T.; Esser-Skala, W.; Rapp, E.; Regl, C.; Huber, C.G. Simultaneous Monitoring of Monoclonal Antibody Variants by Strong Cation-Exchange Chromatography Hyphenated to Mass Spectrometry to Assess Quality Attributes of Rituximab-Based Biotherapeutics. Int. J. Mol. Sci. 2021, 22, 9072. https://doi.org/10.3390/ijms22169072
Di Marco F, Berger T, Esser-Skala W, Rapp E, Regl C, Huber CG. Simultaneous Monitoring of Monoclonal Antibody Variants by Strong Cation-Exchange Chromatography Hyphenated to Mass Spectrometry to Assess Quality Attributes of Rituximab-Based Biotherapeutics. International Journal of Molecular Sciences. 2021; 22(16):9072. https://doi.org/10.3390/ijms22169072
Chicago/Turabian StyleDi Marco, Fiammetta, Thomas Berger, Wolfgang Esser-Skala, Erdmann Rapp, Christof Regl, and Christian G. Huber. 2021. "Simultaneous Monitoring of Monoclonal Antibody Variants by Strong Cation-Exchange Chromatography Hyphenated to Mass Spectrometry to Assess Quality Attributes of Rituximab-Based Biotherapeutics" International Journal of Molecular Sciences 22, no. 16: 9072. https://doi.org/10.3390/ijms22169072
APA StyleDi Marco, F., Berger, T., Esser-Skala, W., Rapp, E., Regl, C., & Huber, C. G. (2021). Simultaneous Monitoring of Monoclonal Antibody Variants by Strong Cation-Exchange Chromatography Hyphenated to Mass Spectrometry to Assess Quality Attributes of Rituximab-Based Biotherapeutics. International Journal of Molecular Sciences, 22(16), 9072. https://doi.org/10.3390/ijms22169072