Abstract
Monocytes play a key role in cardiovascular disease (CVD) as their influx into the vessel wall is necessary for the development of an atherosclerotic plaque. Monocytes are, however, heterogeneous differentiating from classical monocytes through the intermediate subset to the nonclassical subset. While it is recognized that the percentage of intermediate and nonclassical monocytes are higher in individuals with CVD, accompanying changes in inflammatory markers suggest a functional impact on disease development that goes beyond the increased proportion of these ‘inflammatory’ monocyte subsets. Furthermore, emerging evidence indicates that changes in monocyte proportion and function arise in dyslipidemia, with lipid lowering medication having some effect on reversing these changes. This review explores the nature and number of monocyte subsets in CVD addressing what they are, when they arise, the effect of lipid lowering treatment, and the possible implications for plaque development. Understanding these associations will deepen our understanding of the clinical significance of monocytes in CVD.
Keywords:
monocytes; cardiovascular disease; dyslipidemia; atherosclerosis; inflammation; macrophage 1. Introduction
Cardiovascular disease (CVD) is a leading cause of death and disability worldwide [1]. The main etiological factor for CVD is atherosclerosis which principally affects the coronary, as well as the cerebrovascular and peripheral circulation, with risk of heart attack, stroke and limb loss. The clinical presentations of coronary artery disease (CAD) include unstable and stable angina pectoris, myocardial infarction (MI), cardiac arrhythmia and congestive cardiac failure, but CAD may also be asymptomatic and only detected on specific investigations [2]. Atherosclerosis in the cerebrovascular circulation is commonly localized to the carotid artery bifurcation region in the neck [3]. Carotid artery disease can be asymptomatic, but progressive and unstable plaque carries a significant risk of stroke. In peripheral arterial disease (PAD), atherosclerosis may be localized, but it more frequently tends to be diffuse [4], often with long segment occlusions. Arteries predominantly affected are in the aorto-iliac (supra-inguinal) and femoropopliteal (infra-inguinal) segments. Renal arteries to the kidneys and mesenteric arteries supplying the gastro-intestinal tract are much less frequently affected by atherosclerosis. However, renal artery disease can be a factor in hypertension and chronic kidney disease. Regardless of location, atherosclerosis can cause flow-limiting narrowing. More importantly, the erosion or rupture of unstable plaques can cause atherothrombotic material to occlude arteries, and results in an ischemic event in the coronary, cerebrovascular or peripheral circulation.
Risk factors for CVD are broadly considered either non-modifiable (e.g., age, sex, family history of premature CVD) or modifiable (e.g., hypercholesterolemia, hypertension, tobacco smoking, overweight or obesity, insulin resistance or diabetes, lack of physical activity). Addressing these modifiable risk factors could prevent or delay the atherosclerotic process [5]. Among risk factors, dyslipidemias are considered key contributors to the development of atherosclerotic plaque. Dyslipidemia has many clinical presentations, and some of those considered important for CVD risk include high levels of low density lipoprotein (LDL) or its protein component apolipoprotein (Apo)B, low levels of high density lipoprotein (HDL) or its protein component ApoA1, and, more recently, high levels of lipoprotein(a) [Lp(a)] [6]. While CVD risk reduction can be achieved by returning these altered lipids to target levels, this does not affect other facets of risk. Understanding the other risk factors present in a patient allow clinicians to assess absolute CVD risk and formulate suitable therapeutic approaches.
Assessment of absolute CVD risk would be incomplete without considering inflammation. Levels of high sensitivity C-reactive protein (hs-CRP) are used clinically as a marker of inflammation and can reflect the degree of vascular inflammation, which can be induced by dyslipidemia. Within people with the same levels of blood cholesterol, differences in hs-CRP have been implicated as a contributing factor to CVD outcomes [7]. Furthermore, targeting key inflammatory pathways, such as in the CANTOS trial, has been shown to reduce cardiovascular risk, independent of lipid lowering [8].
Of the immune cells involved in inflammatory pathways in CVD, monocytes and macrophages have become a focal point. Macrophages are a predominant cellular component of the atherosclerotic plaque and are a heterogeneous population of different functional phenotypes [9], which have different implications for plaque stability or instability [10]. Inflammatory macrophages, distinguished as the ‘M1’ subset, produce higher levels of inflammatory mediators including cytokines and matrix metalloproteinases which promote degradation of the fibrous cap, destabilizing the plaque structure [10,11]. Clinically, increased proportions of inflammatory M1 macrophages are found in the caps of unstable plaques [12]. Conversely, the ‘M2’ subset of macrophages demonstrate wound-healing functions including increased secretion of anti-inflammatory cytokines and production of collagen in carotid plaques, supporting plaque stabilization [12,13].
Infiltrating monocytes are a key source of plaque macrophages. They arise from precursors in bone marrow and are released into the circulation, where they constitute 3–8% of white blood cells [14]. Mouse studies have shown that blocking monocyte influx into the plaque can reduce development of atherosclerosis [15]. In humans, high levels of monocytes are seen with CVD development, and their numbers are emerging as an indicator of CVD severity. Considering monocyte-derived macrophages play a pivotal role in the inflammatory processes that determine plaque progression and instability, alterations in the monocyte population may influence the inflammatory functions of macrophages and thus promote the progression of an unstable atherosclerotic plaque. Indeed, monocyte phenotypes and functions are emerging as indicators of inflammation in CVD, and furthermore, disease severity. To understand the clinical significance of monocytes in CVD, it is important to delineate both the impact of changes in monocyte number and functional nature. The monocyte alterations of relevance to CVD, and dyslipidemia, will be the focus of this review, with a discussion of more recent insight into how these changes may be promoting plaque progression.
2. Monocyte Subset Proportions in CVD
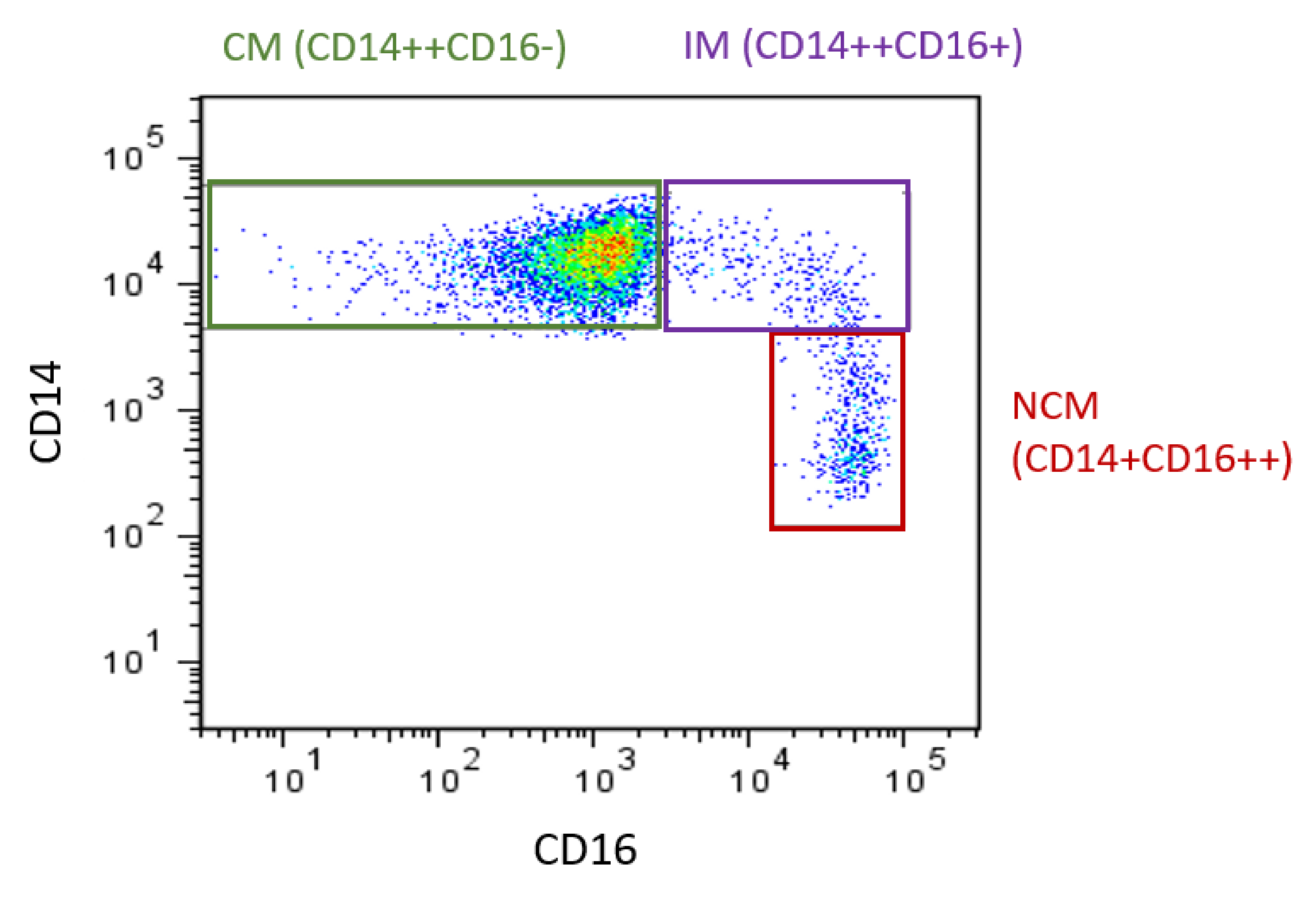
The heterogeneity of monocytes has been increasingly recognized, with this considered to have implications for CVD. Initially, monocytes were classified into two main subsets based on the expression of specific surface markers: both express CD14 but they differ in CD16 expression. These two subsets were referred to as CD16- and CD16+. Of these, CD16+ monocyte counts are elevated in CVD [16]. In 2010, the definition of monocyte subsets was expanded to three subsets. Still based on expression of CD14 and CD16 [17], the three recognized subsets are CD14++CD16- (classical, Mon 1), CD14++CD16+ (intermediate, Mon2), and CD14+CD16++ (nonclassical, Mon3) (Figure 1) [14,17]. Classical monocytes make up ~85% of circulating monocytes, with intermediate and nonclassical being ~5% and 10%, respectively. The use of flow cytometry to identify and enumerate these subsets is widespread. However, as specific methods (in particular the gating steps used to identify the subsets [18]) vary between groups, comparisons between studies can be difficult, as different gating yields different subset percentages. It has been shown that inclusion of a pan monocyte marker, such as CD86 or HLADR in the staining panel, is essential to ensure non-monocyte cells are excluded during the gating steps [19]. Additional monocyte surface markers have also been proposed to allow better separation of the intermediate and nonclassical subsets, such as their respective positive and negative expression of the chemokine receptor CCR2 [20]. Recently, it has been proposed that more than three subsets exist [21,22], which is unsurprising considering monocytes exist on a continuum (of expression of CD14 and CD16: as seen in Figure 1) rather than in discrete populations and that even within each subset, the expression of markers (such as adhesion molecules) is heterogeneous [23]. While classification into three subsets may be simplistic, it has been found to be relevant in studies of monocyte associations with CVD.
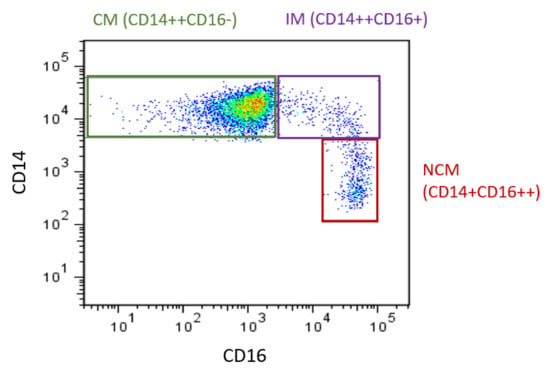
Figure 1.
Monocyte subsets in humans. Flow cytometry plot produced by the authors displaying monocyte subsets from a healthy control. Monocytes are defined using CD14 and CD16. CM: classical monocytes, IM: intermediate monocytes, NCM: nonclassical monocytes.
In CVD, the proportion of intermediate and/or nonclassical monocytes are frequently reported to be elevated (Table 1). There also appears to be further increases in these subsets with increased severity of disease. Patients with atherosclerosis in multiple vessels had significantly more CD16+ (intermediate + nonclassical) monocytes than those with single vessel disease [24]. This relationship was also seen when patients were divided by proportion of monocyte subsets, with those below median for classicals or above median for intermediate or nonclassicals having a higher occurrence of multivessel disease [25]. In PAD patients, the presence of ischemic ulcer (an indicator of greater disease severity) is associated with decreases in classical monocyte proportion and increases in intermediate monocyte proportion [26]. Similarly, high counts of intermediate and nonclassical monocytes have been observed in unstable angina compared to stable coronary heart disease (CHD) [27]. Lastly, when CAD was graded as obstructive, non-obstructive, or minimal, the obstructive and non-obstructive groups had higher intermediate monocytes than the group with minimal CAD [28].

Table 1.
Monocyte proportion changes in CVD.
Occurrence of clinical events is also linked to subset proportion changes. In stroke, patients had higher intermediate monocytes than controls [29]. Furthermore, elevations in monocyte subsets are associated with complications, with high levels of classical monocytes predictive of poor outcome after a stroke [29] and higher levels of intermediate monocytes seen in patients presenting for coronary angiography who subsequently had a cardiovascular event (death, stroke or acute MI) [30].
Could monocyte proportion changes influence plaque stability? There is evidence to suggest they can. In controls, intermediate monocyte count had a strong association with plaque severity [40]. In patients with HIV, a baseline elevation in nonclassical monocytes was linked to coronary artery calcification [41]. Similarly, correcting altered proportions of monocytes could help stabilize plaques, as treatment of unstable angina pectoris patients resulted in an increase in fibrous cap thickness, and this was associated with a reduction in CD16+ (intermediate + nonclassical) monocytes. These studies show monocyte proportion changes have a direct association with the plaque and could influence its stability.
It is clear that despite variations in proportions presented by different groups, there is a consistent pattern of elevated intermediate and/or nonclassical monocytes in CVD, and these are linked to severity of disease and its clinical manifestations. To understand possible mechanisms linking this elevation in proportions to CVD development and severity, consideration of how the functions of these monocyte subsets differ is important.
3. Monocyte Functional Changes in CVD
The elevation in intermediate monocytes in CVD is considered to promote disease progression, through increased inflammation. Intermediate monocytes (compared to the other subsets) have been implicated as major producers of inflammatory cytokines, including Interleukin (IL)-1β, IL-6 and Tumor necrosis factor (TNF)α in response to lipopolysaccharide (LPS: an outer membrane component of bacteria) [42]. Similarly, the monocyte subsets have been shown to have different potential for adhesion and migration [23,43], therefore proportional changes may further impact atherogenesis through altered monocyte entry to the plaque.
There are two caveats of assuming that proportional changes indicate a pro-atherogenic monocyte profile. Firstly, it hinges upon the accurate identification of subsets and secondly, the validity of the functional differences between the subsets may be uncertain due to varying experimental approaches. As mentioned in the previous section, there are differences in the flow cytometry gating approaches used to identify monocyte subsets by different research groups. Such inconsistencies between studies limit comparisons between them. In addition, inaccurate identification of the subsets (through flow analysis gating issues) could cause downstream errors in assigning functions to these subsets. As suitable identification of these subsets has been established [19,44], future studies should be able to be more consistent in identifying the three monocyte subsets. The second concern regarding functional assessment of subsets is due to functions from in vitro studies being assigned to these same cells in vivo. For example, cytokine release is often measured after in vitro stimulation with LPS, which does not replicate the microenvironment monocytes are exposed to in the blood, and thus may not entirely reflect the release of these cytokines in vivo.
While changes in monocyte subset proportions are taken as broadly indicative of overall functional changes in circulating monocytes, this is not the full picture. It is important to consider whether other monocyte changes, either independent of, or in addition to, subset proportions, also occur in CVD. Firstly, considering monocyte entry into the plaque, enhanced adhesion or migration capacity of monocytes could hasten plaque progression. Indeed, monocytes from CAD patients have increased adherence to endothelial cells [45]. Increased adherence could be attributed to elevations in monocyte adhesion molecules, such as the β2-integrin family (CD11a, CD11b, or CD11c complexed with CD18). In CVD, elevations in adhesion markers (CD11b/CD18) have been reported on monocytes [46] (Table 2). That CD18 is increased with greater CVD severity indicates further enhanced adhesion capacity as CVD progresses [28]. Enhanced ability to adhere does not necessarily equate to increased migration into the plaque, but elevated monocyte chemokine receptors indicate enhanced migration capacity in CVD. The increased level of chemokine receptors (CCR2 and CX3CR1) on monocytes in CAD [31] could contribute to enhanced monocyte accumulation in atherosclerotic plaques. Again, advancing severity may see a further increase, as higher CX3CR1 was seen with advancing stenosis [28].
Table 2.
Monocyte functional changes in CVD.
Monocyte inflammatory alterations have also received attention. When stimulated under M1 macrophage polarizing conditions (LPS/IFNγ), monocytes from people with CAD showed a greater gene expression of inflammatory cytokines (IL-6 and IL-1β) compared to controls [35], though another study found little difference in cytokine release between CAD and controls [20] (Table 2). The pathway leading to inflammatory cytokine release is also altered in CVD, with high levels of Toll-like receptor (TLR) 4, an LPS receptor, correlated with serum levels of TNFα [48]. Further activation of this inflammatory pathway is likely to occur with clinical events, as an elevation in TLR4 has been seen in both ST-elevation myocardial infarction (STEMI) and Non-ST-elevation myocardial infarction (NSTEMI) compared to CAD [49]. Apart from direct measurement of cytokine release or factors in this pathway, other indicators of inflammation have been shown to be elevated in CVD. Circulating monocytes in people with CVD resemble inflammatory macrophages (M1), owing to their high CD86/CD163 surface marker expression ratio relative to controls [37]. The CD86/CD163 ratio was associated with LPS-induced expression of IL-1β, indicating this resemblance is accompanied by functional impact [37]. Of interest, resemblance of monocytes to M1 macrophages has also been seen in people with risk factors for CVD such as diabetes and obesity [50,51]. Occurrence of a clinical event has also been linked to inflammatory alterations in monocytes, with patients who had experienced STEMI and NSTEMI having lower levels of the “M2” markers CD163 and CD206 on their monocyte-derived macrophages [39].
Thus, in addition to the implied functional alterations arising from monocyte subset proportion changes, altered adhesion, migration and inflammatory potential of monocytes has been seen in CVD, and are likely to promote disease progression. To understand what may cause these changes, examining alterations in light of risk factors could be beneficial.
4. Monocyte Count/Percentage Changes Relative to Lipids
While monocyte subset proportions are altered in CVD, it is likely that lipid levels are partly responsible for the altered proportions. High levels of atherogenic lipids, such as Lp(a) and small dense (sd)LDL are associated with increased intermediate monocytes [52,53], whereas HDL-C and ApoA1 levels are inversely associated with intermediate monocyte counts [54,55] (Table 3). HDL can impact monocyte development, as it has been shown in murine models that HDL suppresses stem cell proliferation [56,57] and reduces monocytosis [57]. Clinically, there is an inverse correlation between HDL-C level and the level of expression of CD16 on monocytes [58]. The change in CD16 expression would explain the increased proportion of intermediate and nonclassical monocytes in dyslipidemia (Table 3) and in CVD (Table 1) as, by definition, upregulation of CD16 expression would see monocytes no longer classed as ‘classical’ but deemed to be intermediate (or nonclassical) monocytes [58].
Table 3.
Monocyte count/proportion changes relative to lipid levels.
5. Monocyte Functional Changes Associated with Lipid Levels
Aside from changes in monocyte subset proportions, monocyte inflammatory profile and migration is also associated with lipid levels (Table 4 and illustrated in Figure 2). Firstly, monocytes from hyperlipidemic patients display increased cytokine production (post LPS stimulation) [61] and adhesion to endothelial cells [61,62] compared with controls. Secondly, even for generally healthy controls, monocytes are skewed towards an atherogenic state in individuals with a perturbed lipid profile [23,37,42]. Thirdly, high fat feeding increases monocyte adhesion and chemotaxis [63,64]. These all point to there being an immunopathology associated with dyslipidemia, one that begins in generally healthy individuals with perturbed lipids. Dyslipidemia is highly prevalent in adults [65,66]. Monocyte adoption of a pro-atherogenic phenotype is likely occurring largely undetected in many of these individuals, as dyslipidemia is generally asymptomatic [67]. Furthermore, these atherogenic monocytes could be contributing to plaque development, unabated in many, as treatment for dyslipidemia is recommended relative to risk of a cardiovascular event [68] and as such, younger dyslipidemic adults are usually not pharmacologically treated. The increase in monocyte CD11c expression and adherence to VCAM-1 (an adhesion molecule found on endothelial cells) observed after high fat feeding [63] would further promote monocyte migration into the vessel wall. Though of note, the increase in CD11c expression was transient, with its return to baseline matching the change in triglyceride levels [63]. With dyslipidemia being a major risk factor for CVD, the altered monocyte phenotype in dyslipidemia may explain, in part, the increased inflammatory profile of monocytes in CVD (Table 2).
Table 4.
Monocyte functional changes relative to lipid levels.
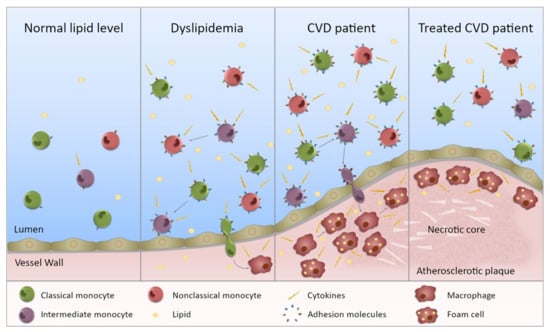
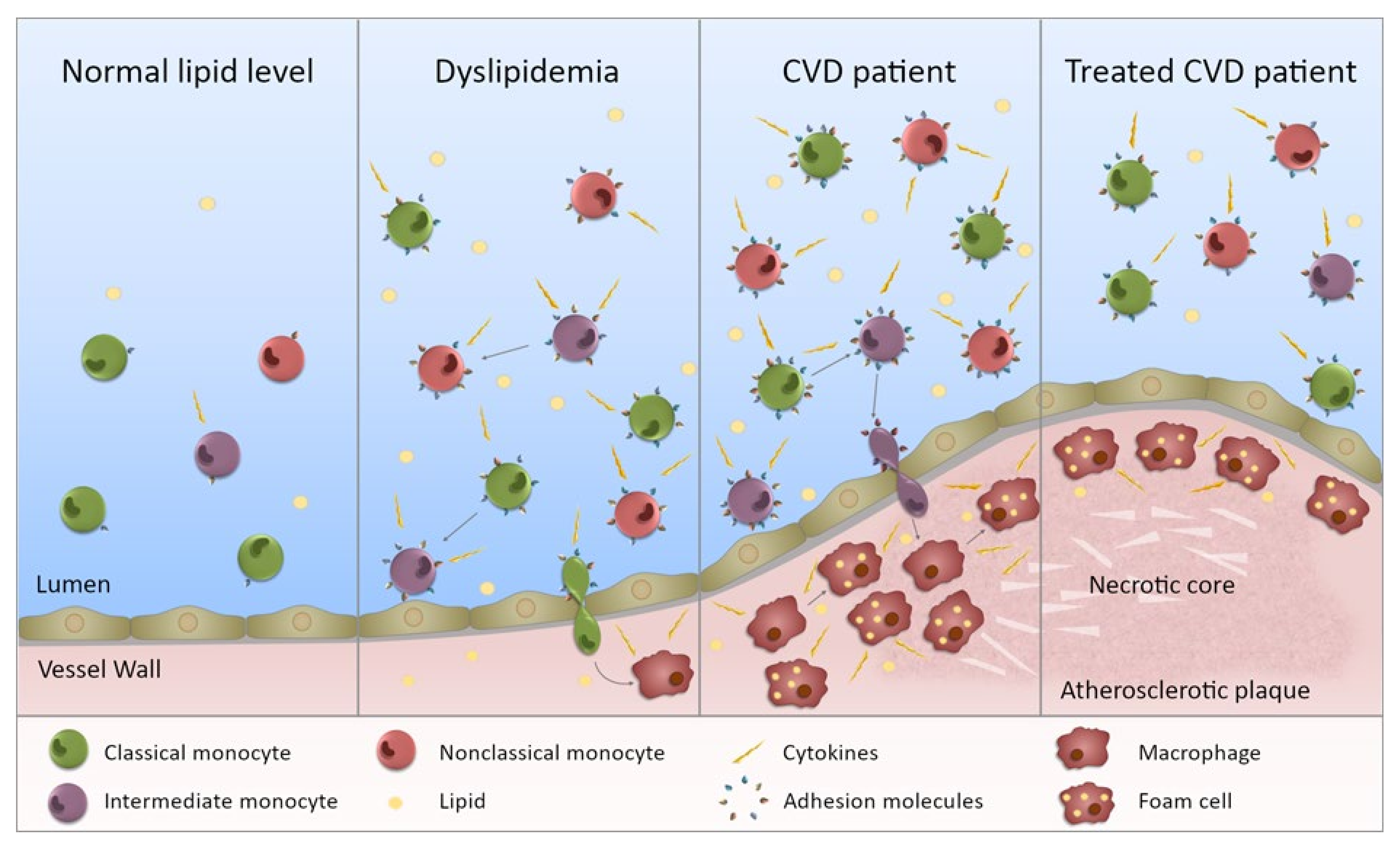
Figure 2.
Monocytes in dyslipidemia and CVD. This figure illustrates the state of monocytes in CVD. Normal lipid levels: in individuals with normal lipid levels, inflammation is low and the different monocyte subsets preferentially express specific adhesion molecules and chemokine receptors. Dyslipidemia: in individuals with dyslipidemia, the monocyte subset proportion is altered with increased intermediate and nonclassical monocytes, presumably through maturation of monocytes from one subset to the next. In addition, all subsets are more inflammatory (not just one) with increased ability to produce inflammatory cytokines. The subsets also express an increased level, and range of, adhesion molecules (and chemokine receptors) giving them a greater potential to bind to the endothelium. Entering the vessel wall, they become macrophages and upon lipid uptake, transform into foam cells. CVD patient: in CVD patients, the inflammatory state of the monocytes is further exacerbated and subset proportions further perturbed. Monocytes are more able to enter the vessel wall and to adopt an inflammatory macrophage phenotype once there. The chronic inflammatory state will contribute to plaque development. Treated CVD patient: upon treatment, the monocyte subsets become less inflammatory, but they do not necessarily return to levels equivalent to controls. Any lipid-associated functional changes of monocyte derived macrophages in the atherosclerotic plaque that have been induced by trained immunity may persist. Thus, plaque progression could continue despite a drop in lipid levels.
Of note, the skewing of monocytes to an atherogenic state in generally healthy individuals with a perturbed lipid profile was evident for all three monocyte subsets [23,42]. Though there were still notable differences between the subsets, these were overshadowed by differences between individuals, such that in some individuals, all three monocyte subsets produced inflammatory cytokines or expressed recruitment markers at a higher level than in other individuals. This suggests that functional distinctions between the monocyte populations are blurred in dyslipidemia, such that the whole monocyte population becomes increasingly pro-atherogenic. The inflammatory state being individual-specific, more so than subset-dependent, explains the finding (that was surprising at the time) that atherosclerosis regression in mice arose from recruitment of classical (Ly6Chi) monocytes that adopted an M2 macrophage phenotype in plaque, even though Ly6Chi monocytes were generally considered to be the ‘inflammatory’ subset [71]. Considering these findings, while monocyte subset count/percentage may be associated with disease state, selective targeting of a specific population to improve clinical outcomes is unlikely to be effective.
The phenotype that monocytes acquire in generally healthy individuals, dyslipidemia or CVD (Table 2, Table 4 and Table 5) is lipid-specific with, for example, TLR2 increased relative to total and LDL cholesterol [42], while most of the adhesion markers examined displayed an inverse relationship with ApoA1 [23]. The inverse correlation of the recruitment markers examined with ApoA1 [23] suggests that monocytes have an increased potential to extravasate when ApoA1 is low. While this is consistent with the findings that high levels of ApoA1 decrease both chemokine receptor levels and monocyte migration [72], it is also recognized that high ApoA1 can, through cholesterol depletion of lipid rafts (lipid-stabilized membrane microdomains in which cell receptors cluster), dampen the signaling pathways required for the reorganization of the actin cytoskeleton that is needed for cell migration [73]. That the level of several recruitment markers was increased on the three monocyte subsets in individuals with low ApoA1 [23] suggests that all subsets may be able to migrate into the vessel wall (or other sites of injury) and do so using a wider repertoire of adhesion molecules than for individuals with normal ApoA1 levels. Indeed, murine models of atherosclerosis are associated with the influx of both classical (Ly6Chi) and nonclassical (Ly6Clow) monocytes, not just one subset [74].
Table 5.
Monocyte functional changes relative to lipid levels: impact of treatment.
Interestingly, in dyslipidemic patients, monocyte TNFα release correlated with total cholesterol and ApoB, and IL-1β release positively correlated with total cholesterol [79], while in contrast in healthy controls, IL-1β production correlated inversely with HDL-C and ApoA1 [42]. The Okopien study [79] was much larger and, notably, a greater proportion of the individuals had elevated cholesterol than in the smaller Patel study [42], which may have enabled more associations with cholesterol to be seen. However, also, ApoA1 was not measured in the Okopien study [79]. As IL-1β production is regulated by lipid rafts, the integrity of which is dependent on cholesterol content [86], then both cholesterol overall and HDL may impact cytokine production through this mechanism. However, as Okopien [79] did not find any correlation between the drop in cytokine levels and extent of lipid lowering, and also found that cytokine levels dropped in those for whom the decrease in LDL-C was slight, it was suggested that the effect of statin treatment was not lipid related but involved other anti-inflammatory pathways. Similarly, while it was found that TLR4 expression was related to lipid levels, the reduction in TLR4 upon statin treatment of ex vivo monocytes was independent of the cholesterol lowering effect of statins [69]. Fibrates were also shown to decrease cytokine production by monocytes of individuals with dyslipidemia [76,77], an effect which is also thought to be unrelated to the lipid lowering effect of fibrates.
Though the drop in monocyte cytokine production (upon lipid lowering) was sometimes to the level of that of the controls [79,84], this was not always the case [82,85] (Table 5). Moreover, in an analysis of monocytes from FH patients’ post-statin treatment [85], Bekkering found that despite there being a large drop in LDL-C level, the monocyte RNA expression profile did not change to a great extent upon statin treatment. Patients’ monocytes were still enriched for metabolic and inflammatory pathways, with evidence of epigenetic modification of the proinflammatory TNFA gene [85].
6. Impact of Functional Changes
These alterations to monocyte function and phenotype, outlined above in both CVD and in dyslipidemia, are likely to have critical implications for atherosclerosis progression (Figure 2), due to the fact that monocytes are a source of plaque macrophages [10,11,12,13]. Previously, the phenotype of plaque macrophages was thought to be solely determined by factors in the plaque microenvironment, such as cytokines [87,88]. However, there is now growing evidence that factors in the circulation can cause functional changes in monocytes that impact the functional state of the differentiated monocyte-derived macrophages [89,90]. Therefore, inflammatory changes to circulating monocytes in CVD or dyslipidemia may persist through differentiation, resulting in an inflammatory macrophage phenotype promoting plaque destabilization. Indeed, there is evidence in CVD of a heightened inflammatory monocyte phenotype persisting in monocyte-derived macrophages [35]. Not only do monocytes isolated from CAD patients show a higher inflammatory cytokine response to LPS stimulation (IL-1β, IL-6, TNFα) compared to controls (Table 2), but this heightened inflammatory response remains present in macrophages derived from their in vitro culture [35]. Further evidence of skewed monocyte–macrophage differentiation is found in the context of obesity, where the peripheral blood mononuclear cells of obese individuals produced higher levels of inflammatory cytokines (IL-1β, TNFα, IFNγ) and had a restricted ability be polarized to an M2 macrophage phenotype (indicated by lower expression of M2 markers (CD163, CD200R, CD204, TGFβ), compared to healthy controls [91].
As it has been shown that monocyte functional changes associate with dyslipidemia, exposure to perturbed lipid levels may subsequently result in the skewed differentiation to inflammatory plaque macrophages. When exposed to LDL or its oxidized form (oxLDL) during culture, M1 monocyte-derived-macrophages display an enhanced inflammatory cytokine response (IL-1β, IL-6, TNFα); while in M2 monocyte-derived-macrophages, LDL exposure reduces M2 surface marker expression (CD206, CD200R) and anti-inflammatory cytokine production (TGFβ, IL-10) [92]. Conversely, the presence of HDL during macrophage culture inhibits polarization to an M1 phenotype (reducing M1-associated expression of CD192, CD64, TNFα, IL-6, MCP-1 and reactive oxygen species generation); while M2 polarization is unaffected [93]. As with monocytes, the effect of HDL on macrophages was due to its interfering impact on the membrane lipid rafts which are involved in activating inflammatory gene expression [93]. Specifically, this was mediated through the ApoA1 component of HDL inducing cholesterol efflux from the cells, subsequently reducing the cholesterol content of these lipid rafts [94].
There is also evidence that some inflammatory changes in monocyte–macrophage differentiation associated with CVD and dyslipidemia may be the result of a more stable cellular reprogramming, which would persistently promote vascular inflammation despite lipid-lowering treatment. Trained immunity is a phenomenon whereby monocytes form a metabolic and epigenetic memory that enables them to mount a heightened inflammatory response to secondary stimuli, even after the initial stimuli are removed [90]. This effect is indeed evident in atherosclerosis, as monocytes isolated from patients with symptomatic coronary atherosclerosis demonstrate an increased inflammatory cytokine response to LPS stimulation (TNFα, IL-6, IL-1β, IL-8, MCP-1). This response was associated with the mechanisms of trained immunity, specifically increased expression of glycolytic enzymes and epigenetic modifications to inflammatory genes [95]. As in vitro studies demonstrate that a trained response persists after macrophage differentiation [90], then it can be expected that these changes will result in inflammatory macrophage differentiation in the plaque. However, this effect was not observed in patients with asymptomatic atherosclerosis [95], suggesting that, clinically, trained immunity may only be evident in progressed disease.
Furthermore, the finding of Bekkering (mentioned above) that despite significant LDL lowering, statin treatment in FH patients was unable to reverse inflammatory monocyte changes, provides evidence of trained immunity in dyslipidemia [85]. While statins could reverse inflammatory changes in some cases of dyslipidemia (Table 5), they were unable to reverse the increased expression of metabolic genes and epigenetic modifications to inflammatory genes that may have been induced by the extensive duration of elevated lipids in FH [85]. Such functional reprogramming suggests that despite lipid-lowering by statin treatment, any lipid-associated functional changes to monocytes in CVD or dyslipidemia that are induced via trained immunity will persist and likely drive their differentiation to an inflammatory macrophage phenotype in the plaque.
While it has been demonstrated that native LDL does not induce a trained immune response, oxLDL is established as a key stimulus for the induction of trained immunity in monocyte–macrophage differentiation [96]. Monocytes treated with oxLDL, washed, cultured into macrophages and restimulated with LPS show increased inflammatory cytokine and chemokine production and foam cell formation. This is dependent on increased glycolysis and associated with epigenetic modifications to the promoters of inflammatory genes [96,97]. Lp(a), both isolated and in serum from an individual with elevated levels, has also been shown to induce trained immunity (increased TNFα and IL-6 response) through a similar in vitro protocol, whereas an increased cytokine response was not observed for LDL or HDL [98].
If trained immunity in response to certain lipids ‘locks’ monocyte differentiation to an inflammatory macrophage phenotype, then macrophages may be unable to demonstrate the wound-healing functions necessary for plaque stabilization in CVD. Normal wound healing involves a switch in macrophage phenotype from M1 to M2, however, this switch is dysfunctional in chronic wounds such as the atherosclerotic plaque [99]. While some studies suggest there is plasticity in macrophage phenotypes [11], it has been demonstrated that the metabolic changes associated with M1 macrophage polarization, many of which are also evident in trained immunity by oxLDL and in atherosclerosis [95,96,97], prevent inflammatory macrophages from being able to respond to M2 stimulus [100]. As such, inflammatory monocyte changes evident in CVD and dyslipidemia which result from trained immunity may restrict these cells from becoming wound-healing macrophages in the plaque, so that plaque instability persists, even after lipid-lowering treatments. This may contribute to the residual risk of a clinical event that CVD patients face, despite optimal therapy.
Considering the ongoing COVID-19 pandemic, a brief word on links between this condition and CVD is pertinent. A bidirectional relationship between CVD and COVID-19 has been proposed, with presence of CVD increasing risk of poor COVID-19 outcomes, and the presence of COVID-19 increasing the risk of cardiovascular outcomes [101]. Similar to in CVD, monocyte alterations could be one contributor to this relationship, as CD16+ monocytes have been reported to increase in COVID-19, along with inflammatory cytokine levels in the serum [102], and as we have shown here these types of changes could be mechanistically associated with CVD. Furthermore, COVID-19 outcomes are more severe in individuals with dyslipidemia, in particular, low HDL-C [103]. As outlined here, these individuals have a heightened cytokine response when challenged with LPS; a similar response when challenged with SARS-CoV-2 may perhaps contribute to the cytokine storm, particularly as HDL-C levels decrease even further in COVID-19 [104]. The heightened inflammatory response may be further exacerbated if also coupled with a trained immune response.
7. Conclusions
In investigating the question of monocyte nature versus number, it is evident that both the functional nature and distribution of subset numbers are not only altered in CVD, but may also contribute to disease progression. Assessment of monocyte numbers reveals that the proportions of the three subsets are perturbed in CVD and associate with disease severity, the occurrence and outcomes of clinical events, and plaque progression. While elevation of the intermediate and nonclassical subsets suggests that their ascribed inflammatory functions contribute to atherosclerosis, it is important to consider the broader functional changes both within the subsets and across the whole monocyte population, that occur in CVD. Of specific relevance to atherosclerosis are increased inflammation and migration.
These cellular changes are observed in association with specific lipids, particularly (low levels of) HDL-C, indicating that lipids may influence both monocyte subset proportions and function. As these functional changes in cytokine production, inflammatory surface marker expression and recruitment markers are evident in all three subsets, there is an overall skewing of monocytes to a pro-atherogenic profile in dyslipidemia. While lipid-lowering treatments demonstrate some ability to reduce these inflammatory functional changes, this is not always restored to control levels and some inflammatory functions may persist through differentiation into plaque macrophages. Persistent skewing of plaque macrophages to an inflammatory function, left untreated, will promote plaque instability. As such, both the proportional and functional changes to monocytes evident with increased CVD risk in dyslipidemia, and further exacerbated in CVD itself, are likely to contribute to the development and progression of atherosclerosis, and moreover, limit the ability of lipid lowering treatment to stabilize atherosclerotic plaques.
Author Contributions
Conceptualization, H.W. and H.J.M.; writing—original draft preparation was by all authors (H.W., C.D.M., S.C.H.L., J.P.F. and H.J.M.); writing—review and editing was done by H.W., H.J.M., C.D.M. All authors have read and agreed to the published version of the manuscript.
Funding
The APC and work from which this paper arose was funded by the Clinical Chemistry Research and Education Fund, ICPMR-NSWHP.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Cardiovascular Diseases (CVDs); WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- American Diabetes Association. Peripheral arterial disease in people with diabetes. Diabetes Care 2003, 26, 3333–3341. [Google Scholar] [CrossRef] [Green Version]
- Khot, U.N.; Khot, M.B.; Bajzer, C.T.; Sapp, S.K.; Ohman, E.M.; Brener, S.J.; Ellis, S.G.; Lincoff, A.M.; Topol, E.J. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA 2003, 290, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Stoger, J.L.; Gijbels, M.J.; van der Velden, S.; Manca, M.; van der Loos, C.M.; Biessen, E.A.; Daemen, M.J.; Lutgens, E.; de Winther, M.P. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef]
- Medbury, H.J.; James, V.; Ngo, J.; Hitos, K.; Wang, Y.; Harris, D.C.; Fletcher, J.P. Differing association of macrophage subsets with atherosclerotic plaque stability. Int. Angiol. 2013, 32, 74–84. [Google Scholar]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Shantsila, E.; Hristov, M.; Caligiuri, G.; Guzik, T.; Heine, G.H.; Hoefer, I.E.; Monaco, C.; Peter, K.; Rainger, E.; et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thromb. Haemost. 2016, 116, 626–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poupel, L.; Boissonnas, A.; Hermand, P.; Dorgham, K.; Guyon, E.; Auvynet, C.; Charles, F.S.; Lesnik, P.; Deterre, P.; Combadiere, C. Pharmacological inhibition of the chemokine receptor, CX3CR1, reduces atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2297–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlitt, A.; Heine, G.H.; Blankenberg, S.; Espinola-Klein, C.; Dopheide, J.F.; Bickel, C.; Lackner, K.J.; Iz, M.; Meyer, J.; Darius, H.; et al. CD14+CD16+ monocytes in coronary artery disease and their relationship to serum TNF-alpha levels. Thromb. Haemost. 2004, 92, 419–424. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Zawada, A.M.; Fell, L.H.; Untersteller, K.; Seiler, S.; Rogacev, K.S.; Fliser, D.; Ziegler-Heitbrock, L.; Heine, G.H. Comparison of two different strategies for human monocyte subsets gating within the large-scale prospective CARE FOR HOMe Study. Cytom. A 2015, 87, 750–758. [Google Scholar] [CrossRef]
- Zawada, A.M.; Rogacev, K.S.; Schirmer, S.H.; Sester, M.; Bohm, M.; Fliser, D.; Heine, G.H. Monocyte heterogeneity in human cardiovascular disease. Immunobiology 2012, 217, 1273–1284. [Google Scholar] [CrossRef]
- Shantsila, E.; Tapp, L.D.; Wrigley, B.J.; Pamukcu, B.; Apostolakis, S.; Montoro-Garcia, S.; Lip, G.Y. Monocyte subsets in coronary artery disease and their associations with markers of inflammation and fibrinolysis. Atherosclerosis 2014, 234, 4–10. [Google Scholar] [CrossRef]
- Hamers, A.A.J.; Dinh, H.Q.; Thomas, G.D.; Marcovecchio, P.; Blatchley, A.; Nakao, C.S.; Kim, C.; McSkimming, C.; Taylor, A.M.; Nguyen, A.T.; et al. Human Monocyte Heterogeneity as Revealed by High-Dimensional Mass Cytometry. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 25–36. [Google Scholar] [CrossRef]
- Merah-Mourah, F.; Cohen, S.O.; Charron, D.; Mooney, N.; Haziot, A. Identification of Novel Human Monocyte Subsets and Evidence for Phenotypic Groups Defined by Interindividual Variations of Expression of Adhesion Molecules. Sci. Rep. 2020, 10, 4397. [Google Scholar] [CrossRef]
- Patel, V.K.; Williams, H.; Li, S.C.H.; Fletcher, J.P.; Medbury, H.J. Monocyte Subset Recruitment Marker Profile Is Inversely Associated with Blood ApoA1 Levels. Front. Immunol. 2021, 12, 616305. [Google Scholar] [CrossRef]
- Ozaki, Y.; Imanishi, T.; Taruya, A.; Aoki, H.; Masuno, T.; Shiono, Y.; Komukai, K.; Tanimoto, T.; Kitabata, H.; Akasaka, T. Circulating CD14+CD16+ monocyte subsets as biomarkers of the severity of coronary artery disease in patients with stable angina pectoris. Circ. J. 2012, 76, 2412–2418. [Google Scholar] [CrossRef] [Green Version]
- Afanasieva, O.I.; Filatova, A.Y.; Arefieva, T.I.; Klesareva, E.A.; Tyurina, A.V.; Radyukhina, N.V.; Ezhov, M.V.; Pokrovsky, S.N. The Association of Lipoprotein(a) and Circulating Monocyte Subsets with Severe Coronary Atherosclerosis. J. Cardiovasc. Dev. Dis 2021, 8, 63. [Google Scholar] [CrossRef]
- Wildgruber, M.; Aschenbrenner, T.; Wendorff, H.; Czubba, M.; Glinzer, A.; Haller, B.; Schiemann, M.; Zimmermann, A.; Berger, H.; Eckstein, H.H.; et al. The “Intermediate” CD14(++)CD16(+) monocyte subset increases in severe peripheral artery disease in humans. Sci. Rep. 2016, 6, 39483. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Zhou, X.; Ge, L.; Ji, W.J.; Shi, R.; Lu, R.Y.; Sun, H.Y.; Guo, Z.Z.; Zhao, J.H.; Jiang, T.M.; et al. Monocyte subsets and monocyte-platelet aggregates in patients with unstable angina. J. Thromb. Thrombol. 2014, 38, 439–446. [Google Scholar] [CrossRef]
- Sbrana, S.; Campolo, J.; Clemente, A.; Bastiani, L.; Cecchettini, A.; Ceccherini, E.; Caselli, C.; Neglia, D.; Parodi, O.; Chiappino, D.; et al. Blood Monocyte Phenotype Fingerprint of Stable Coronary Artery Disease: A Cross-Sectional Substudy of SMARTool Clinical Trial. Biomed. Res. Int. 2020, 2020, 8748934. [Google Scholar] [CrossRef]
- Urra, X.; Villamor, N.; Amaro, S.; Gomez-Choco, M.; Obach, V.; Oleaga, L.; Planas, A.M.; Chamorro, A. Monocyte subtypes predict clinical course and prognosis in human stroke. J. Cereb. Blood Flow Metab. 2009, 29, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Rogacev, K.S.; Cremers, B.; Zawada, A.M.; Seiler, S.; Binder, N.; Ege, P.; Grosse-Dunker, G.; Heisel, I.; Hornof, F.; Jeken, J.; et al. CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. J. Am. Coll. Cardiol. 2012, 60, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallone, T.; Turconi, G.; Soldati, G.; Pedrazzini, G.; Moccetti, T.; Vassalli, G. Heterogeneity of human monocytes: An optimized four-color flow cytometry protocol for analysis of monocyte subsets. J. Cardiovasc. Transl. Res. 2011, 4, 211–219. [Google Scholar] [CrossRef]
- Dopheide, J.F.; Obst, V.; Doppler, C.; Radmacher, M.C.; Scheer, M.; Radsak, M.P.; Gori, T.; Warnholtz, A.; Fottner, C.; Daiber, A.; et al. Phenotypic characterisation of pro-inflammatory monocytes and dendritic cells in peripheral arterial disease. Thromb. Haemost. 2012, 108, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Tapp, L.D.; Shantsila, E.; Wrigley, B.J.; Pamukcu, B.; Lip, G.Y. The CD14++CD16+ monocyte subset and monocyte-platelet interactions in patients with ST-elevation myocardial infarction. J. Thromb. Haemost. 2012, 10, 1231–1241. [Google Scholar] [CrossRef]
- Sturhan, H.; Ungern-Sternberg, S.N.; Langer, H.; Gawaz, M.; Geisler, T.; May, A.E.; Seizer, P. Regulation of EMMPRIN (CD147) on monocyte subsets in patients with symptomatic coronary artery disease. Thromb. Res. 2015, 135, 1160–1164. [Google Scholar] [CrossRef]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Han, Y.; Xu, D.; Zhu, G.; Singh, S.; Chen, L.; Zhu, M.; Chen, W.; Xu, Y.; Li, X. Comparison of circulating dendritic cell and monocyte subsets at different stages of atherosclerosis: Insights from optical coherence tomography. BMC Cardiovasc. Disord. 2017, 17, 270. [Google Scholar] [CrossRef]
- Williams, H.; Cassorla, G.; Pertsoulis, N.; Patel, V.; Vicaretti, M.; Marmash, N.; Hitos, K.; Fletcher, J.P.; Medbury, H.J. Human classical monocytes display unbalanced M1/M2 phenotype with increased atherosclerotic risk and presence of disease. Int. Angiol. 2017, 36, 145–155. [Google Scholar] [CrossRef]
- Brown, R.A.; Lip, G.Y.H.; Varma, C.; Shantsila, E. Impact of Mon2 monocyte-platelet aggregates on human coronary artery disease. Eur. J. Clin. Investig. 2018, 48, e12911. [Google Scholar] [CrossRef]
- Eligini, S.; Cosentino, N.; Fiorelli, S.; Fabbiocchi, F.; Niccoli, G.; Refaat, H.; Camera, M.; Calligaris, G.; De Martini, S.; Bonomi, A.; et al. Biological profile of monocyte-derived macrophages in coronary heart disease patients: Implications for plaque morphology. Sci. Rep. 2019, 9, 8680. [Google Scholar] [CrossRef]
- Lo, S.C.; Lee, W.J.; Chen, C.Y.; Lee, B.C. Intermediate CD14(++)CD16(+) monocyte predicts severe coronary stenosis and extensive plaque involvement in asymptomatic individuals. Int. J. Cardiovasc. Imaging 2017, 33, 1223–1236. [Google Scholar] [CrossRef]
- Zungsontiporn, N.; Tello, R.R.; Zhang, G.; Mitchell, B.I.; Budoff, M.; Kallianpur, K.J.; Nakamoto, B.K.; Keating, S.M.; Norris, P.J.; Ndhlovu, L.C.; et al. Non-Classical Monocytes and Monocyte Chemoattractant Protein-1 (MCP-1) Correlate with Coronary Artery Calcium Progression in Chronically HIV-1 Infected Adults on Stable Antiretroviral Therapy. PLoS ONE 2016, 11, e0149143. [Google Scholar] [CrossRef]
- Patel, V.K.; Williams, H.; Li, S.C.H.; Fletcher, J.P.; Medbury, H.J. Monocyte inflammatory profile is specific for individuals and associated with altered blood lipid levels. Atherosclerosis 2017, 263, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marimuthu, R.; Francis, H.; Dervish, S.; Li, S.C.H.; Medbury, H.; Williams, H. Characterization of Human Monocyte Subsets by Whole Blood Flow Cytometry Analysis. J. Vis. Exp. 2018, 140, 57941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.H.; Hung, C.H.; Shih, J.Y.; Chu, P.M.; Cheng, Y.H.; Lin, H.C.; Tsai, K.L. SIRT1 inhibition causes oxidative stress and inflammation in patients with coronary artery disease. Redox. Biol. 2017, 13, 301–309. [Google Scholar] [CrossRef]
- Mazzone, A.; De Servi, S.; Mazzucchelli, I.; Fossati, G.; Gritti, D.; Canale, C.; Cusa, C.; Ricevuti, G. Increased expression of CD11b/CD18 on phagocytes in ischaemic disease: A bridge between inflammation and coagulation. Eur. J. Clin. Investig. 1997, 27, 648–652. [Google Scholar] [CrossRef]
- Kassirer, M.; Zeltser, D.; Prochorov, V.; Schoenman, G.; Frimerman, A.; Keren, G.; Shapira, I.; Miller, H.; Roth, A.; Arber, N.; et al. Increased expression of the CD11b/CD18 antigen on the surface of peripheral white blood cells in patients with ischemic heart disease: Further evidence for smoldering inflammation in patients with atherosclerosis. Am. Heart J. 1999, 138, 555–559. [Google Scholar] [CrossRef]
- Kashiwagi, M.; Imanishi, T.; Ozaki, Y.; Satogami, K.; Masuno, T.; Wada, T.; Nakatani, Y.; Ishibashi, K.; Komukai, K.; Tanimoto, T.; et al. Differential expression of Toll-like receptor 4 and human monocyte subsets in acute myocardial infarction. Atherosclerosis 2012, 221, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Tapp, L.D.; Shantsila, E.; Wrigley, B.J.; Montoro-Garcia, S.; Lip, G.Y. TLR4 expression on monocyte subsets in myocardial infarction. J. Intern. Med. 2013, 273, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadini, G.P.; de Kreutzenberg, S.V.; Boscaro, E.; Albiero, M.; Cappellari, R.; Krankel, N.; Landmesser, U.; Toniolo, A.; Bolego, C.; Cignarella, A.; et al. An unbalanced monocyte polarisation in peripheral blood and bone marrow of patients with type 2 diabetes has an impact on microangiopathy. Diabetologia 2013, 56, 1856–1866. [Google Scholar] [CrossRef] [Green Version]
- Satoh, N.; Shimatsu, A.; Himeno, A.; Sasaki, Y.; Yamakage, H.; Yamada, K.; Suganami, T.; Ogawa, Y. Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients: Effect of pioglitazone. Diabetes Care 2010, 33, e7. [Google Scholar] [CrossRef] [Green Version]
- Krychtiuk, K.A.; Kastl, S.P.; Hofbauer, S.L.; Wonnerth, A.; Goliasch, G.; Ozsvar-Kozma, M.; Katsaros, K.M.; Maurer, G.; Huber, K.; Dostal, E.; et al. Monocyte subset distribution in patients with stable atherosclerosis and elevated levels of lipoprotein(a). J. Clin. Lipidol. 2015, 9, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krychtiuk, K.A.; Kastl, S.P.; Pfaffenberger, S.; Lenz, M.; Hofbauer, S.L.; Wonnerth, A.; Koller, L.; Katsaros, K.M.; Pongratz, T.; Goliasch, G.; et al. Association of small dense LDL serum levels and circulating monocyte subsets in stable coronary artery disease. PLoS ONE 2015, 10, e0123367. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.S.; Chiang, B.L. Correlation between serum lipid profiles and the ratio and count of the CD16+ monocyte subset in peripheral blood of apparently healthy adults. J. Formos. Med. Assoc. 2002, 101, 11–17. [Google Scholar] [PubMed]
- Rogacev, K.S.; Zawada, A.M.; Emrich, I.; Seiler, S.; Bohm, M.; Fliser, D.; Woollard, K.J.; Heine, G.H. Lower Apo A-I and lower HDL-C levels are associated with higher intermediate CD14++CD16+ monocyte counts that predict cardiovascular events in chronic kidney disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2120–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Liang, B.; Zhang, X.; Zheng, F. Lower HDL-C levels are associated with higher expressions of CD16 on monocyte subsets in coronary atherosclerosis. Int. J. Med. Sci. 2020, 17, 2171–2179. [Google Scholar] [CrossRef]
- Krychtiuk, K.A.; Kastl, S.P.; Pfaffenberger, S.; Pongratz, T.; Hofbauer, S.L.; Wonnerth, A.; Katsaros, K.M.; Goliasch, G.; Gaspar, L.; Huber, K.; et al. Small high-density lipoprotein is associated with monocyte subsets in stable coronary artery disease. Atherosclerosis 2014, 237, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Dai Perrard, X.Y.; Lian, Z.; Bobotas, G.; Dicklin, M.R.; Maki, K.C.; Wu, H. Effects of n-3 fatty acid treatment on monocyte phenotypes in humans with hypertriglyceridemia. J. Clin. Lipidol. 2017, 11, 1361–1371. [Google Scholar] [CrossRef]
- Devaraj, S.; Jialal, I. Validation of the circulating monocyte being representative of the cholesterol-loaded macrophage: Biomediator activity. Arch. Pathol. Lab. Med. 2008, 132, 1432–1435. [Google Scholar] [CrossRef]
- Jongkind, J.F.; Verkerk, A.; Hoogerbrugge, N. Monocytes from patients with combined hypercholesterolemia-hypertriglyceridemia and isolated hypercholesterolemia show an increased adhesion to endothelial cells in vitro: II. Influence of intrinsic and extrinsic factors on monocyte binding. Metabolism 1995, 44, 374–378. [Google Scholar] [CrossRef]
- Gower, R.M.; Wu, H.; Foster, G.A.; Devaraj, S.; Jialal, I.; Ballantyne, C.M.; Knowlton, A.A.; Simon, S.I. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Short, J.D.; Tavakoli, S.; Nguyen, H.N.; Carrera, A.; Farnen, C.; Cox, L.A.; Asmis, R. Dyslipidemic Diet-Induced Monocyte “Priming” and Dysfunction in Non-Human Primates Is Triggered by Elevated Plasma Cholesterol and Accompanied by Altered Histone Acetylation. Front. Immunol. 2017, 8, 958. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-F.; Chang, Y.-H.; Chien, S.-C.; Lin, Y.-H.; Yeh, H.-Y. Epidemiology of Dyslipidemia in the Asia Pacific Region. Int. J. Gerontol. 2018, 12, 2–6. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Jonas, J.B.; You, Q.S.; Wang, Y.X.; Yang, H. Prevalence and associated factors of dyslipidemia in the adult Chinese population. PLoS ONE 2011, 6, e17326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talpur, M.T.H.; Katbar, M.T.; Shabir, K.U.; Shabir, K.U.; Yaqoob, U.; Jabeen, S.; Zia, D. Prevalence of dyslipidemia in young adults. Prof. Med. J. 2020, 27, 987–993. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Foldes, G.; von Haehling, S.; Okonko, D.O.; Jankowska, E.A.; Poole-Wilson, P.A.; Anker, S.D. Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. Int. J. Cardiol. 2008, 124, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Simoni, F.; Cappellari, R.; Vitturi, N.; Galasso, S.; Vigili de Kreutzenberg, S.; Previato, L.; Avogaro, A. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis 2014, 237, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Vengrenyuk, Y.; Ramsey, S.A.; Vila, N.R.; Girgis, N.M.; Liu, J.; Gusarova, V.; Gromada, J.; Weinstock, A.; Moore, K.J.; et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Investig. 2017, 127, 2904–2915. [Google Scholar] [CrossRef]
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.J.; Barrett, T.J.; Taylor, L.; McNeill, E.; Manmadhan, A.; Recio, C.; Carmineri, A.; Brodermann, M.H.; White, G.E.; Cooper, D.; et al. Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. Elife 2016, 5, e15190. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.H.; Han, K.O.; Green, S.R.; Quehenberger, O. Expression of the monocyte chemoattractant protein-1 receptor CCR2 is increased in hypercholesterolemia. Differential effects of plasma lipoproteins on monocyte function. J. Lipid Res. 1999, 40, 1053–1063. [Google Scholar] [CrossRef]
- Okopien, B.; Huzarska, M.; Kulach, A.; Stachura-Kulach, A.; Madej, A.; Belowski, D.; Zielinski, M.; Herman, Z.S. Hypolipidemic drugs affect monocyte IL-1beta gene expression and release in patients with IIa and IIb dyslipidemia. J. Cardiovasc. Pharmacol. 2005, 45, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Okopien, B.; Kowalski, J.; Krysiak, R.; Labuzek, K.; Stachura-Kulach, A.; Kulach, A.; Zielinski, M.; Herman, Z.S. Monocyte suppressing action of fenofibrate. Pharmacol. Rep. 2005, 57, 367–372. [Google Scholar] [PubMed]
- Okopien, B.; Krysiak, R.; Haberka, M.; Herman, Z.S. Effect of monthly atorvastatin and fenofibrate treatment on monocyte chemoattractant protein-1 release in patients with primary mixed dyslipidemia. J. Cardiovasc. Pharmacol. 2005, 45, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Okopien, B.; Krysiak, R.; Kowalski, J.; Madej, A.; Belowski, D.; Zielinski, M.; Herman, Z.S. Monocyte release of tumor necrosis factor-alpha and interleukin-1beta in primary type IIa and IIb dyslipidemic patients treated with statins or fibrates. J. Cardiovasc. Pharmacol. 2005, 46, 377–386. [Google Scholar] [CrossRef]
- Serrano, C.V., Jr.; Pesaro, A.E.; de Lemos, J.A.; Rached, F.; Segre, C.A.; Gomes, F.; Ribeiro, A.F.; Nicolau, J.C.; Yoshida, V.M.; Monteiro, H.P. Native LDL-cholesterol mediated monocyte adhesion molecule overexpression is blocked by simvastatin. Cardiovasc. Drugs Ther. 2009, 23, 215–220. [Google Scholar] [CrossRef]
- Krysiak, R.; Okopien, B. Different effects of simvastatin on ex vivo monocyte cytokine release in patients with hypercholesterolemia and impaired glucose tolerance. J. Physiol. Pharmacol. 2010, 61, 725–732. [Google Scholar] [PubMed]
- Krysiak, R.; Stachura-Kulach, A.; Okopien, B. Metabolic and monocyte-suppressing actions of fenofibrate in patients with mixed dyslipidemia and early glucose metabolism disturbances. Pharmacol. Rep. 2010, 62, 120–130. [Google Scholar] [CrossRef]
- Krysiak, R.; Okopien, B. The effect of ezetimibe and simvastatin on monocyte cytokine release in patients with isolated hypercholesterolemia. J. Cardiovasc. Pharmacol. 2011, 57, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, R.; Okopien, B. Effect of bezafibrate on monocyte cytokine release and systemic inflammation in patients with impaired fasting glucose. J. Clin. Pharmacol. 2011, 51, 1459–1467. [Google Scholar] [CrossRef]
- Bekkering, S.; Stiekema, L.C.A.; Bernelot Moens, S.; Verweij, S.L.; Novakovic, B.; Prange, K.; Versloot, M.; Roeters van Lennep, J.E.; Stunnenberg, H.; de Winther, M.; et al. Treatment with Statins Does Not Revert Trained Immunity in Patients with Familial Hypercholesterolemia. Cell Metab. 2019, 30, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef]
- Wolfs, I.M.; Donners, M.M.; de Winther, M.P. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb. Haemost. 2011, 106, 763–771. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Bories, G.; Caiazzo, R.; Derudas, B.; Copin, C.; Raverdy, V.; Pigeyre, M.; Pattou, F.; Staels, B.; Chinetti-Gbaguidi, G. Impaired alternative macrophage differentiation of peripheral blood mononuclear cells from obese subjects. Diab. Vasc. Dis. Res. 2012, 9, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sharea, A.; Lee, M.K.; Moore, X.L.; Fang, L.; Sviridov, D.; Chin-Dusting, J.; Andrews, K.L.; Murphy, A.J. Native LDL promotes differentiation of human monocytes to macrophages with an inflammatory phenotype. Thromb. Haemost. 2016, 115, 762–772. [Google Scholar] [CrossRef]
- Lee, M.K.; Moore, X.L.; Fu, Y.; Al-Sharea, A.; Dragoljevic, D.; Fernandez-Rojo, M.A.; Parton, R.; Sviridov, D.; Murphy, A.J.; Chin-Dusting, J.P. High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br. J. Pharmacol. 2016, 173, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.J.; Woollard, K.J.; Hoang, A.; Mukhamedova, N.; Stirzaker, R.A.; McCormick, S.P.; Remaley, A.T.; Sviridov, D.; Chin-Dusting, J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2071–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.; Netea, M.G.; Gomes, M.E.; et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.T.; Groh, L.; Thiem, K.; Bekkering, S.; Li, Y.; Matzaraki, V.; van der Heijden, C.; van Puffelen, J.H.; Lachmandas, E.; Jansen, T.; et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J. Mol. Med. 2020, 98, 819–831. [Google Scholar] [CrossRef]
- Valk, F.M.v.d.; Bekkering, S.; Kroon, J.; Yeang, C.; Bossche, J.V.d.; Buul, J.D.v.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, Y.; Wei, X.; Liu, X.; Guan, J.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z.; Lin, X. Dynamic changes in monocytes subsets in COVID-19 patients. Hum. Immunol. 2021, 82, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Masana, L.; Correig, E.; Ibarretxe, D.; Anoro, E.; Arroyo, J.A.; Jerico, C.; Guerrero, C.; Miret, M.; Naf, S.; Pardo, A.; et al. Low HDL and high triglycerides predict COVID-19 severity. Sci. Rep. 2021, 11, 7217. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. The bidirectional link between HDL and COVID-19 infections. J. Lipid Res. 2021, 62, 100067. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).