
Directional Persistence of Cell Migration in Schizophrenia Patient-Derived Olfactory Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
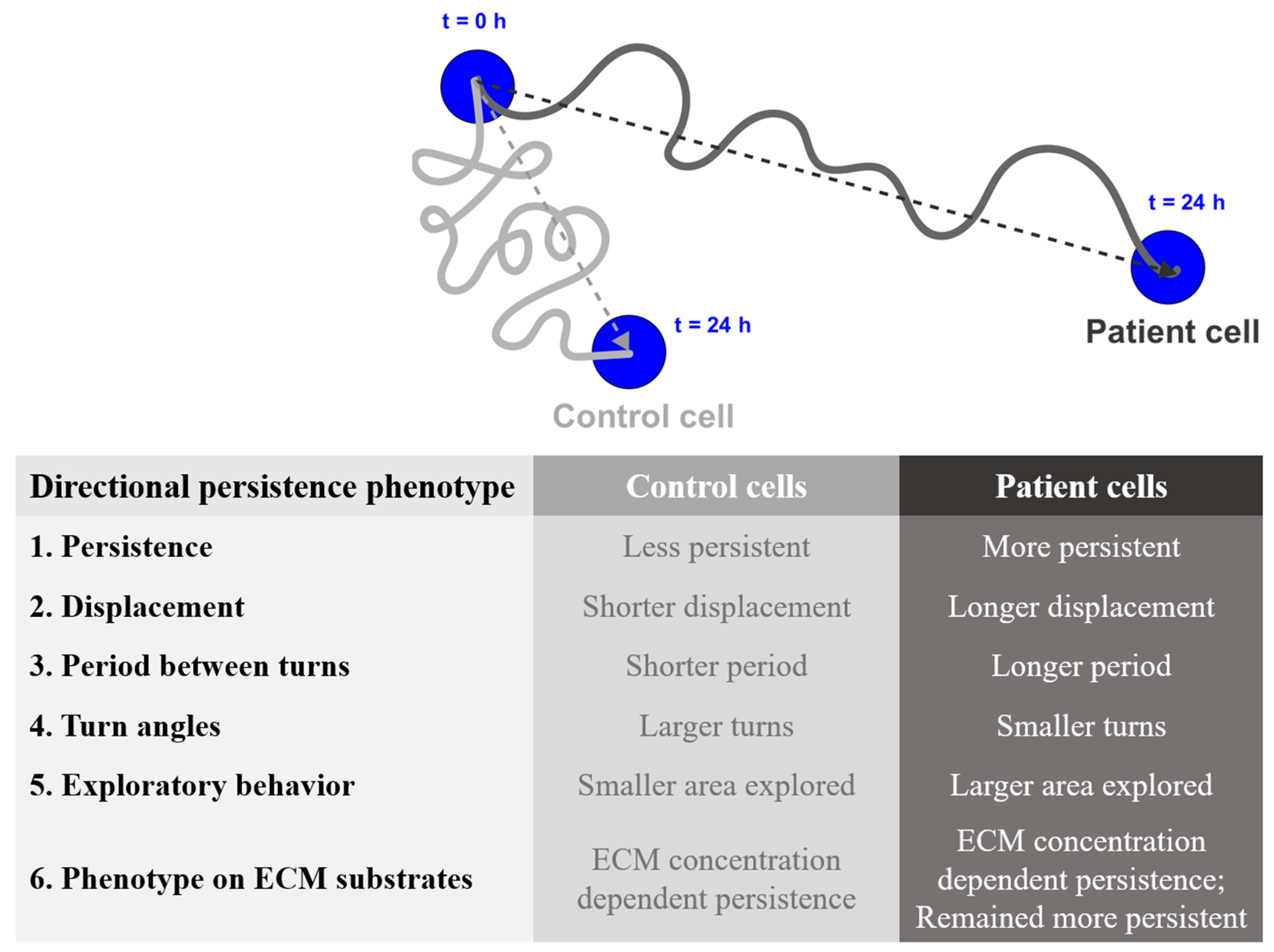
:1. Introduction
2. Results
2.1. Patient Cells Moved in Straighter Trajectories than Control Cells at Baseline
2.2. Patient Cell Migration Covered Greater Surface Areas than Control Cells at Baseline
2.3. Patient Cells Spent More Time in Persistent Movement than Control Cells at Baseline
2.4. Patient Cells Spent More Time in Active Movement and Less Time Idling than Control Cells at Baseline
2.5. Patient Cells Made Smaller Turns during the Active Phase of Cell Migration than Control Cells at Baseline
2.6. Patient Cell Trajectories Were Straighter on Most Concentrations of Extracellular Matrix Proteins
2.7. Patient Cells Explored Larger Surface Areas on Most Concentrations of ECM Proteins
2.8. Patient Cells Persisted for Longer on Most Concentrations of Extracellular Matrix Proteins
2.9. Patient Cell Active and Idling Times Were Similar on Most Extracellular Matrix Proteins
2.10. Patient Cell Turn Angles Were Similar on Most Extracellular Matrix Proteins
3. Discussion
4. Materials and Methods
4.1. Olfactory Neurosphere-Derived Cells
4.2. High-Throughput Single Cell Tracking
4.3. In Silico Single Cell Directional Persistence Analysis
4.4. In Silico Single Cell Pausing and Turning Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cliffe, A.; Doupe, D.P.; Sung, H.; Lim, I.K.; Ong, K.H.; Cheng, L.; Yu, W. Quantitative 3D analysis of complex single border cell behaviors in coordinated collective cell migration. Nat. Commun. 2017, 8, 14905. [Google Scholar] [CrossRef] [Green Version]
- Aman, A.; Piotrowski, T. Cell migration during morphogenesis. Dev. Biol. 2010, 341, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Woods, M.L.; Carmona-Fontaine, C.; Barnes, C.P.; Couzin, I.D.; Mayor, R.; Page, K.M. Directional collective cell migration emerges as a property of cell interactions. PLoS ONE 2014, 9, e104969. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, P.; Rupprecht, J.F.; Wieser, S.; Ruprecht, V.; Benichou, O.; Carpi, N.; Coppey, M.; De Beco, S.; Gov, N.; Heisenberg, C.P.; et al. Actin flows mediate a universal coupling between cell speed and cell persistence. Cell 2015, 161, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Franco, S.J.; Muller, U. Extracellular matrix functions during neuronal migration and lamination in the mammalian central nervous system. Dev. Neurobiol. 2011, 71, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, F.J.; Valiente, M.; Lopez Bendito, G.; Szabo, G.; Moya, F.; Valdeolmillos, M.; Marin, O. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development 2009, 136, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.E.; Jiang, H.; Rao, Y. Regulated formation and selection of neuronal processes underlie directional guidance of neuronal migration. Mol. Cell. Neurosci. 2005, 30, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Wagstyl, K.; Ronan, L.; Whitaker, K.J.; Goodyer, I.M.; Roberts, N.; Crow, T.J.; Fletcher, P.C. Multiple markers of cortical morphology reveal evidence of supragranular thinning in schizophrenia. Transl. Psychiatry 2016, 6, e780. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Hyman, B.T.; Van Hoesen, G.W.; Damasio, A.R. Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch. Gen. Psychiatry 1991, 48, 625–632. [Google Scholar] [CrossRef]
- Yang, Y.; Fung, S.J.; Rothwell, A.; Tianmei, S.; Weickert, C.S. Increased interstitial white matter neuron density in the dorsolateral prefrontal cortex of people with schizophrenia. Biol. Psychiatry 2011, 69, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Roussos, P.; Katsel, P.; Davis, K.L.; Siever, L.J.; Haroutunian, V. A system-level transcriptomic analysis of schizophrenia using postmortem brain tissue samples. Arch. Gen. Psychiatry 2012, 69, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst. Biol. 2016, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Narla, S.T.; Lee, Y.W.; Benson, C.A.; Sarder, P.; Brennand, K.J.; Stachowiak, E.K.; Stachowiak, M.K. Common developmental genome deprogramming in schizophrenia—Role of Integrative Nuclear FGFR1 Signaling (INFS). Schizophr. Res. 2017, 185, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, E.K.; Benson, C.A.; Narla, S.T.; Dimitri, A.; Chuye, L.E.B.; Dhiman, S.; Harikrishnan, K.; Elahi, S.; Freedman, D.; Brennand, K.J.; et al. Cerebral organoids reveal early cortical maldevelopment in schizophrenia-computational anatomy and genomics, role of FGFR1. Transl. Psychiatry 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, W.; Feron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent stem cells from adult olfactory mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef]
- Fan, Y.; Abrahamsen, G.; Mills, R.; Calderon, C.C.; Tee, J.Y.; Leyton, L.; Murrell, W.; Cooper-White, J.; McGrath, J.J.; Mackay-Sim, A. Focal adhesion dynamics are altered in schizophrenia. Biol. Psychiatry 2013, 74, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Tee, J.Y.; Sutharsan, R.; Fan, Y.; Mackay-Sim, A. Cell migration in schizophrenia: Patient-derived cells do not regulate motility in response to extracellular matrix. Mol. Cell. Neurosci. 2017, 80, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.L.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Model. Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, R.; Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nat. Protoc. 2014, 9, 1931–1943. [Google Scholar] [CrossRef]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Gorelik, R.; Gautreau, A. The Arp2/3 inhibitory protein arpin induces cell turning by pausing cell migration. Cytoskeleton 2015, 72, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- English, J.A.; Fan, Y.; Focking, M.; Lopez, L.M.; Hryniewiecka, M.; Wynne, K.; Dicker, P.; Matigian, N.; Cagney, G.; Mackay-Sim, A.; et al. Reduced protein synthesis in schizophrenia patient-derived olfactory cells. Transl. Psychiatry 2015, 5, e663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunty, J.M.; Gabarra-Niecko, V.; King, M.L.; Ceccarelli, D.F.; Eck, M.J.; Schaller, M.D. FERM domain interaction promotes FAK signaling. Mol. Cell. Biol. 2004, 24, 5353–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, I.; Gorelik, R.; Sousa-Blin, C.; Derivery, E.; Guerin, C.; Linkner, J.; Nemethova, M.; Dumortier, J.G.; Giger, F.A.; Chipysheva, T.A.; et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature 2013, 503, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Watanabe, N. A new link between the retrograde actin flow and focal adhesions. J. Biochem. 2014, 156, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Datta, D.; Arion, D.; Roman, K.M.; Volk, D.W.; Lewis, D.A. Altered Expression of ARP2/3 Complex Signaling Pathway Genes in Prefrontal Layer 3 Pyramidal Cells in Schizophrenia. Am. J. Psychiatry 2017, 174, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Tivodar, S.; Kalemaki, K.; Kounoupa, Z.; Vidaki, M.; Theodorakis, K.; Denaxa, M.; Kessaris, N.; de Curtis, I.; Pachnis, V.; Karagogeos, D. Rac-GTPases Regulate Microtubule Stability and Axon Growth of Cortical GABAergic Interneurons. Cereb. Cortex 2015, 25, 2370–2382. [Google Scholar] [CrossRef] [Green Version]
- Gardel, M.L.; Sabass, B.; Ji, L.; Danuser, G.; Schwarz, U.S.; Waterman, C.M. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J. Cell Biol. 2008, 183, 999–1005. [Google Scholar] [CrossRef]
- Prentice-Mott, H.V.; Meroz, Y.; Carlson, A.; Levine, M.A.; Davidson, M.W.; Irimia, D.; Charras, G.T.; Mahadevan, L.; Shah, J.V. Directional memory arises from long-lived cytoskeletal asymmetries in polarized chemotactic cells. Proc. Natl. Acad. Sci. USA 2016, 113, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyer, S.R.; Singh, A.; Dumbauld, D.W.; Calderwood, D.A.; Craig, S.W.; Delamarche, E.; Garcia, A.J. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J. Cell Sci. 2012, 125, 5110–5123. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Lee, O.; Win, Z.; Edwards, R.M.; Alford, P.W.; Kim, D.H.; Provenzano, P.P. Anisotropic forces from spatially constrained focal adhesions mediate contact guidance directed cell migration. Nat. Commun. 2017, 8, 14923. [Google Scholar] [CrossRef]
- Shafaq-Zadah, M.; Gomes-Santos, C.S.; Bardin, S.; Maiuri, P.; Maurin, M.; Iranzo, J.; Gautreau, A.; Lamaze, C.; Caswell, P.; Goud, B.; et al. Persistent cell migration and adhesion rely on retrograde transport of beta(1) integrin. Nat. Cell Biol. 2016, 18, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Dembo, M.; Hanks, S.K.; Wang, Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA 2001, 98, 11295–11300. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Estrada, J.; Benitez-King, G.; Berlanga, C.; Meza, I. Altered subcellular distribution of the 75-kDa DISC1 isoform, cAMP accumulation, and decreased neuronal migration in schizophrenia and bipolar disorder: Implications for neurodevelopment. CNS Neurosci. Ther. 2015, 21, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Akamatsu, W.; Okada, Y.; Ohnishi, T.; Balan, S.; Hisano, Y.; Iwayama, Y.; Toyota, T.; Matsumoto, T.; Itasaka, N.; et al. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl. Psychiatry 2016, 6, e934. [Google Scholar] [CrossRef] [Green Version]
- Tee, J.Y.; Sutharsan, R.; Fan, Y.; Mackay-Sim, A. Schizophrenia patient-derived olfactory neurosphere-derived cells do not respond to extracellular reelin. NPJ Schizophr. 2016, 2, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luccardini, C.; Hennekinne, L.; Viou, L.; Yanagida, M.; Murakami, F.; Kessaris, N.; Ma, X.; Adelstein, R.S.; Mege, R.M.; Metin, C. N-cadherin sustains motility and polarity of future cortical interneurons during tangential migration. J. Neurosci. 2013, 33, 18149–18160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, H.A.; Ito, S.; Corfas, G. Extracellular signals that regulate the tangential migration of olfactory bulb neuronal precursors: Inducers, inhibitors, and repellents. J. Neurosci. 2001, 21, 7654–7663. [Google Scholar] [CrossRef]
- Polleux, F.; Whitford, K.L.; Dijkhuizen, P.A.; Vitalis, T.; Ghosh, A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 2002, 129, 3147–3160. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.; Zhou, L.; Wu, J.Y.; Rao, Y. Cellular and molecular guidance of GABAergic neuronal migration from an extracortical origin to the neocortex. Neuron 1999, 23, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Molina, N.; Kim, Y.; Hockberger, P.; Szele, F.G. Rostral migratory stream neuroblasts turn and change directions in stereotypic patterns. Cell Adhes. Migr. 2011, 5, 83–95. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tee, J.Y.; Mackay-Sim, A. Directional Persistence of Cell Migration in Schizophrenia Patient-Derived Olfactory Cells. Int. J. Mol. Sci. 2021, 22, 9177. https://doi.org/10.3390/ijms22179177
Tee JY, Mackay-Sim A. Directional Persistence of Cell Migration in Schizophrenia Patient-Derived Olfactory Cells. International Journal of Molecular Sciences. 2021; 22(17):9177. https://doi.org/10.3390/ijms22179177
Chicago/Turabian StyleTee, Jing Yang, and Alan Mackay-Sim. 2021. "Directional Persistence of Cell Migration in Schizophrenia Patient-Derived Olfactory Cells" International Journal of Molecular Sciences 22, no. 17: 9177. https://doi.org/10.3390/ijms22179177