
Understanding the Potential of Genome Editing in Parkinson’s Disease

 , , and
, , and
Abstract
:1. Introduction
2. The Genetic Basis of PD
2.1. Genes at the Basis of PD
2.2. Autophagic Pathway
2.3. Mitochondrial Pathway
2.4. Lysosomal Pathway
3. Advances in CRISPR/Cas Systems and Delivery Strategies
3.1. CRISPR/Cas Component Design for Precise Gene Editing
3.2. Delivery Strategies for CRISPR Applications
3.3. Delivery Carriers to Improve the Performance of CRISPR/Cas9-Based Therapies
3.4. Perspectives on Delivery Carriers and Potential for Future Research
4. Potential of Genetic Therapies as Treatment Alternatives for Parkinson’s Disease
4.1. Therapeutic Approaches Based on the Stimulation of Dopamine Production
4.2. Therapeutic Approaches Targeting Neurotrophic Genes
4.3. Other Gene Therapy Approaches
4.3.1. Approaches Targeting Mitochondrial Genes
4.3.2. Approaches Focused on α-Synuclein
4.4. Stem Cell-Derived Therapies and Stem Cell In Vitro Models
4.5. Future of Gene Therapy for Parkinson’s Disease
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease—Repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Correia Guedes, L.; Mestre, T.; Outeiro, T.F.; Ferreira, J.J. Are genetic and idiopathic forms of Parkinson’s disease the same disease? J. Neurochem. 2020, 152, 515–522. [Google Scholar] [CrossRef]
- Valdés, P.; Schneider, B.L. Gene Therapy: A Promising Approach for Neuroprotection in Parkinson’s Disease? Front. Neuroanat. 2016, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, D.; Mäkelä, J.; Di Liberto, V.; Mudò, G.; Belluardo, N.; Eriksson, O.; Saarma, M. Current disease modifying approaches to treat Parkinson’s disease. Cell. Mol. Life Sci. 2016, 73, 1365–1379. [Google Scholar] [CrossRef]
- Axelsen, T.M.; Woldbye, D.P.D. Gene Therapy for Parkinson’s Disease, An Update. J. Park. Dis. 2018, 8, 195–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberio, T.; Lopiano, L.; Fasano, M. Cellular models to investigate biochemical pathways in Parkinson’s disease. FEBS J. 2012, 279, 1146–1155. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Jamebozorgi, K.; Taghizadeh, E.; Rostami, D.; Pormasoumi, H.; Barreto, G.E.; Hayat, S.M.G.; Sahebkar, A. Cellular and Molecular Aspects of Parkinson Treatment: Future Therapeutic Perspectives. Mol. Neurobiol. 2019, 56, 4799–4811. [Google Scholar] [CrossRef]
- Foster, H.D.; Hoffer, A. The two faces of L-DOPA: Benefits and adverse side effects in the treatment of Encephalitis lethargica, Parkinson’s disease, multiple sclerosis and amyotrophic lateral sclerosis. Med. Hypotheses 2004, 62, 177–181. [Google Scholar] [CrossRef]
- Bjorklund, A.; Kordower, J.H. Cell Therapy for Parkinson’s Disease: What Next? Mov. Disord. 2013, 28, 110–115. [Google Scholar] [CrossRef]
- O’Connor, D.M.; Boulis, N.M. Gene therapy for neurodegenerative diseases. Trends Mol. Med. 2015, 21, 504–512. [Google Scholar] [CrossRef]
- Chen, W.; Hu, Y.; Ju, D. Gene therapy for neurodegenerative disorders: Advances, insights and prospects. Acta Pharm Sin. B 2020, 10, 1347–1359. [Google Scholar] [CrossRef]
- Fan, H.-C.; Chi, C.-S.; Lee, Y.-J.; Tsai, J.-D.; Lin, S.-Z.; Harn, H.-J. The Role of Gene Editing in Neurodegenerative Diseases. Cell Transplant. 2018, 27, 364–378. [Google Scholar] [CrossRef]
- Baker, M. Gene-editing nucleases. Nat. Methods 2012, 9, 23–26. [Google Scholar] [CrossRef]
- Zych, A.O.; Bajor, M.; Zagozdzon, R. Application of Genome Editing Techniques in Immunology. Arch. Immunol. Ther. Exp. 2018, 66, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 125809. [Google Scholar] [CrossRef]
- Persaud, A.; Desine, S.; Blizinsky, K.; Bonham, V.L. A CRISPR focus on attitudes and beliefs toward somatic genome editing from stakeholders within the sickle cell disease community. Genet. Med. 2019, 21, 1726–1734. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.; Yi, L.; Hou, Z.; Chen, J.; Kou, X.; Zhao, Y.; Wang, H.; Sun, X.-F.; Jiang, C.; et al. Naïve Induced Pluripotent Stem Cells Generated From β-Thalassemia Fibroblasts Allow Efficient Gene Correction With CRISPR/Cas9. Stem Cells Transl. Med. 2016, 5, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.; Lin, T.-Y.; Chang, Y.-C.; Lai, H.I.-A.M.; Lin, S.-C.; Ma, C.; Yarmishyn, A.A.; Lin, S.-C.; Chang, K.-J.; Chou, Y.-B.; et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int. J. Mol. Sci. 2021, 22, 4534. [Google Scholar] [CrossRef]
- Wu, S.-S.; Li, Q.-C.; Yin, C.-Q.; Xue, W.; Song, C.-Q. Advances in CRISPR/Cas-based Gene Therapy in Human Genetic Diseases. Theranostics 2020, 10, 4374–4382. [Google Scholar] [CrossRef]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; Oliveira, S.D.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, A.; Melosh, N. Nanostructured Materials for Intracellular Cargo Delivery. Acc. Chem. Res. 2019, 52, 2462–2471. [Google Scholar] [CrossRef] [PubMed]
- Twelves, D.; Perkins, K.S.M.; Counsell, C. Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. 2003, 18, 19–31. [Google Scholar] [CrossRef]
- Niethammer, M.; Tang, C.C.; Vo, A.; Nguyen, N.; Spetsieris, P.; Dhawan, V.; Ma, Y.; Small, M.; Feigin, A.; During, M.J.; et al. Gene therapy reduces Parkinson’s disease symptoms by reorganizing functional brain connectivity. Sci. Transl. Med. 2018, 10, eaau0713. [Google Scholar] [CrossRef] [Green Version]
- Maraganore, D.M.; Andrade, M.D.; Lesnick, T.C.; Strain, K.J.; Farrer, M.J.; Rocca, W.A.; Pant, P.V.K.; Frazer, K.A.; Cox, D.R.; Ballinger, D.C. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005, 77, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Wider, C.; Lincoln, S.J.; Heckman, M.G.; Diehl, N.N.; Stone, J.T.; Haugarvoll, K.; Aasly, J.O.; Gibson, J.M.; Lynch, T.; Rajput, A.; et al. Phactr2 and Parkinson’s disease. Neurosci. Lett. 2009, 453, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E. Prevention of Alzheimer’s Disease: Lessons Learned and Applied. J. Am. Geriatr. Soc. 2017, 65, 2128–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Casacuberta, I.; Juárez-Flores, D.L.; Morén, C.; Garrabou, G. Bioenergetics and Autophagic Imbalance in Patients-Derived Cell Models of Parkinson Disease Supports Systemic Dysfunction in Neurodegeneration. Front. Neurosci. 2019, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Daida, K.; Nishioka, K.; Li, Y.; Yoshino, H.; Kikuchi, A.; Hasegawa, T.; Funayama, M.; Hattori, N. Mutation analysis of LRP10 in Japanese patients with familial Parkinson’s disease, progressive supranuclear palsy, and frontotemporal dementia. Neurobiol. Aging 2019, 84, 235.e11–235.e16. [Google Scholar] [CrossRef]
- Barbeau, A.; Roy, M. Familial Subsets in Idiopathic Parkinson’s Disease. Can. J. Neurol. Sci. 1984, 11, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, G.; Vitale, C.; Trojano, L.; Errico, D.; Amboni, M.; Barbarulo, M.; Grossi, D.; Barone, P. Neurophysological correlates of theory of mind in patients with early Parkinson’s Disease. Mov. Disord. 2011, 27, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Reetz, K.; Gaser, C.; Klein, C.; Hagenah, J.; Büchel, C.; Gottschalk, S.; Pramstaller, P.P.; Siebner, H.R.; Binkofski, F. Structural findings in the basal ganglia in genetically determined and idiopathic Parkinson’s disease. Mov. Disord. 2009, 24, 99–103. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Kim, Y.A.; Yoo, Y.H. Loss of Pigment Epithelial Cells Is Prevented by Autophagy. In Autophagy: Cancer Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 105–117. [Google Scholar]
- Hou, X.; Watzlawik, J.; Fiesel, F.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Stricher, F.; Macri, C.; Ruff, M.; Muller, S. HSPA8/HSC70 chaperone protein. Autophagy 2013, 9, 1937–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of Protein Quality Control in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The Role of Autophagy in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Z.H.; Ip, N.Y. The emerging role of autophagy in Parkinson’s disease. Mol. Brain 2009, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Engelender, S. Ubiquitination of α-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef] [Green Version]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre-Abarrategui, J.; Wade-Martins, R. Parkinson disease, LRRK2 and the endocytic-autophagic pathway. Autophagy 2009, 5, 1208–1210. [Google Scholar] [CrossRef] [Green Version]
- Volta, M.; Milnerwood, A.J.; Farrer, M.J. Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson’s disease. Lancet Neurol. 2015, 14, 1054–1064. [Google Scholar] [CrossRef]
- Maraganore, D.M.; Lesnick, T.G.; Elbaz, A.; Chartier-Harlin, M.-C.; Gasser, T.; Krüger, R.; Hattori, N.; Mellick, G.D.; Quattrone, A.; Satoh, J.-I.; et al. UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 2004, 55, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Brug, M.v.d.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Jinn, S.; Blauwendraat, C.; Toolan, D.; Gretzula, C.A.; Drolet, R.E.; Smith, S.; Nalls, M.A.; Marcus, J.; Singleton, A.B.; Stone, D.J. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet. 2019, 28, 3244–3254. [Google Scholar] [CrossRef] [Green Version]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in Lower Motor Neuron Predominant Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef]
- Rusten, T.E.; Filimonenko, M.; Rodahl, L.M.; Stenmark, H.; Simonsen, A. ESCRTing autophagic clearance of aggregating proteins. Autophagy 2008, 4, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Tanikawa, S.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in α-synucleinopathy. Neurosci. Lett. 2012, 527, 16–21. [Google Scholar] [CrossRef]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef] [Green Version]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Yang, Y.-P.; Mao, C.-J.; Zhang, X.-Q.; Wang, C.-T.; Yang, J.; Lv, D.-J.; Wang, F.; Hu, L.-F.; Liu, C.-F. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol. Aging 2017, 60, 104–115. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, K.; Vinet, J.; den Dunnen, W.F.A.; Brunt, E.R.; Meister, M.; Boncoraglio, A.; Zijlstra, M.P.; Boddeke, H.W.G.M.; Rüb, U.; Kampinga, H.H.; et al. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 2012, 38, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.R.; Le Scouarnec, S.; Schott, J.-J.; Mohler, P.J. Exon organization and novel alternative splicing of the human ANK2 gene: Implications for cardiac function and human cardiac disease. J. Mol. Cell. Cardiol. 2008, 45, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Weiner, A.T.; Seebold, D.Y.; Michael, N.L.; Guignet, M.; Feng, C.; Follick, B.; Yusko, B.A.; Wasilko, N.P.; Torres-Gutierrez, P.; Rolls, M.M. Identification of Proteins Required for Precise Positioning of Apc2 in Dendrites. G3 Genes Genomes Genet. 2018, 8, 1841–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auburger, G.; Gispert, S.; Torres-Odio, S.; Jendrach, M.; Brehm, N.; Canet-Pons, J.; Key, J.; Sen, N.-E. SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits as Hub between Autophagy and Synapse Changes. Int. J. Mol. Sci. 2019, 20, 3284. [Google Scholar] [CrossRef] [Green Version]
- Hale, C.M.; Cheng, Q.; Ortuno, D.; Huang, M.; Nojima, D.; Kassner, P.D.; Wang, S.; Ollmann, M.M.; Carlisle, H.J. Identification of modulators of autophagic flux in an image-based high content siRNA screen. Autophagy 2016, 12, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Füllgrabe, J.; Lynch-Day, M.A.; Heldring, N.; Li, W.; Struijk, R.B.; Ma, Q.; Hermanson, O.; Rosenfeld, M.G.; Klionsky, D.J.; Joseph, B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013, 500, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.M.; Melandri, D.; Annuario, E.; Monaghan, A.E.; Welsh, N.J.; D’Sa, K.; Guelfi, S.; Zhang, D.; Pittman, A.; Trabzuni, D.; et al. Regulation of mitophagy by the NSL complex underlies genetic risk for Parkinson’s disease at Chr16q11.2 and on the MAPT H1 allele. boiRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy 2013, 9, 1621–1623. [Google Scholar] [CrossRef] [Green Version]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Tapia, C.; Pérez, M.J. Possible role of mitochondrial permeability transition pore in the pathogenesis of Huntington disease. Biochem. Biophys. Res. Commun. 2017, 483, 1078–1083. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Holden, S.; Maksoud, R.; Eaton-Fitch, N.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. A systematic review of mitochondrial abnormalities in myalgic encephalomyelitis/chronic fatigue syndrome/systemic exertion intolerance disease. J. Transl. Med. 2020, 18, 1–16. [Google Scholar]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The multi-faceted role of mitochondria in the pathology of Parkinson’s disease. J. Neurochem. 2021, 156, 715–752. [Google Scholar] [CrossRef]
- Toulorge, D.; Schapira, A.H.V.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016, 139, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Genetic Analysis of Pathways to Parkinson Disease. Neuron 2010, 68, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Walker, D.; Goldstein, J.; de Laat, R.; Banducci, K. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy Body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.; Duda, J.; Murray, I.; Chen, Q.; Souza, J.M.; Hurting, H.I.; Ischiropolous, H.; Trajanowski, J.Q.; Lee, V.M.-Y. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome. J. Biol. Chem. 2003, 278, 44404–44411. [Google Scholar] [CrossRef] [Green Version]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Nakajima, Y.-I.; Kuranaga, E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017, 24, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.-P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3. A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, A.; Troadec, J.-D.; Hunot, S.; Kikly, K.; Faucheux, B.A.; Mouatt-Prigent, A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Caspase-8 Is an Effector in Apoptotic Death of Dopaminergic Neurons in Parkinson’s Disease, But Pathway Inhibition Results in Neuronal Necrosis. J. Neurosci. 2001, 21, 2247–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Nguyen, L.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef] [Green Version]
- Brahmachari, S.; Lee, S.; Kim, S.; Yuan, C.; Karuppagounder, S.S.; Ge, P.; Shi, R.; Kim, E.J.; Liu, A.; Kim, D.; et al. Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson’s disease. Brain 2019, 142, 2380–2401. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef]
- Sunico, C.R.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.; Newmeyer, T.; Masliah, E.; Nakanishi, N.; Lipton, S.A. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 2013, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin Localizes to the Lewy Bodies of Parkinson Disease and Dementia with Lewy Bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.S.; von Coelln, R.; Sriram, S.R.; Kim, S.W.; Chung, K.K.K.; Pletnikova, O.; Troncoso, J.; Johnson, B.; Saffary, R.; Goh, E.L.; et al. Accumulation of the Authentic Parkin Substrate Aminoacyl-tRNA Synthetase Cofactor, p38/JTV-1, Leads to Catecholaminergic Cell Death. J. Neurosci. 2005, 25, 7968–7978. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Lee, K.; Panda, S.; Gonzales-Rojas, R.; Chong, A.; Bugay, V.; Park, H.M.; Brenner, R.; Murthy, N.; Lee, H.Y. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat. Biomed. Eng. 2018, 2, 497–507. [Google Scholar] [CrossRef]
- Klein, A.D.; Mazzulli, J.R. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, L.; Lu, Z.-J.; Sun, X.-Y.; Li, J.-Y.; Peng, R. Association of three candidate genetic variants in RAB7L1/NUCKS1 MCCC1 and STK39 with sporadic Parkinson’s disease in Han Chinese. J. Neural Transm. 2016, 123, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Li, Y.; Niu, M.; Li, G.; Luo, N.; Zhou, L.; Kang, W.; Liu, J. SNPs in SNCA, MCCC1, DLG2, GBF1 and MBNL2 are associated with Parkinson’s disease in southern Chinese population. J. Cell. Mol. Med. 2020, 24, 8744–8752. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M.; Chen, Y.-C.; Fung, H.-C.; Wu, Y.-R. Polymorphisms of ACMSD—TMEM163, MCCC1, and BCKDK—STX1B Are Not Associated with Parkinson’s Disease in Taiwan. Parkinsons. Dis. 2019, 2019, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, B.; Yu, R.; Li, K.; Liu, Z.; Xu, Q.; Sun, Q.; Yan, X.; Guo, J. Association analysis of STK39 MCCC1/LAMP3 and sporadic PD in the Chinese Han population. Neurosci. Lett. 2014, 566, 206–209. [Google Scholar] [CrossRef]
- Brás, J.; Guerreiro, R.; Hardy, J. SnapShot: Genetics of Parkinson’s Disease. Cell 2015, 160, 570.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial Iron Metabolism and Its Role in Neurodegeneration. J. Alzheimers Dis. 2010, 20, S551–S568. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.G.; Raven, E.L.; Chernova, T. The regulatory role of heme in neurons. Metallomics 2011, 3, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiao, Y.; Guo, W.; Zhou, M.; Huang, S.; Mo, M.; Li, Z.; Li, G.; Liu, H.; Peng, G.; et al. Relationship between variants of 17 newly loci and Parkinson’s disease in a Chinese population. Neurobiol. Aging 2019, 73, 230.e1–230.e4. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sanchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef] [Green Version]
- Ebadi, M.; Govitrapong, P.; Sharma, S.; Muralikrishnan, D.; Shavali, S.; Pellett, L.; Schafer, R.; Albano, C.; Eken, J. Ubiquinone (coenzyme q10) and mitochondria in oxidative stress of Parkinson’s disease. Neurosignals 2001, 10, 224–253. [Google Scholar] [CrossRef]
- Krohn, L.; Öztürk, T.N.; Vanderperre, B.; Ouled Amar Bencheikh, B.; Ruskey, J.A.; Laurent, S.B.; Spiegelman, D.; Postuma, R.B.; Arnulf, I.; Hu, M.T.M.; et al. Genetic, Structural, and Functional Evidence Link TMEM175 to Synucleinopathies. Ann. Neurol. 2020, 87, 139–153. [Google Scholar] [CrossRef]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef]
- Fernández, E.; García-Moreno, J.-M.; Martín de Pablos, A.; Chacón, J. May the Evaluation of Nitrosative Stress Through Selective Increase of 3-Nitrotyrosine Proteins Other Than Nitroalbumin and Dominant Tyrosine-125/136 Nitrosylation of Serum α-Synuclein Serve for Diagnosis of Sporadic Parkinson’s Disease? Antioxid. Redox Signal. 2013, 19, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef]
- Pitcairn, C.; Wani, W.Y.; Mazzulli, J.R. Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. 2019, 122, 72–82. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Schapira, A.H.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, B.S.; Magalhaes, J.; Beavan, M.S.; McNeill, A.; Gegg, M.E.; Cleeter, M.W.J.; Bloor-Young, D.; Churchill, G.C.; Duchen, M.R.; Schapira, A.H.; et al. Endoplasmic reticulum and lysosomal Ca2+ stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium 2016, 59, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.-K.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Kanneganti, T.-D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Levy, O.A.; Waters, C.H.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Contu, R.; Condorelli, G. ATP6V0A1 Polymorphism and MicroRNA-637. Circ. Cardiovasc. Genet. 2011, 4, 337–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Nüsslein-Volhard, C. Live Imaging of Neuronal Degradation by Microglia Reveals a Role for v0-ATPase a1 in Phagosomal Fusion In Vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Larson, M.; Gilbert, L.; Doudna, J.; Weissman, J.; Arkin, A.; Lim, W. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanenbaum, M.; Gilbert, L.; Qi, L.; Weissman, J.; Vale, R. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Kang, J.G.; Park, J.S.; Ko, J.-H.; Kim, Y.-S. Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci. Rep. 2019, 9, 11960. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.; Pi, M.; Cox, J.V.; Nishimoto, S.K.; Quarles, L.D. CRISPR/Cas9 targeting of GPRC6A suppresses prostate cancer tumorigenesis in a human xenograft model. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef]
- Tycko, J.; Myer, V.; Hsu, P. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol. Cell 2016, 63, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Hanna, R.E.; Doench, J.G. Design and analysis of CRISPR–Cas experiments. Nat. Biotechnol. 2020, 38, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, S.E.; Hu, Y.; Ewen-Campen, B.; Housden, B.E.; Viswanatha, R.; Perrimon, N. CRISPR guide RNA design for research applications. FEBS J. 2016, 283, 3232–3238. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xing, H.L.; Wang, Z.P.; Zhang, H.Y.; Yang, F.; Wang, X.C.; Chen, Q.J. Potential high-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention. Plant Mol. Biol. 2018, 96, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Joung, J.K. 731. High-Fidelity CRISPR-Cas9 Nucleases with No Detectable Genome-Wide Off-Target Effects. Mol. Ther. 2016, 24, S288. [Google Scholar] [CrossRef]
- Mateus, A.; Treyer, A.; Wegler, C.; Karlgren, M.; Matsson, P.; Artursson, P. Intracellular drug bioavailability: A new predictor of system dependent drug disposition. Sci. Rep. 2017, 7, 43047. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, S.; Sharda, N.; Chakraborti, A.K. New Findings on Degradation of Famotidine under Basic Conditions: Identification of a Hitherto Unknown Degradation Product and the Condition for Obtaining the Propionamide Intermediate in Pure Form. J. Pharm. Sci. 2002, 91, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Hogg, P.J. Disulfide bonds as switches for protein function. Trends Biochem. Sci. 2003, 28, 210–214. [Google Scholar] [CrossRef]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef]
- Perez, J.; Rueda, J.; Cuellar, M.; Suarez-Arnedo, A.; Cruz, J.C.; Muñoz-Camargo, C. Cell-Penetrating And Antibacterial BUF-II Nanobioconjugates: Enhanced Potency Via Immobilization On Polyetheramine-Modified Magnetite Nanoparticles. Int. J. Nanomed. 2019, 14, 8483–8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Moyo, B.; Lee, C.M.; Leong, K.; Bao, G. Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 2019, 4, 726–737. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, M.A.; Corn, J.E.; Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods 2017, 121–122, 9–15. [Google Scholar] [CrossRef]
- Zhang, Z.; Wan, T.; Chen, Y.; Chen, Y.; Sun, H.; Cao, T.; Songyang, Z.; Tang, G.; Wu, C.; Ping, Y.; et al. Cationic Polymer-Mediated CRISPR/Cas9 Plasmid Delivery for Genome Editing. Macromol. Rapid Commun. 2019, 40, 1800068. [Google Scholar] [CrossRef]
- Hamilton, T.A.; Pellegrino, G.M.; Therrien, J.A.; Ham, D.T.; Bartlett, P.C.; Karas, B.J.; Gloor, G.B.; Edgell, D.R. Efficient inter-species conjugative transfer of a CRISPR nuclease for targeted bacterial killing. Nat. Commun. 2019, 10, 4544. [Google Scholar] [CrossRef]
- Wang, H.-X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.-W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Jeon, S.; Kim, S.; Chang, Y.K.; Kim, Y.-C. Development of a pVEC peptide-based ribonucleoprotein (RNP) delivery system for genome editing using CRISPR/Cas9 in Chlamydomonas reinhardtii. Sci. Rep. 2020, 10, 22158. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimd-Burgk, J.L.; Gao, L.; Li, D.; Gardner, Z.; Strecker, J.; Lash, B.; Zhang, F. Highly parallel Profiling of Cas9 Variant Specificity. Mol. Cell 2020, 78, 794–800. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.-R.; Kim, H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020, 38, 1328–1336. [Google Scholar] [CrossRef]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Cereseto, A. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Niu, D.; Wu, C.; Xu, F.-J.; Church, G.; Ping, Y. Material solutions for delivery of CRISPR/Cas-based genome editing tools: Current status and future outlook. Mater. Today 2019, 26, 40–66. [Google Scholar] [CrossRef]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef]
- Ablain, J.; Durand, E.; Yang, S.; Zhou, Y.; Zon, L. A CRISPR/Cas9 Vector System for Tissue-Specific Gene Disruption in Zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhou, X.; Chenga, M.; Xing, D. Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing. Nanoscale 2018, 10, 1063–1071. [Google Scholar] [CrossRef]
- Wang, H.X.; Song, Z.; Lao, Y.H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D.; et al. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef] [Green Version]
- Rueda-Gensini, L.; Cifuentes, J.; Castellanos, M.C.; Puentes, P.R.; Serna, J.A.; Muñoz-Camargo, C.; Cruz, J.C. Tailoring iron oxide nanoparticles for efficient cellular internalization and endosomal escape. Nanomaterials 2020, 10, 1816. [Google Scholar] [CrossRef] [PubMed]
- Tiana, W.; Ma, Y. Insights into the endosomal escape mechanism via investigation of dendrimer–membrane interactions. Soft Matter 2012, 8, 6378–6384. [Google Scholar] [CrossRef]
- Mout, R.; Ray, M.; Lee, Y.W.; Scaletti, F.; Rotello, V.M. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjug Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Liu, Z.; Jo, M.C.; Zhang, K.; Li, Y.; Zeng, Z.; Li, N.; Zu, Y.; Qin, L. CRISPR-Cas9 delivery to hard-to-transfect cells via membrane deformation. Sci. Adv. 2015, 1, e1500454. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Imani, R.; Emami, S.H.; Faghihi, S. Synthesis and characterization of an octaarginine functionalized graphene oxide nano-carrier for gene delivery applications. Phys. Chem. Chem. Phys. 2015, 17, 6328–6339. [Google Scholar] [CrossRef]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of Staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Barbosa, N.; Suárez-Arnedo, A.; Cifuentes, J.; Gonzalez Barrios, A.F.; Silvera Batista, C.A.; Osma, J.F.; Muñoz-Camargo, C.; Cruz, J.C. Magnetite–OmpA Nanobioconjugates as Cell-Penetrating Vehicles with Endosomal Escape Abilities. ACS Biomater. Sci. Eng. 2020, 6, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Song, L.; Liu, X.; Yang, X.; Li, X.; He, T.; Wei, Y. Artificial Virus Delivers CRISPR-Cas9 System for Genome Editing of Cells in Mice. ACS Nano 2016, 11, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Z.Y.; Wei, X.W.; Gao, G.P.; Wei, Y.Q. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum. Gene Ther. 2015, 26, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Schmidt, F.; Grimm, D. CRISPR genome engineering and viral gene delivery: A case of mutual attraction. Biotechnol. J. 2015, 10, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Lööv, C.; Zaborowski, M.P.; Takeda, S.; Kleinstiver, B.P.; Commins, C.; Kastanenka, K.; Mu, D.; Volak, A.; Giedraitis, V.; et al. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2018, 11, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef]
- Senís, E.; Fatouros, C.; Große, S.; Wiedtke, E.; Niopek, D.; Mueller, A.K.; Börner, K.; Grimm, D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014, 9, 1402–1412. [Google Scholar] [CrossRef]
- Choi, J.-H.; Yu, N.-K.; Baek, G.-C.; Bakes, J.; Seo, D.; Nam, H.; Baek, S.; Lim, C.-S.; Lee, Y.-S.; Kaang, B.-K. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol. Brain 2014, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Kaeppel, C.; Beattie, S.G.; Fronza, R.; van Logtenstein, R.; Salmon, F.; Schmidt, S.; Wolf, S.; Nowrouzi, A.; Glimm, H.; von Kalle, C.; et al. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 2013, 19, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Yang, L.; Slone, J.; Li, Z.; Lou, X.; Hu, Y.-C.; Queme, L.F.; Jankowski, M.P.; Huang, T. Systemic administration of AAV-Slc25a46 mitigates mitochondrial neuropathy in Slc25a46−/− mice. Hum. Mol. Genet. 2020, 29, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Cerutti, R.; Luna-Sanchez, M.; Zeviani, M.; Viscomi, C. A Single Intravenous Injection of AAV-PHP.B-hNDUFS4 Ameliorates the Phenotype of Ndufs4 Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Burning, H.; Srivastava, A. Capsid modification for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.J.; Turunen, H.T.; Vandenberghe, L.H.; Wolfe, J.H. Controlling AAV Tropism in the Nervous System with Natural and Engineered Capsids. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; pp. 133–149. [Google Scholar]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS Transduction of Adult Mice by Intravenously Delivered rAAVrh.8 and rAAVrh.10 and Nonhuman Primates by rAAVrh.10. Mol. Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; McCarty, D.M. Crossing the blood–brain-barrier with viral vectors. Curr. Opin. Virol. 2016, 21, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Park, S.J.; Lee, S.; Cho, C.S.; Cho, M.H. Aerosol gene delivery using viral vectors and cationic carriers for in vivo lung cancer therapy. Expert Opin. Drug Deliv. 2015, 12, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.R.; Ho, S.-M.; Schrode, N.; Brennand, K.J. Integration of CRISPR-engineering and hiPSC-based models of psychiatric genomics. Mol. Cell. Neurosci. 2020, 107, 103532. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Fricano-Kugler, C.J.; Getz, S.A.; Skelton, P.D.; Lee, J.; Rizzuto, C.P.; Geller, J.S.; Li, M.; Luikart, B.W. A Retroviral CRISPR-Cas9 System for Cellular Autism-Associated Phenotype Discovery in Developing Neurons. Sci. Rep. 2016, 6, 25611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Solati, N.; Ghasemi, A.; Estiar, M.A.; Hashemkhani, M.; Kiani, P.; Mohamed, E.; Saeidi, A.; Taheri, M.; Avci, P.; et al. Carbon nanotubes part II: A remarkable carrier for drug and gene delivery. Expert Opin. Drug Deliv. 2015, 12, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, S.; Moonen, D.; Lee, C.; Lee, A.Y.; Schaffer, D.V.; He, L. CRISPR-READI: Efficient Generation of Knockin Mice by CRISPR RNP Electroporation and AAV Donor Infection. Cell Rep. 2019, 27, 3780–3789. [Google Scholar] [CrossRef] [Green Version]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application. Adv. Pharm. Bull. 2015, 5, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. Nano-emulsions and Micro-emulsions: Clarifications of the Critical Differences. Pharm. Res. 2011, 28, 978–985. [Google Scholar] [CrossRef]
- Lao, Y.H.; Li, M.; Gao, M.A.; Shao, D.; Chi, C.W.; Huang, D.; Chakraborty, S.; Ho, T.C.; Jiang, W.; Wang, H.X.; et al. HPV Oncogene Manipulation Using Nonvirally Delivered CRISPR/Cas9 or Natronobacterium gregoryi Argonaute. Adv. Sci. 2018, 5, 1700540. [Google Scholar] [CrossRef]
- Röhrborn, D. DPP4 in diabetes. Front. Immunol. 2015, 6, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Sun, Z.; Jiang, H.; Vaidya, A.M.; Xin, R.; Ayat, N.R.; Schilb, A.L.; Qiao, P.L.; Han, Z.; Naderi, A.; et al. Synthesis and Evaluation of pH-Sensitive Multifunctional Lipids for Efficient Delivery of CRISPR/Cas9 in Gene Editing. Bioconjug Chem. 2019, 30, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhang, L.; Zheng, W.; Cong, L.; Guo, Z.; Xie, Y.; Wang, L.; Tang, R.; Feng, Q.; Hamada, Y.; et al. Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy. Angew. Chem. Int. Ed. 2018, 57, 1491–1496. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery—Liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Chikh, G.G.; Li, W.M.; Schutze-Redelmeier, M.-P.; Meunier, J.-C.; Bally, M.B. Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ion-chelating lipids. Biochim. Biophys. Acta Biomembr. 2002, 1567, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tian, X.; Zhang, J.; Zhao, X.; Chen, X.; Jiang, Y.; Wang, D.; Pan, W. In vitro and in vivo evaluation of folate receptor-targeting amphiphilic copolymer-modified liposomes loaded with docetaxel. Int. J. Nanomed. 2011, 6, 1167–1184. [Google Scholar] [CrossRef] [Green Version]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liao, J.; Shao, X.; Li, Q.; Lin, Y. The Effect of shape on Cellular Uptake of Gold Nanoparticles in the forms of Stars, Rods, and Triangles. Sci. Rep. 2017, 7, 3827. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR\Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Wei, T.; Cheng, Q.; Min, Y.-L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef]
- Podstawczyk, D.; Nizioł, M.; Szymczyk, P.; Wiśniewski, P.; Guiseppi-Elie, A. 3D printed stimuli-responsive magnetic nanoparticle embedded alginate-methylcellulose hydrogel actuators. Addit. Manuf. 2020, 34, 101275. [Google Scholar] [CrossRef]
- Zhuang, J.; Tan, J.; Wu, C.; Zhang, J.; Liu, T.; Fan, C.; Li, J.; Zhang, Y. Extracellular vesicles engineered with valency-controlled DNA nanostructures deliver CRISPR/Cas9 system for gene therapy. Nucleic Acids Res. 2020, 48, 8870–8882. [Google Scholar] [CrossRef]
- Xie, J.; Zhao, C.; Lin, Z.; Gu, P.; Zhang, Q. Nanostructured Conjugated Polymers for Energy-Related Applications beyond Solar Cells. Chem. Asian J. 2016, 11, 1489–1511. [Google Scholar] [CrossRef] [PubMed]
- Abdolahpour, S.; Toliyat, T.; Omidfar, K.; Modjtahedi, H.; Wong, A.J.; Rasaee, M.J.; Kashanian, S.; Paknejad, M. Targeted delivery of doxorubicin into tumor cells by nanostructured lipid carriers conjugated to anti-EGFRvIII monoclonal antibody. Artif. Cells Nanomed. Biotechnol. 2018, 46, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.; Munawar, A.; Khan, W.S.; Mujahid, A.; Ihsan, A.; Rehman, A.; Ahmed, I.; Bajwa, S.Z. Assessing manganese nanostructures based carbon nanotubes composite for the highly sensitive determination of vitamin C in pharmaceutical formulation. Biosens. Bioelectron. 2017, 89, 822–828. [Google Scholar] [CrossRef]
- Ramírez-Acosta, C.M.; Cifuentes, J.; Castellanos, M.C.; Moreno, R.J.; Muñoz-Camargo, C.; Cruz, J.C.; Reyes, L.H. PH-Responsive, Cell-Penetrating, Core/Shell Magnetite/Silver Nanoparticles for the Delivery of Plasmids: Preparation, Characterization, and Preliminary In Vitro Evaluation. Pharmaceutics 2020, 12, 561. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhao, Y. Technological breakthroughs in generating transgene-free and genetically stable CRISPR-edited plants. aBIOTECH 2020, 1, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Noureddine, A.; Maestas-Olguin, A.; Saada, E.A.; LaBauve, A.E.; Agola, J.O.; Baty, K.E.; Howard, T.; Sabo, J.K.; Espinoza, C.R.S.; Doudna, J.A.; et al. Engineering of monosized lipid-coated mesoporous silica nanoparticles for CRISPR delivery. Acta Biomater. 2020, 114, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Rejinold, N.S.; Piao, H.; Choy, J.-H. Inorganic–inorganic nanohybrids for drug delivery, imaging and photo-therapy: Recent developments and future scope. Chem. Sci. 2021. [Google Scholar] [CrossRef]
- Farcas, C.G.; Dehelean, C.; Pinzaru, I.A.; Mioc, M.; Socoliuc, V.; Moaca, E.-A.; Avram, S.; Ghiulai, R.; Coricovac, D.; Pavel, I.; et al. Thermosensitive Betulinic Acid-Loaded Magnetoliposomes: A Promising Antitumor Potential for Highly Aggressive Human Breast Adenocarcinoma Cells Under Hyperthermic Conditions. Int. J. Nanomed. 2020, 15, 8175–8200. [Google Scholar] [CrossRef]
- Shen, K.; Wang, X.; Zhang, Y.; Zhu, H.; Chen, Z.; Huang, C.; Mai, Y. Insights into the role of interface modification in performance enhancement of ZnTe:Cu contacted CdTe thin film solar cells. Sol. Energy 2020, 201, 55–62. [Google Scholar] [CrossRef]
- Paterlini, V.; Bettinelli, M.; Rizzi, R.; El Khouri, A.; Rossi, M.; Della Ventura, G.; Capitelli, F. Characterization and Luminescence of Eu3+- and Gd3+-Doped Hydroxyapatite Ca10(PO4)6(OH)2. Crystals 2020, 10, 806. [Google Scholar] [CrossRef]
- Mout, R.; Ray, M.; Yesilbag, T.G.; Lee, Y.W.; Tay, T.; Sasaki, K.; Rotello, V.M. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano 2017, 11, 2452–2458. [Google Scholar] [CrossRef] [Green Version]
- Fonte, P.; Reis, S.; Sarmento, B. Facts and evidences on the lyophilization of polymeric nanoparticles for drug delivery. J. Control. Release 2016, 225, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Masood, F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater. Sci. Eng. C 2016, 60, 569–578. [Google Scholar] [CrossRef]
- Chertok, B.; David, A.E.; Yang, V.C. Polyethyleneimine-modified iron oxide nanoparticles for brain tumor drug delivery using magnetic targeting and intra-carotid administration. Biomaterials 2010, 31, 6317–6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Moscoso, A.; Guilloteau, N.; Bienvenu, C.; Méndez-Ardoy, A.; Jiménez Blanco, J.L.; Benito, J.M.; Le Gourriérec, L.; Di Giorgio, C.; Vierling, P.; Defaye, J.; et al. Mannosyl-coated nanocomplexes from amphiphilic cyclodextrins and pDNA for site-specific gene delivery. Biomaterials 2011, 32, 7263–7273. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, J.P.; Sami, H.; Rajalekshmi, D.C.; Sivakumar, S.; Abraham, A. Immunocompatibility and Toxicity Studies of Poly-L-Lysine Nanocapsules in Sprague–Dawley Rats for Drug-Delivery Applications. Chem. Biol. Drug Des. 2014, 84, 292–299. [Google Scholar] [CrossRef]
- Fornaguera, C.; Dols-Perez, A.; Calderó, G.; García-Celma, M.J.; Camarasa, J.; Solans, C. PLGA nanoparticles prepared by nano-emulsion templating using low-energy methods as efficient nanocarriers for drug delivery across the blood–brain barrier. J. Control. Release 2015, 211, 134–143. [Google Scholar] [CrossRef]
- Llop, J.; Jiang, P.; Marradi, M.; Gómez-Vallejo, V.; Echeverría, M.; Yu, S.; Puigivila, M.; Baz, Z.; Szczupak, B.; Pérez-Campaña, C.; et al. Visualisation of dual radiolabelled poly(lactide-co-glycolide) nanoparticle degradation in vivo using energy-discriminant SPECT. J. Mater. Chem. B 2015, 3, 6293–6300. [Google Scholar] [CrossRef] [Green Version]
- Kreyling, W.G.; Abdelmonem, A.M.; Ali, Z.; Alves, F.; Geiser, M.; Haberl, N.; Hartmann, R.; Hirn, S.; de Aberasturi, D.J.; Kantner, K.; et al. In vivo integrity of polymer-coated gold nanoparticles. Nat. Nanotechnol. 2015, 10, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liu, C.; Pang, Z. Dendrimer-based drug delivery systems for brain targeting. Biomolecules 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauro, R.; Nandave, M.; Jain, V.K.; Jain, K. Advances in dendrimer-mediated targeted drug delivery to the brain. J. Nanopart. Res. 2021, 23, 1–20. [Google Scholar] [CrossRef]
- Moscariello, P.; Ng, D.Y.W.; Jansen, M.; Weil, T.; Luhmann, H.J.; Hedrich, J. Brain delivery of multifunctional dendrimer protein bioconjugates. Adv. Sci. 2018, 5, 1700897. [Google Scholar] [CrossRef] [PubMed]
- Janaszewska, A.; Lazniewska, J.; Trzepiński, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Taharabaru, T.; Yokoyama, R.; Higashi, T.; Mohammed, A.F.A.; Inoue, M.; Maeda, Y.; Niidome, T.; Onodera, R.; Motoyama, K. Genome editing in a wide area of the brain using dendrimer-based ternary polyplexes of Cas9 ribonucleoprotein. ACS Appl. Mater. Interfaces 2020, 12, 21386–21397. [Google Scholar] [CrossRef]
- Ke, W.; Shao, K.; Huang, R.; Han, L.; Liu, Y.; Li, J.; Kuang, Y.; Ye, L.; Lou, J.; Jiang, C. Gene delivery targeted to the brain using an Angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials 2009, 30, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, R.; Han, L.; Ke, W.; Shao, K.; Ye, L.; Lou, J.; Jiang, C. Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials 2009, 30, 4195–4202. [Google Scholar] [CrossRef]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.A.; Feng, S.-S. Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Florence, A.T.; Hillery, A.M.; Hussain, N.; Jani, P.U. Nanoparticles as carriers for oral peptide absorption: Studies on particle uptake and fate. J. Control. Release 1995, 36, 39–46. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. In Nanoscience and Technology; Co-Published with Macmillan Publishers Ltd.: London, UK, 2009; pp. 239–250. [Google Scholar]
- Duan, X.; Li, Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small 2013, 9, 1521–1532. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Krishnan, V.; Mitragotri, S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv. Drug Deliv. Rev. 2019, 143, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Rescignano, N.; Tarpani, L.; Romani, A.; Bicchi, I.; Mattioli, S.; Emiliani, C.; Torre, L.; Kenny, J.M.; Martino, S.; Latterini, L.; et al. In-vitro degradation of PLGA nanoparticles in aqueous medium and in stem cell cultures by monitoring the cargo fluorescence spectrum. Polym. Degrad. Stab. 2016, 134, 296–304. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Y.; Ma, Y.; Liu, Y.; Yan, B.; Wang, L. Smart nanocarrier based on PEGylated hyaluronic acid for deacetyl mycoepoxydience: High stability with enhanced bioavailability and efficiency. Carbohydr. Polym. 2019, 203, 356–368. [Google Scholar] [CrossRef]
- Adamo, G.; Campora, S.; Ghersi, G. Functionalization of nanoparticles in specific targeting and mechanism release. In Nanostructures for Novel Therapy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 57–80. [Google Scholar]
- Adamo, G.; Grimaldi, N.; Campora, S.; Bulone, D.; Bondì, M.; Al-Sheikhly, M.; Sabatino, M.; Dispenza, C.; Ghersi, G. Multi-Functional Nanogels for Tumor Targeting and Redox-Sensitive Drug and siRNA Delivery. Molecules 2016, 21, 1594. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, L.; Tong, S.; Lee, C.M.; Deshmukh, H.; Bao, G. Spatial control of in vivo CRISPR–Cas9 genome editing via nanomagnets. Nat. Biomed. Eng. 2019, 3, 126–136. [Google Scholar] [CrossRef]
- Mondal, S.; Dorozhkin, S.V.; Pal, U. Recent progress on fabrication and drug delivery applications of nanostructured hydroxyapatite. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1504. [Google Scholar] [CrossRef]
- Chuang, C.K.; Chen, C.H.; Huang, C.L.; Su, Y.H.; Peng, S.H.; Lin, T.Y.; Tai, H.C.; Yang, T.S.; Tu, C.F. Generation of GGTA1 Mutant Pigs by Direct Pronuclear Microinjection of CRISPR/Cas9 Plasmid Vectors. Anim. Biotechnol. 2017, 28, 174–181. [Google Scholar] [CrossRef]
- Love, J.; Gribbin, C.; Mather, C.; Sang, H. Transgenic Birds by DNA Microinjection. Bio/Technology 1994, 12, 60–63. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lee, B.; Lee, A.Y.; Modzelewski, A.J.; He, L. Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J. Biol. Chem. 2016, 291, 14457–14467. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Oh, J.; Shim, G.; Cho, B.; Chang, Y.; Kim, S.; Baek, S.; Kim, H.; Shin, J.; Choi, H.; et al. In vivo neuronal gene editing via CRISPR–Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Höijer, I.; Tsai, Y.-C.; Clark, T.A.; Kotturi, P.; Dahl, N.; Stattin, E.-L.; Bondeson, M.-L.; Feuk, L.; Gyllensten, U.; Ameur, A. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum. Mutat. 2018, 39, 1262–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.-L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Gorjizadeh, N.; Alemi, F.; Asemi, Z.; Azizian, K.; Soleimanpour, J.; Malakouti, F.; Targhazeh, N.; Majidinia, M.; Yousefi, B. CRISPR/Cas9 novel therapeutic road for the treatment of neurodegenerative diseases. Life Sci. 2020, 259, 118165. [Google Scholar] [CrossRef]
- Hitti, F.L.; Yang, A.I.; Gonzalez-Alegre, P.; Baltuch, G.H. Human gene therapy approaches for the treatment of Parkinson’s disease: An overview of current and completed clinical trials. Parkinsonism Relat. Disord. 2019, 66, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Khodr, C.E.; Sapru, M.K.; Pedapati, J.; Bohn, M.C. A microRNA embedded AAV alpha-synuclein gene silencing vector for dopaminergic neurons. Brain Res. 2011, 1386, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; Melrose, H.; Bumcrot, D.; Hope, A.; Zehr, C.; Lincoln, S.; Braithwaite, A.; He, Z.; Ogholikhan, S.; Hinkle, K.; et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 2008, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirik, D.; Georgievska, B.; Burger, C.; Winkler, C.; Muzyczka, N.; Mandel, R.J.; Bjorklund, A. Reversal of motor impairments in parkinsonian rats by continuous intrastriatal delivery of L-dopa using rAAV-mediated gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 4708–4713. [Google Scholar] [CrossRef] [Green Version]
- Leriche, L.; Bjorklund, T.; Breysse, N.; Besret, L.; Gregoire, M.-C.; Carlsson, T.; Dolle, F.; Mandel, R.J.; Deglon, N.; Hantraye, P.; et al. Positron Emission Tomography Imaging Demonstrates Correlation between Behavioral Recovery and Correction of Dopamine Neurotransmission after Gene Therapy. J. Neurosci. 2009, 29, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Cederfjäll, E.; Broom, L.; Kirik, D. Controlled Striatal DOPA Production From a Gene Delivery System in a Rodent Model of Parkinson’s Disease. Mol. Ther. 2015, 23, 896–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederfjäll, E.; Nilsson, N.; Sahin, G.; Chu, Y.; Nikitidou, E.; Björklund, T.; Kordower, J.H.; Kirik, D. Continuous DOPA synthesis from a single AAV: Dosing and efficacy in models of Parkinson’s disease. Sci. Rep. 2013, 3, 2157. [Google Scholar] [CrossRef] [Green Version]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009, 73, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, S.-I.; Fujimoto, K.-I.; Kato, S.; Mizukami, H.; Asari, S.; Ikeguchi, K.; Kawakami, T.; Urabe, M.; Kume, A.; Sato, T.; et al. A Phase I Study of Aromatic L-Amino Acid Decarboxylase Gene Therapy for Parkinson’s Disease. Mol. Ther. 2010, 18, 1731–1735. [Google Scholar] [CrossRef] [Green Version]
- Ciesielska, A.; Samaranch, L.; San Sebastian, W.; Dickson, D.W.; Goldman, S.; Forsayeth, J.; Bankiewicz, K.S. Depletion of AADC activity in caudate nucleus and putamen of Parkinson’s disease patients; implications for ongoing AAV2-AADC gene therapy trial. PLoS ONE 2017, 12, e0169965. [Google Scholar] [CrossRef] [Green Version]
- Christine, C.W.; Bankiewicz, K.S.; Van Laar, A.D.; Richardson, R.M.; Ravina, B.; Kells, A.P.; Boot, B.; Martin, A.J.; Nutt, J.; Thompson, M.E.; et al. Magnetic resonance imaging–guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann. Neurol. 2019, 85, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Muramatsu, S.; Lu, Y.; Ikeguchi, K.; Fujimoto, K.; Okada, T.; Mizukami, H.; Hanazono, Y.; Kume, A.; Urano, F.; et al. Delayed delivery of AAV-GDNF prevents nigral neurodegeneration and promotes functional recovery in a rat model of Parkinson’s disease. Gene Ther. 2002, 9, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.-Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration Prevented by Lentiviral Vector Delivery of GDNF in Primate Models of Parkinson’s Disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Ge, G.; Liu, J.; Biju, K.C.; Laing, S.D.; Qian, Y.; Ballard, C.; He, Z.; Masliah, E.; et al. GDNF-expressing macrophages mitigate loss of dopamine neurons and improve Parkinsonian symptoms in MitoPark mice. Sci. Rep. 2018, 8, 5460. [Google Scholar] [CrossRef]
- Chen, C.; Guderyon, M.J.; Li, Y.; Ge, G.; Bhattacharjee, A.; Ballard, C.; He, Z.; Masliah, E.; Clark, R.A.; O’Connor, J.C.; et al. Non-toxic HSC transplantation-based macrophage/microglia-mediated GDNF delivery for Parkinson’s disease. Mol. Ther. Clin. Dev. 2020, 17, 83–98. [Google Scholar] [CrossRef] [Green Version]
- Lindvall, O.; Wahlberg, L.U. Encapsulated cell biodelivery of GDNF: A novel clinical strategy for neuroprotection and neuroregeneration in Parkinson’s disease? Exp. Neurol. 2008, 209, 82–88. [Google Scholar] [CrossRef]
- Fan, C.-H.; Ting, C.-Y.; Lin, C.; Chan, H.-L.; Chang, Y.-C.; Chen, Y.-Y.; Liu, H.-L.; Yeh, C.-K. Noninvasive, Targeted and Non-Viral Ultrasound-Mediated GDNF-Plasmid Delivery for Treatment of Parkinson’s Disease. Sci. Rep. 2016, 6, 19579. [Google Scholar] [CrossRef]
- Mead, B.P.; Kim, N.; Miller, G.W.; Hodges, D.; Mastorakos, P.; Klibanov, A.L.; Mandell, J.W.; Hirsh, J.; Suk, J.S.; Hanes, J.; et al. Novel Focused Ultrasound Gene Therapy Approach Noninvasively Restores Dopaminergic Neuron Function in a Rat Parkinson’s Disease Model. Nano Lett. 2017, 17, 3533–3542. [Google Scholar] [CrossRef]
- Berke, J.D. What does dopamine mean? Nat. Neurosci. 2018, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Eban-Rothschild, A.; Rothschild, G.; Giardino, W.J.; Jones, J.R.; de Lecea, L. VTA dopaminergic neurons regulate ethologically relevant sleep–wake behaviors. Nat. Neurosci. 2016, 19, 1356–1366. [Google Scholar] [CrossRef]
- Martinez-Martín, P.; Rodriguez-Blazquez, C.; Paz, S.; Forjaz, M.J.; Frades-Payo, B.; Cubo, E.; Pedro-Cuesta, J.d.; Lizán, L.; ELEP Group. Parkinson symptoms and health related quality of life as predictors of costs: A longitudinal observational study with linear mixed model analysis. PLoS ONE 2015, 10, e0145310. [Google Scholar] [CrossRef] [Green Version]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and Levodopa Prodrugs for the Treatment of Parkinson’s Disease. Molecules 2018, 23, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porras, G.; De Deurwaerdere, P.; Li, Q.; Marti, M.; Morgenstern, R.; Sohr, R.; Bezard, E.; Morari, M.; Meissner, W.G. L-dopa-induced dyskinesia: Beyond an excessive dopamine tone in the striatum. Sci. Rep. 2015, 4, 3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberling, J.L.; Jagust, W.J.; Christine, C.W.; Starr, P.; Larson, P.; Bankiewicz, K.S.; Aminoff, M.J. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 2008, 70, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- Bankiewicz, K.S.; Forsayeth, J.; Eberling, J.L.; Sanchez-Pernaute, R.; Pivirotto, P.; Bringas, J.; Herscovitch, P.; Carson, R.E.; Eckelman, W.; Reutter, B.; et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol. Ther. 2006, 14, 564–570. [Google Scholar] [CrossRef]
- Merkel, S.F.; Andrews, A.M.; Lutton, E.M.; Mu, D.; Hudry, E.; Hyman, B.T.; Maguire, C.A.; Ramirez, S.H. Trafficking of adeno-associated virus vectors across a model of the blood–brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J. Neurochem. 2017, 140, 21. [Google Scholar] [CrossRef] [Green Version]
- Mittermeyer, G.; Christine, C.W.; Rosenbluth, K.H.; Baker, S.L.; Starr, P.; Larson, P.; Kaplan, P.L.; Forsayeth, J.; Aminoff, M.J.; Bankiewicz, K.S. Long-term evaluation of a phase 1 study of AADC gene therapy for parkinson’s disease. Hum. Gene Ther. 2011, 23, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Witt, J.; Marks, W.J. An Update on Gene Therapy in Parkinson’s Disease. Curr. Neurol. Neurosci. Rep. 2011, 11, 362–370. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Winkler, C.; Sauer, H.; Lee, C.S.; Björklund, A. Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1996, 16, 72. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.E.D.; Hua, K.; Monis, S.; Saxena, V.; Norazit, A.; Noor, S.M.; Ekker, M. Gdnf affects early diencephalic dopaminergic neuron development through regulation of differentiation-associated transcription factors in zebrafish. J. Neurochem. 2020, 156, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line–derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line–derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Ai, Y.; Fischer, B.; Zhang, A.; Grondin, R.; Zhang, Z.; Gerhardt, G.; Gash, D. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006, 202, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended Treatment with Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Garbayo, E.; Montero-Menei, C.N.; Ansorena, E.; Lanciego, J.L.; Aymerich, M.S.; Blanco-Prieto, M.J. Effective GDNF brain delivery using microspheres—a promising strategy for Parkinson’s disease. J. Control. Release 2009, 135, 11. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Herzog, C.D.; Dass, B.; Bakay, R.A.E.; Stansell, J.; Gasmi, M.; Bartus, R.T. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann. Neurol. 2006, 60, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, S.; Scarpato, M.; Damiani, D.; Managò, F.; Mereu, M.; Contestabile, A.; Peruzzo, O.; Carninci, P.; Santoro, C.; Papaleo, F.; et al. SINEUP Non-coding RNA Targeting GDNF Rescues Motor Deficits and Neurodegeneration in a Mouse Model of Parkinson’s Disease. Mol. Ther. 2020, 28, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Herzog, C.D.; Dass, B.; Holden, J.E.; Stansell, J.; Gasmi, M.; Tuszynski, M.H.; Bartus, R.T.; Kordower, J.H. Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov. Disord. 2007, 22, 1124–1132. [Google Scholar] [CrossRef]
- Marks, W.J.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2–neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Olanow, C.W.; Bartus, R.T.; Baumann, T.L.; Factor, S.; Boulis, N.; Stacy, M.; Turner, D.A.; Marks, W.; Larson, P.; Starr, P.A.; et al. Gene delivery of neurturin to putamen and Substantia nigra in P arkinson disease: A double-blind, randomized, controlled trial. Ann. Neurol. 2015, 78, 248. [Google Scholar] [CrossRef]
- Chu, Y.; Bartus, R.T.; Manfredsson, F.P.; Olanow, C.W.; Kordower, J.H. Long-term post-mortem studies following neurturin gene therapy in patients with advanced Parkinson’s disease. Brain 2020, 143, 960–975. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Désaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Choi, Y.; Park, J.; Nah, W.; Park, J.; Jung, Y.; Lee, J.; Lee, H.; Park, S.; Hwang, S.; et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological α-synuclein. Sci. Adv. 2020, 6, eaba1193. [Google Scholar] [CrossRef] [PubMed]
- Khodr, C.E.; Sapru, M.K.; Pedapati, J.; Han, Y.; West, N.C.; Kells, A.P.; Bankiewicz, K.S.; Bohn, M.C. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res. 2011, 1395, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Khodr, C.E.; Becerra, A.; Han, Y.; Bohn, M.C. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat Substantia nigra: Positive and negative effects. Brain Res. 2014, 1550, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Cheung, H. Stem cell-based therapies for Parkinson disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef]
- Coccia, E.; Ahfeldt, T. Towards physiologically relevant human pluripotent stem cell (hPSC) models of Parkinson’s disease. Stem Cell Res. Ther. 2021, 12, 253. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embyonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Arora, N.; Huo, H. Disease-specfic induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldner, F.; Hockemeyer, D.; Beard, C. Parkinson’s Disease Patient-Derived Induced Pluripotent Stem Cells Free of Viral Reprogramming Factors. Cell 2009, 135, 964–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, W.; Tan, S.; Lin, T. Stem cells for modeling and therapy of Parkinson’s disease. Hum. Gene Ther. 2017, 28, 85–98. [Google Scholar] [CrossRef]
- Safari, F.; Hatam, G.; Behbahani, A.B.; Rezaei, V.; Barekati-Mowahed, M.; Petramfar, P.; Khademi, F. CRISPR system: A high-throughput toolbox for research and treatment of Parkinson’s disease. Cell. Mol. Neurobiol. 2020, 40, 477–493. [Google Scholar] [CrossRef]
- Sison, S.L.; Vermilyea, S.C.; Emborg, M.E.; Ebert, A.D. Using patient-derived induced pluripotent stem cells to identify Parkinson’s disease-relevant phenotypes. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ke, M.; Chong, C.M.; Su, H. Using induced pluripotent stem cells for modeling Parkinson’s disease. World J. Stem Cells 2019, 11, 634. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Mao, C.; Fan, L.; Luo, H.; Hu, Z.; Zhang, S.; Yang, Z.; Zheng, H.; Sun, H.; Fan, Y.; et al. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Stem Cells Int. 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Barker, R.A.; Studer, L.; Cattaneo, E.; Takahashi, J. G-Force PD: A global initiative in coordinating stem cell-based dopamine treatments for Parkinson’s disease. Parkinsons. Dis. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahfeldt, T.; Ordureau, A.; Bell, C.; Sarrafha, L.; Sun, C.; Piccinotti, S.; Grass, T.; Parfitt, G.M.; Paulo, J.A.; Yanagawa, F.; et al. Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. 2020, 14, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Calatayud, C.; Carola, G.; Consiglio, A.; Raya, A. Modeling the genetic complexity of Parkinson’s disease by targeted genome edition in iPS cells. Curr. Opin. Genet. Dev. 2017, 46, 123–131. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Consiglio, A. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, C.C.; Luo, X.G.; Cui, H.G.; Li, F.R.; Li, P.; Jiang, E.Z.; Ren, Y.; Pang, H. Screening of polymorphisms located in the FGF20 and TMEM175 genes in North Chinese Parkinson’s disease patients. Genet. Mol. Res. 2015, 14, 13679–13687. [Google Scholar] [CrossRef] [PubMed]
- Harjuhaahto, S.; Rasila, T.S.; Molchanova, S.M.; Woldegebriel, R.; Kvist, J.; Konovalova, S.; Tyynismaa, H. ALS and Parkinson’s disease genes CHCHD10 and CHCHD2 modify synaptic transcriptomes in human iPSC-derived motor neurons. Neurobiol. Dis. 2020, 14, 104940. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.H.; Kao, K.C. Growth-Coupled Carotenoids Production Using Adaptive Laboratory Evolution. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; pp. 319–330. [Google Scholar]




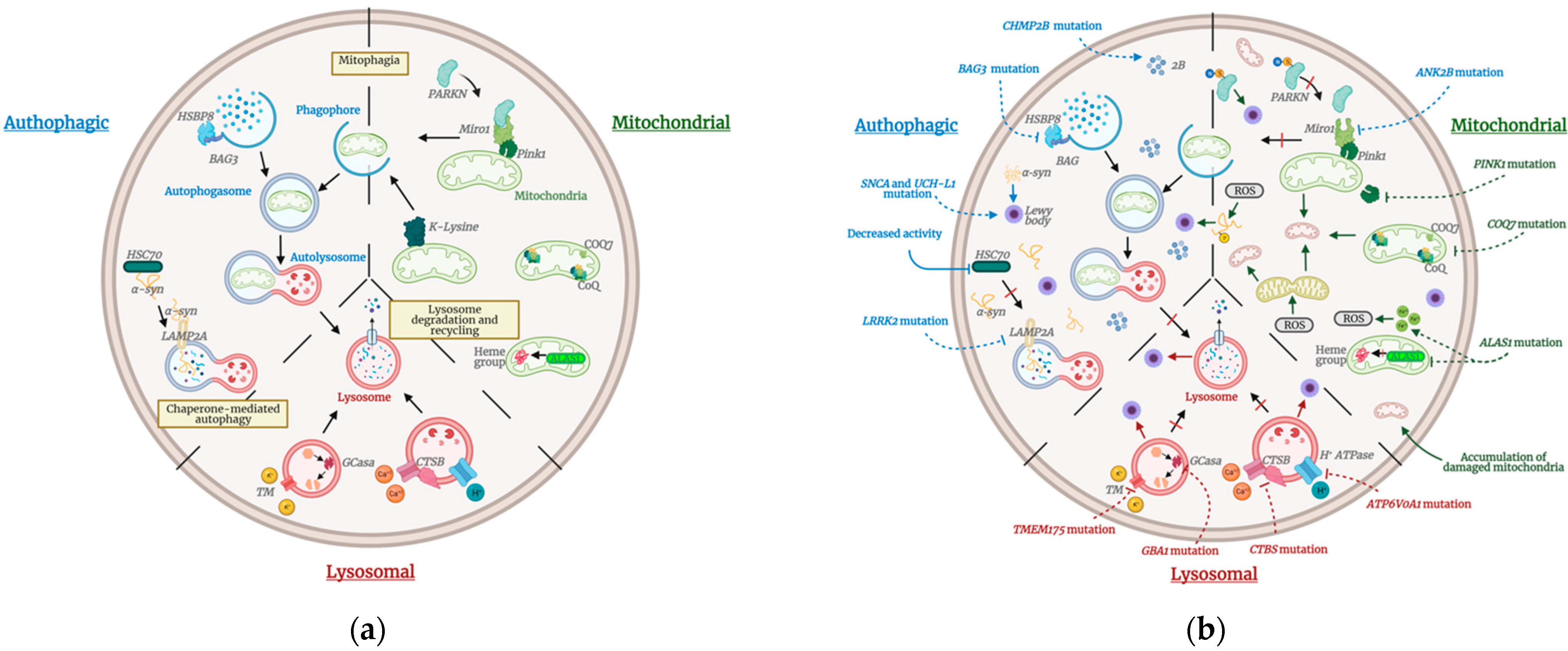{kind=link}
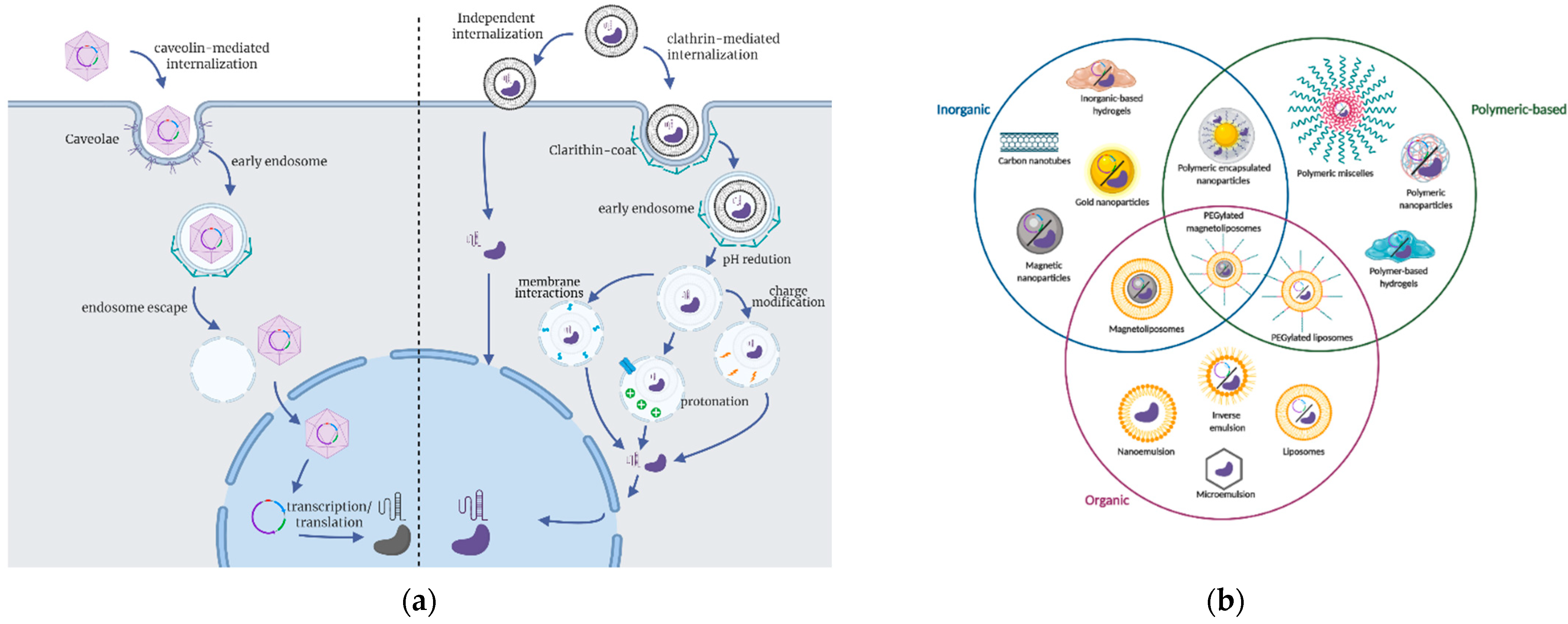{kind=link}
{kind=link}
| Gene | Alternative Gene Names | Gene Locus | Protein | Protein Function and Cell Pathway Governed | Onset of Familial PD |
|---|---|---|---|---|---|
| SNCA | PARK 1 or PARK4 | 4q22.1 | α-synuclein | Synaptic vesicles trafficking | Early |
| Unknown | PARK3 | 2p13 | Unknown | Unknown | Late |
| UCHL1 | PARK5 | 4p13 | Ubiquitin C-terminal hydrolase L1 | Proteasome system | Late |
| LRRK2 | PARK8 | 12q12 | Leucine-rich repeat kinase 2 | Autophagy processing | Late |
| HTRA2 | PARK13 | 2p13.1 | HtrA serine peptidase 2 | Mitophagy development | Unknown |
| VPS35 | PARK17 | 16q12 | Vacuolar protein sorting 35 | Endosome regulation | Late |
| EIF4G1 | PARK18 | 3q27.1 | Eukaryotic translation initiation factor 4 gamma 1 | Protein translation | Late |
| DNAJC13 | PARK21 | 3q22.1 | DnaJ heat shock protein family (Hsp40) member C13 | Endosome regulation | Late |
| CHCHD2 | PARK22 | 7p11.2 | Coiled-coil-helix-coiled-coil-helix domain containing 2 | Mitochondria-mediated apoptosis and metabolism | Late/Early |
| PRKN | PARK2 | 6q26 | Parkin | Mitophagy development | Early |
| PINK1 | PARK6 | 1p36.12 | PTEN-induced putative kinase 1 | Mitophagy development | Early |
| DJ-1 | PARK7 | 1p36.23 | DJ-1 | Mitophagy development | Early |
| ATP13A2 | PARK9 | 1p36.13 | ATPase cation transporting 13A2 | Lysosomal function | Early |
| GIGYF2 | PARK11 | 2q36-7 | GRB10 interacting GYF protein 2 | Insulin-like growth factors (IGFs) signaling | Early |
| PLA2G6 | PARK14 | 22q13.1 | Phospholipase A2 group VI | Lipids metabolism | Early |
| FBXO7 | PARK15 | 22q12.3 | F-box protein 7 | Mitophagy development | Early |
| DNAJC6 | PARK19 | 1p31.3 | DnaJ heat shock protein family (Hsp40) member C6 | Endosome regulation | Early |
| SYNJ1 | PARK20 | 21q22.11 | Synaptojanin 1 | Endosome regulation | Early |
| VPS13C | PARK23 | 15q22.2 | Vacuolar protein sorting 13 homolog C | Mitophagy development | Early |
| Unknown | PARK10 | 1p32 | Unknown | Unknown | Unknown |
| Unknown | PARK12 | Xq21–q22 | Unknown | Unknown | Unknown |
| Unknown | PARK16 | 1q32 | Unknown | Unknown | Unknown |
| GBA | - | 1q22 | Glucosylceramidase beta | Lysosomal function | Late |
| LRP10 | - | - | LDLR-related protein 10 | Retinoid metabolism and transport | Late |
| Approach | Vector | Phase of Clinical Study | Reference |
|---|---|---|---|
| DP activity (TH) | AAV-TH | Animal Model: murine | [270,271,272,273] |
| DP activity (AADC) | AAV-AADC | Animal Model: murine (Phase I) | [274,275,276,277] |
| Neurotrophic genes (GDNF) | AAV-GDNF | Animal Model: murine and primate (Phase I and II) | [278,279] |
| Neurotrophic genes (GNDF) | Hematopoietic stem cell macrophages | Animal Model: murine | [280,281] |
| Neurotrophic genes (GNDF) | Encapsulated GDNF-secreting cells | Animal Model: murine | [282] |
| Neurotrophic genes (GNDF) | Cationic microbubbles | Animal Model: murine | [283] |
| Neurotrophic genes (GNDF) | Brain penetrating nanoparticles | Animal Model: murine | [284] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arango, D.; Bittar, A.; Esmeral, N.P.; Ocasión, C.; Muñoz-Camargo, C.; Cruz, J.C.; Reyes, L.H.; Bloch, N.I. Understanding the Potential of Genome Editing in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 9241. https://doi.org/10.3390/ijms22179241
Arango D, Bittar A, Esmeral NP, Ocasión C, Muñoz-Camargo C, Cruz JC, Reyes LH, Bloch NI. Understanding the Potential of Genome Editing in Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(17):9241. https://doi.org/10.3390/ijms22179241
Chicago/Turabian StyleArango, David, Amaury Bittar, Natalia P. Esmeral, Camila Ocasión, Carolina Muñoz-Camargo, Juan C. Cruz, Luis H. Reyes, and Natasha I. Bloch. 2021. "Understanding the Potential of Genome Editing in Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 17: 9241. https://doi.org/10.3390/ijms22179241