
Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder

 , , ,
, , , 
Abstract
:1. Introduction
2. Environmental Pollution and ASD
3. Aryl Hydrocarbon Receptor Pathway and ASD
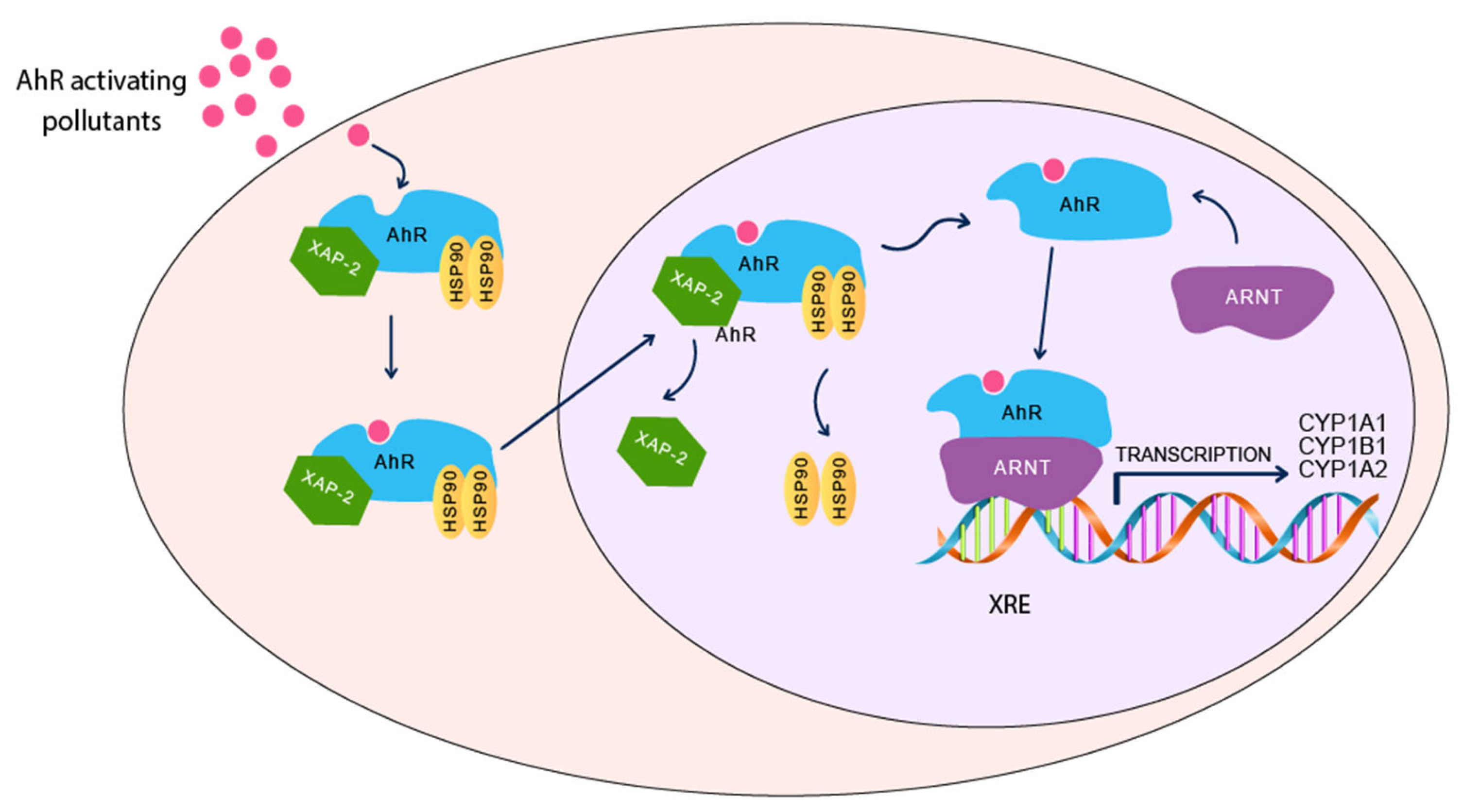
3.1. Aryl Hydrocarbon Receptor Pathway
3.2. Evidence of Involvement of AhR/CYP1A Pathway in Autism Development
3.2.1. Human and Epidemiological Studies
3.2.2. Experimental Animal Studies

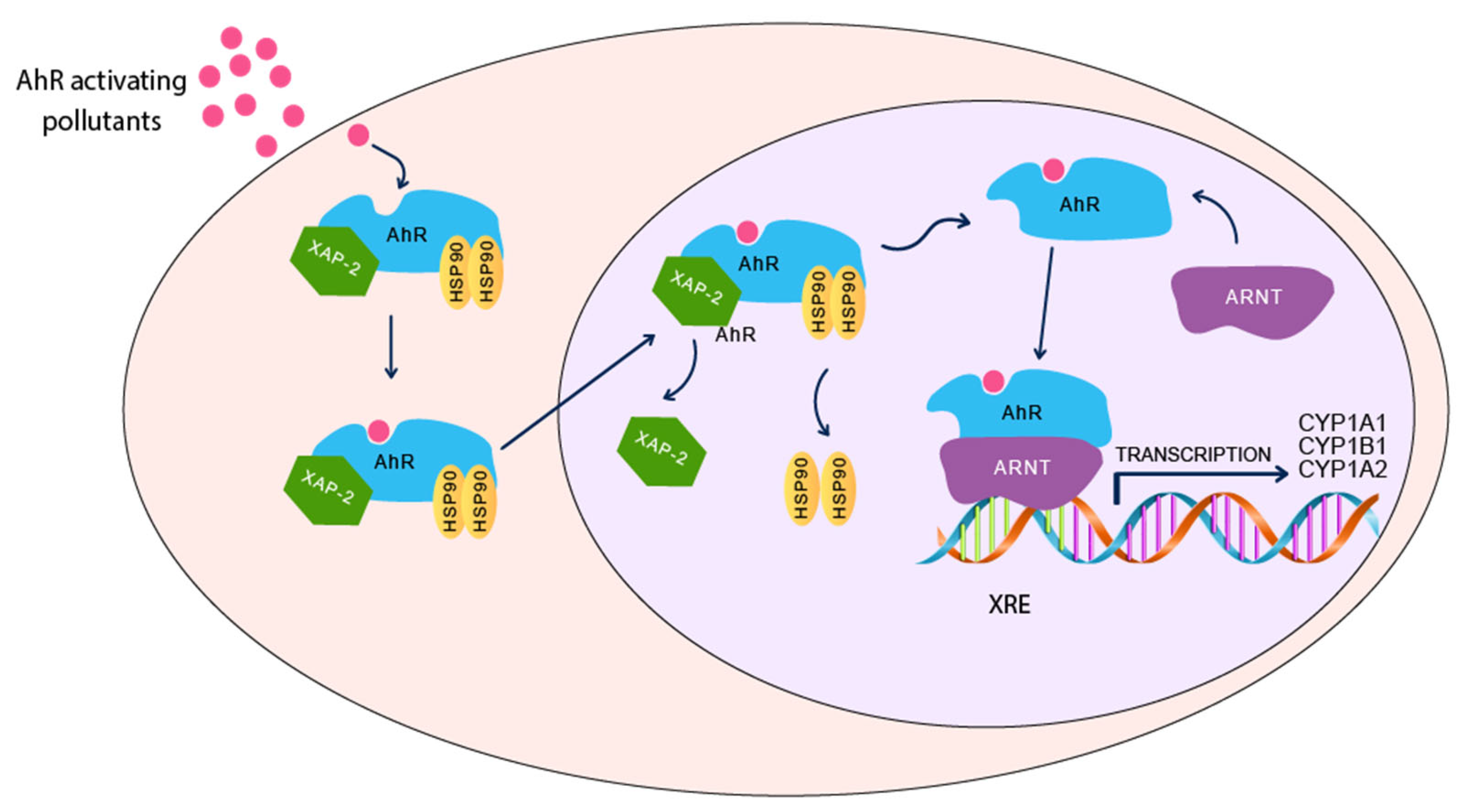{kind=link}
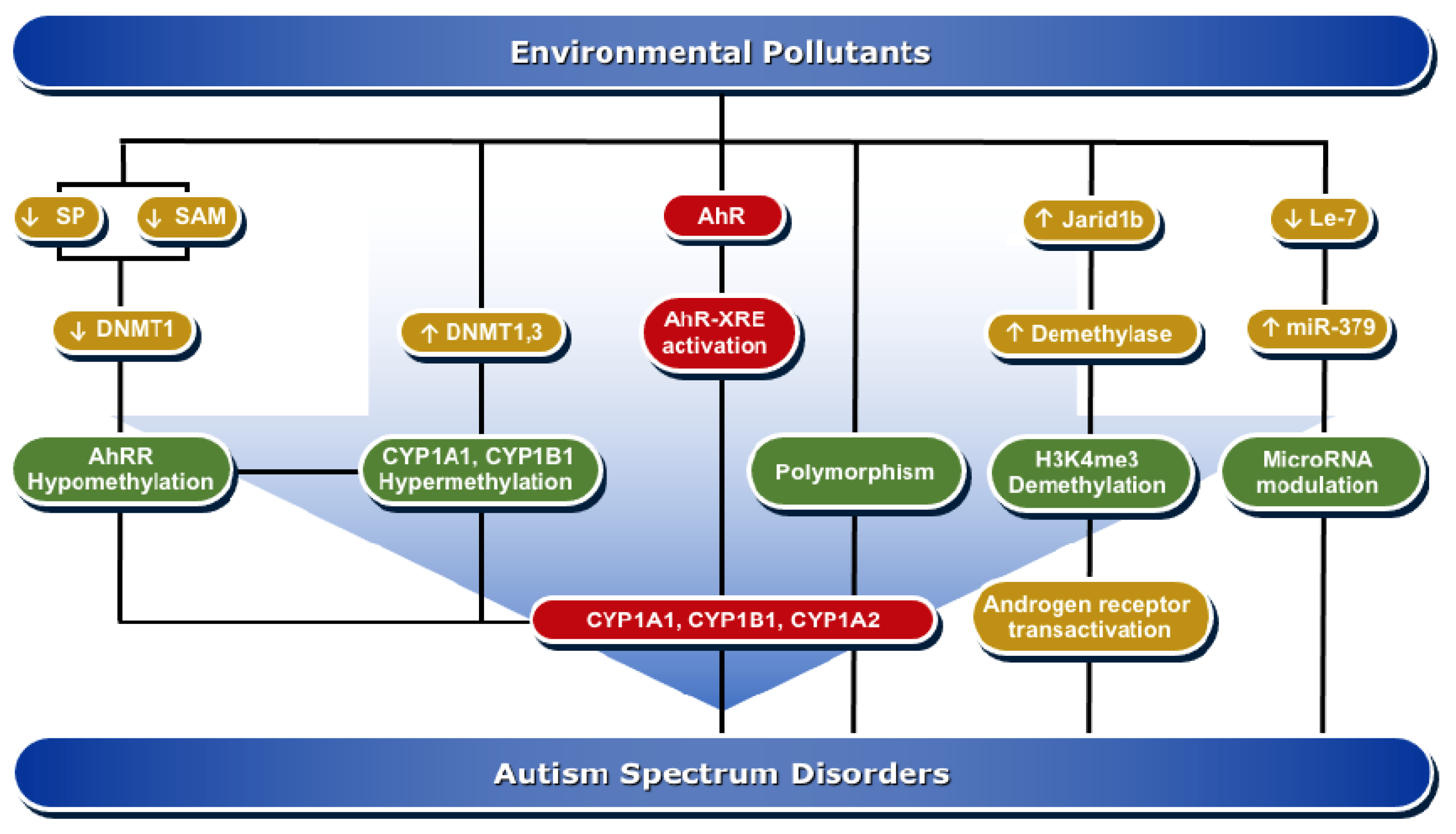{kind=link}
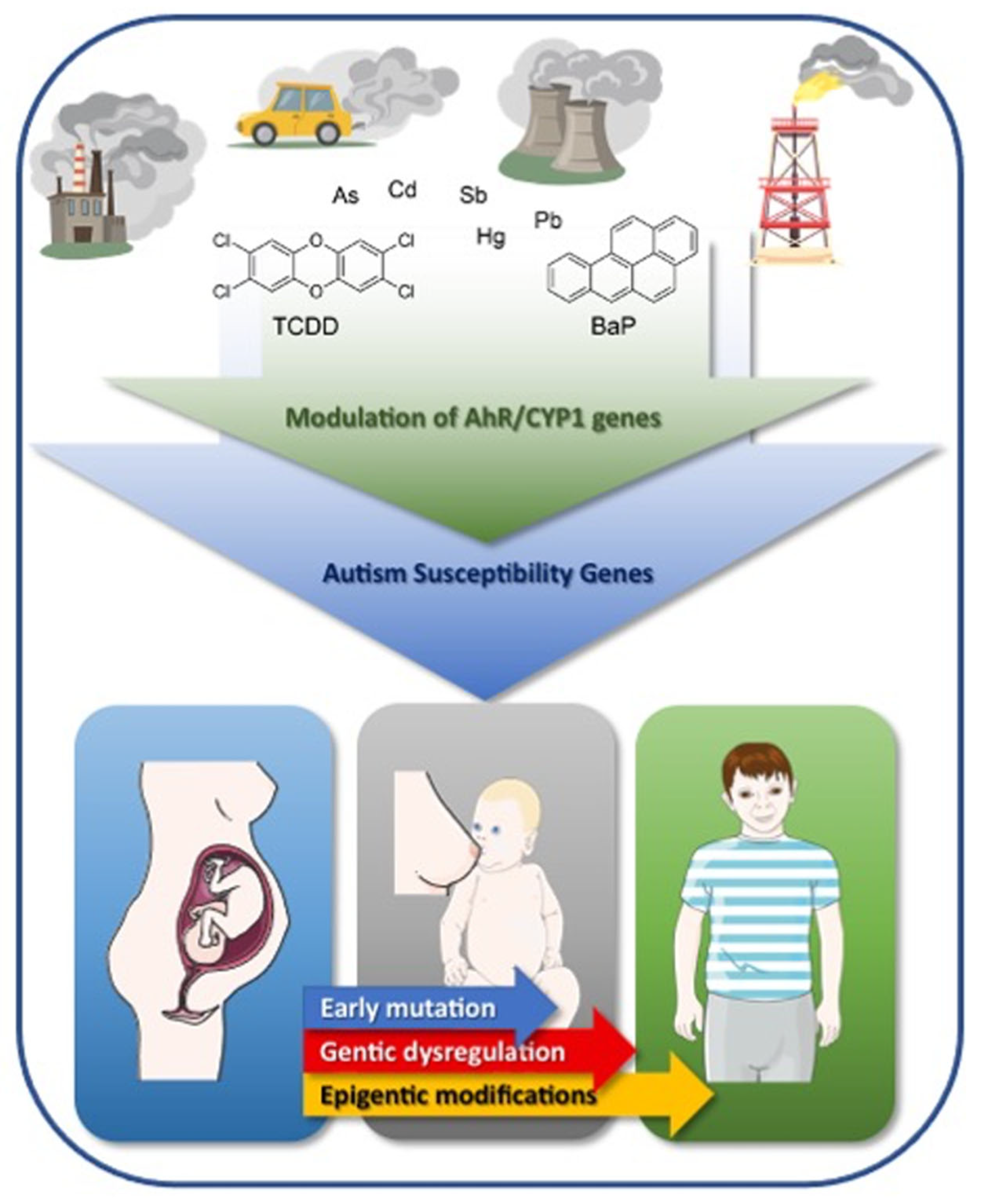{kind=link}
| Gene | Study Model | Study Design | AhR/CYP1 Modulator | Effect on AhR/CYP1 Pathway | Effect on Autism Incidence & Development | References |
|---|---|---|---|---|---|---|
| AhR | Human | SK-N-SH human-derived neurons | TCDD | AhR activation → ↓AChE | ↓ Neuronal activity | [90] |
| CH223191 | AhR inhibitor, CH223191 → ↑ AChE | ↑ Neuronal activity | ||||
| Pregnant amniotic fluids | PCBs & heavy metals | ↑ AhR transactivation | ↓ The levels of PFAS were lower in ASD cases compared to control. | [78] | ||
| Rats | Perinatal exposure to TCDD | TCDD | Activation of AhR → ↓ AChE, monoamines, ↑ stimulation of GABA ↓ thyroid hormones, increase in TSH, decrease growth hormones in cerebellum of offspring | ↑ Permanent brain damage. Impaired the development of cerebellum of their offspring | [89] | |
| CYP1A | Human | Autistic subject | ↓ CYP1A1 gene expression in umbilical cord blood | ↑ ASD incidence compared to control | [82] | |
| Pregnant | Dioxin | ↑ dioxin levels in maternal blood | ↑ Neurodevelopmental deficits and autistic traits in the children with ASD | [63,83] | ||
| PCDFs | ↑ PCDFs levels in maternal blood | ↑ Autistic traits in middle to late childhood using SRS | [61] | |||
| Zebrafish | Developmental exposure to PCB126 on early- and later-life behavioral | PCB126 | ↑ CYP1A1 in early stages of development, with no significant upregulation at adulthood | Impaired short-term and long-term habituation to unfamiliar environment ↑ Anxiety-related behavior with no change in the larval locomotor activity | [91,92] | |
| Mice | Pregnant C57BL/6N | AhR plasmid transfection | Constitutive AhR activation | Affects neuronal migration during hippocampal development. | [84] | |
| High affinity Cyp1a2(−/−) | PCBs | CYP1A2 Knockout | ↑ Motor dysfunction compared to wild-type mouse. ↑ Susceptibility to nigrostriatal dysfunction and motor deficit, and toxicity of the cerebellum and cortex. | [93,94] | ||
| CYP1B1 | Human | Autistic children | Vitamin D deficiency | ↓ CYP1B1 plasma levels by 70% through epigenetic silencing of CYP1B1 | ↓ Vitamin D by 60% → positively correlates with ASD | [81] |
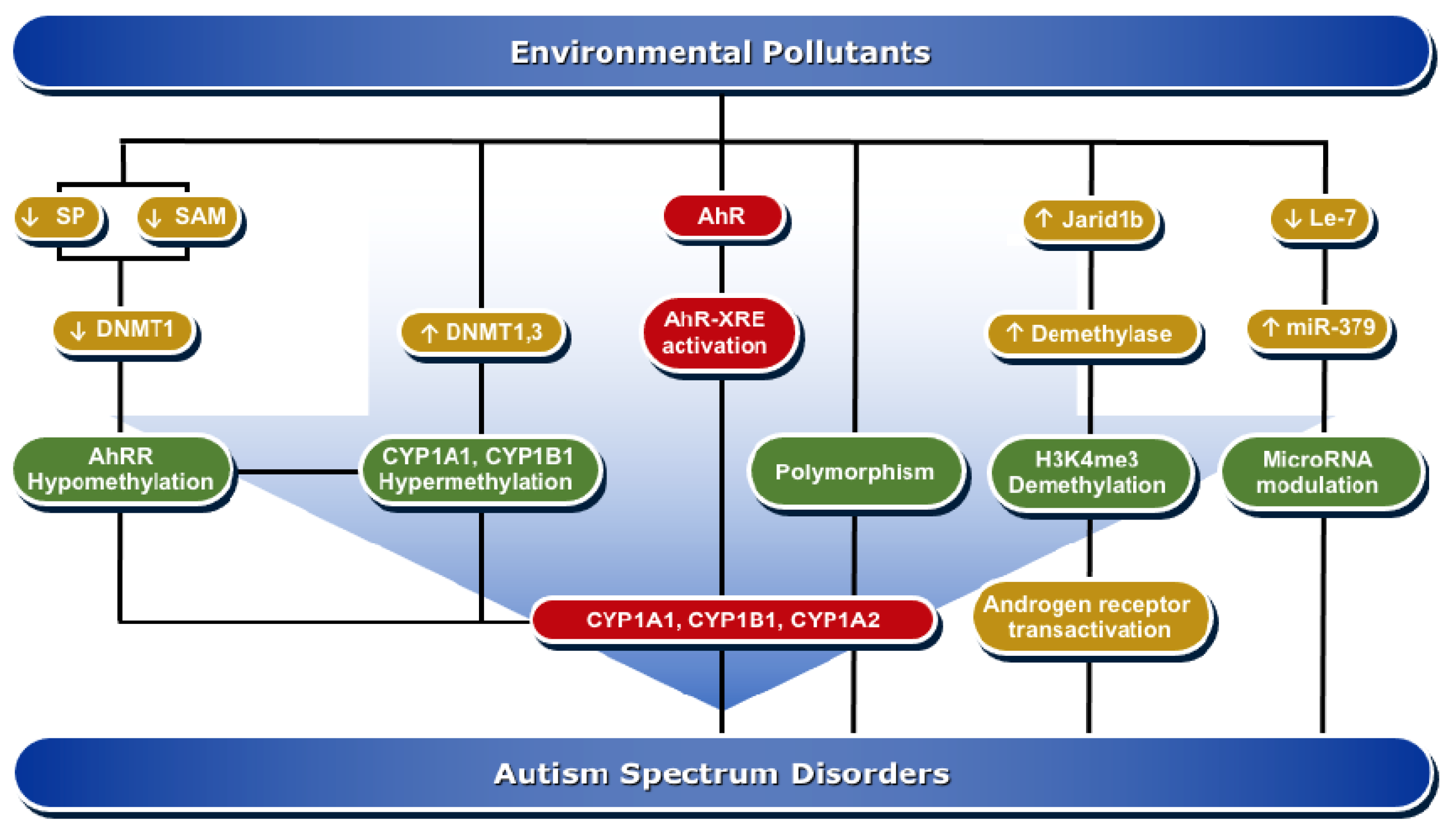
4. Molecular Mechanisms of AhR/CYP1A Regulation in ASD Development
4.1. Epigenesis
4.1.1. DNA Methylation
4.1.2. Histone Modifications
4.1.3. MicroRNAs
4.2. Genetic Polymorphism
| Mechanisms | Study Model | Study Design | Gene/Enzyme Changes | Effect on Autism | References |
|---|---|---|---|---|---|
| DNA methylation | Human | Autistic subject | ↑ DNMT3a, b | ↑ DNA damage | [100] |
| Autistic men exposed prenatally to PCBs | ↑ Methylation of CYP1B1/Cyp1A1 | ↑ Incidence of autism | [102] | ||
| ↓ Methylation of AhRR | [102] | ||||
| Newborns with maternal smoking during pregnancy | ↑ Methylation of Cyp1A1 | ↑ Incidence of autism | [103,106] | ||
| ↓ Methylation of AhRR | hypomethylation in the early development period can persist for a long period | [103,104,106] | |||
| Rats | Pre- & postnatal exposure to AhR activators | ↓ DNA methylation ↓ DNMT1, 3a, 3b mRNA levels in brain | ↓ Sp1↓ S-adenosyl methionine levels. | [107,108,109] | |
| ↑ DNMT1 and ↑ ARNT | Delay in onset of puberty in males and affecting estrous cyclicity in females | [111] | |||
| Mice | BTBR T+tf/J mice | ↑ DNMT3a, b | ↑ DNA damage and autism risk | [100] | |
| Histone modifications | Human | HEK293 cells exposed to PCB Human post-mortem cerebellum autistic individuals | ↑ Demethylation of H3K4me3 by Jarid1b ↑ DNMT3a and 3b | ↑ Activation of androgen receptor transcriptional activity ↑ DNA oxidative damage genes 8-oxodG → ↑ autism incidence | [100,121] |
| Rats | Valproic acid -induced autism in rats | ↓ HDAC → hyperacetylation of histone | ↑ Neurological birth defect | [115] | |
| Mice | BTBR T+tf/J mice | ↑ DNMT3a and 3b ↔ H3K9ac, H3K56ac, H3K4me3, H3K9me3, H3K27me3, and H4K20me3 compared to the control | ↑ DNA oxidative damage genes 8-oxodG → ↑ autism incidence | [100] | |
| MicroRNAs | Mice | Prenatal exposure to TCDD | ↑ miR-379 | ↓ Brain neuronal development → ↑ hypo-social behavior observed | [128] |
| ↓ let-7 | Regulated neuronal stem cell proliferation | [128] | |||
| Gene polymorphism | Human | Autistic subjects | ↑ ARNT gene (rs2228099), but not AhR rs2066853, | Significant difference in the severity, particularly social communication | [130] |
| ↑ ARNT2 (rs17225178) | ↑ Association with Asperger syndrome | [131] | |||
| Autistic children | CYP1A1 rs1048943 and rs4646422
CYP1A2*1C (rs2069514) CYP1A2*1F (rs762551) | Associated in Thai children and adolescents with autism spectrum disorder | [132] | ||
| ↑ CYP1A2*1C (rs2069514), CYP1A2*4 (rs72547516), and CYP1A2*6 (rs28399424) → ↓ CYP1A2 activity → ↑ levels of melatonin | Loss of circadian rhythms and loss of supplemental melatonin effectiveness | [134] |
5. The Involvement of AhR/CYP1 Pathways in Autism Development and Treatment by Drugs
5.1. Sulforaphane
5.2. Valproic Acid
5.3. Resveratrol
5.4. Metformin
5.5. Haloperidol
6. Conclusions and Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tebartz van Elst, L.; Pick, M.; Biscaldi, M.; Fangmeier, T.; Riedel, A. High-functioning autism spectrum disorder as a basic disorder in adult psychiatry and psychotherapy: Psychopathological presentation, clinical relevance and therapeutic concepts. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, S189–S196. [Google Scholar] [CrossRef]
- Kim, S.K. Recent update of autism spectrum disorders. Korean J. Pediatr. 2015, 58, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.N.; Schendel, D.E.; Parner, E.T. Explaining the increase in the prevalence of autism spectrum disorders: The proportion attributable to changes in reporting practices. JAMA Pediatr. 2015, 169, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doernberg, E.; Hollander, E. Neurodevelopmental Disorders (ASD and ADHD): DSM-5, ICD-10, and ICD-11. CNS Spectr. 2016, 21, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posar, A.; Resca, F.; Visconti, P. Autism according to diagnostic and statistical manual of mental disorders 5(th) edition: The need for further improvements. J. Pediatr. Neurosci. 2015, 10, 146–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Nardone, S.; Elliott, E. The Interaction between the Immune System and Epigenetics in the Etiology of Autism Spectrum Disorders. Front. Neurosci. 2016, 10, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, G.; Rice, C.E. The Complex Etiology of Autism Presents Challenges in Risk Communication. Pediatrics 2016, 137, e20152703. [Google Scholar] [CrossRef] [Green Version]
- Bourgeron, T. Current knowledge on the genetics of autism and propositions for future research. C. R. Biol. 2016, 339, 300–307. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Crespi, B.J. Autism, psychosis, and genomic imprinting: Recent discoveries and conundrums. Curr. Opin. Behav. Sci. 2019, 25, 1–7. [Google Scholar] [CrossRef]
- Duffney, L.J.; Valdez, P.; Tremblay, M.W.; Cao, X.; Montgomery, S.; McConkie-Rosell, A.; Jiang, Y.H. Epigenetics and autism spectrum disorder: A report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, P.M.; Matte, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsuzaki, H.; Iwata, K.; Kameno, Y.; Shimmura, C.; Kawai, S.; Yoshihara, Y.; Wakuda, T.; Takebayashi, K.; Takagai, S.; et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS ONE 2011, 6, e20470. [Google Scholar] [CrossRef]
- Napolioni, V.; Ober-Reynolds, B.; Szelinger, S.; Corneveaux, J.J.; Pawlowski, T.; Ober-Reynolds, S.; Kirwan, J.; Persico, A.M.; Melmed, R.D.; Craig, D.W.; et al. Plasma cytokine profiling in sibling pairs discordant for autism spectrum disorder. J. Neuroinflamm. 2013, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Qasem, H.; Al-Ayadhi, L.; Bjorklund, G.; Chirumbolo, S.; El-Ansary, A. Impaired lipid metabolism markers to assess the risk of neuroinflammation in autism spectrum disorder. Metab. Brain Dis. 2018, 33, 1141–1153. [Google Scholar] [CrossRef]
- Levie, D.; Korevaar, T.I.M.; Bath, S.C.; Dalmau-Bueno, A.; Murcia, M.; Espada, M.; Dineva, M.; Ibarluzea, J.M.; Sunyer, J.; Tiemeier, H.; et al. Thyroid Function in Early Pregnancy, Child IQ, and Autistic Traits: A Meta-Analysis of Individual Participant Data. J. Clin. Endocrinol. Metab. 2018, 103, 2967–2979. [Google Scholar] [CrossRef]
- Akhtar, S.; Hourani, S.; Therachiyil, L.; Al-Dhfyan, A.; Agouni, A.; Zeidan, A.; Uddin, S.; Korashy, H.M. Epigenetic Regulations of Cancer Stem Cells by the Aryl Hydrocarbon Receptor Pathway. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Korashy, H.M.; El-Kadi, A.O. Differential effects of mercury, lead and copper on the constitutive and inducible expression of aryl hydrocarbon receptor (AHR)-regulated genes in cultured hepatoma Hepa 1c1c7 cells. Toxicology 2004, 201, 153–172. [Google Scholar] [CrossRef]
- Lee, B.J.; Kim, B.; Lee, K. Air pollution exposure and cardiovascular disease. Toxicol. Res. 2014, 30, 71–75. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Al-Kindi, S.G.; Brook, R.D. Air Pollution and Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2054–2070. [Google Scholar] [CrossRef]
- Kim, H.B.; Shim, J.Y.; Park, B.; Lee, Y.J. Long-Term Exposure to Air Pollutants and Cancer Mortality: A Meta-Analysis of Cohort Studies. Int. J. Environ. Res. Public Health 2018, 15, 2608. [Google Scholar] [CrossRef] [Green Version]
- Boffetta, P. Human cancer from environmental pollutants: The epidemiological evidence. Mutat. Res. 2006, 608, 157–162. [Google Scholar] [CrossRef]
- Kurt, O.K.; Zhang, J.; Pinkerton, K.E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 2016, 22, 138–143. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Brook, R.D. Air pollution and type 2 diabetes: Mechanistic insights. Diabetes 2012, 61, 3037–3045. [Google Scholar] [CrossRef] [Green Version]
- Genc, S.; Zadeoglulari, Z.; Fuss, S.H.; Genc, K. The adverse effects of air pollution on the nervous system. J. Toxicol. 2012, 2012, 782462. [Google Scholar] [CrossRef] [Green Version]
- Webb, E.; Moon, J.; Dyrszka, L.; Rodriguez, B.; Cox, C.; Patisaul, H.; Bushkin, S.; London, E. Neurodevelopmental and neurological effects of chemicals associated with unconventional oil and natural gas operations and their potential effects on infants and children. Rev. Environ. Health 2018, 33, 3–29. [Google Scholar] [CrossRef] [Green Version]
- McGuinn, L.; Windham, G.; Kalkbrenner, A.; Di, Q.; Schwartz, J.; Rappold, A.; Neas, L.; Richardson, D.; Schieve, L.; Daniels, J. Pre- and Postnatal Air Pollution Exposure and Autism Spectrum Disorder: Findings from the Study to Explore Early Development. Environ. Epidemiol. 2019, 3, 267. [Google Scholar] [CrossRef]
- Volk, H.E.; Kerin, T.; Lurmann, F.; Hertz-Picciotto, I.; McConnell, R.; Campbell, D.B. Autism spectrum disorder: Interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology 2014, 25, 44–47. [Google Scholar] [CrossRef] [Green Version]
- Ritz, B.; Liew, Z.; Yan, Q.; Cuia, X.; Virk, J.; Ketzel, M.; Raaschou-Nielsen, O. Air pollution and autism in Denmark. Environ. Epidemiol. 2018, 2, e028. [Google Scholar] [CrossRef]
- Carter, C.J.; Blizard, R.A. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochem. Int. 2016. [Google Scholar] [CrossRef]
- Macedoni-Luksic, M.; Gosar, D.; Bjorklund, G.; Orazem, J.; Kodric, J.; Lesnik-Musek, P.; Zupancic, M.; France-Stiglic, A.; Sesek-Briski, A.; Neubauer, D.; et al. Levels of metals in the blood and specific porphyrins in the urine in children with autism spectrum disorders. Biol. Trace Elem. Res. 2015, 163, 2–10. [Google Scholar] [CrossRef]
- Adams, K.; Greenbaum, D.S.; Shaikh, R.; van Erp, A.M.; Russell, A.G. Particulate matter components, sources, and health: Systematic approaches to testing effects. J. Air Waste Manag. Assoc. 2015, 65, 544–558. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Pokorna, P.; Hovorka, J.; Krouzek, J.; Hopke, P.K. Particulate matter source apportionment in a village situated in industrial region of Central Europe. J. Air Waste Manag. Assoc. 2013, 63, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Garciduenas, L.; Kulesza, R.J.; Doty, R.L.; D’Angiulli, A.; Torres-Jardon, R. Megacities air pollution problems: Mexico City Metropolitan Area critical issues on the central nervous system pediatric impact. Environ. Res. 2015, 137, 157–169. [Google Scholar] [CrossRef]
- Chang, Y.C.; Cole, T.B.; Costa, L.G. Prenatal and early-life diesel exhaust exposure causes autism-like behavioral changes in mice. Part Fibre Toxicol. 2018, 15, 18. [Google Scholar] [CrossRef]
- Talbott, E.O.; Arena, V.C.; Rager, J.R.; Clougherty, J.E.; Michanowicz, D.R.; Sharma, R.K.; Stacy, S.L. Fine particulate matter and the risk of autism spectrum disorder. Environ. Res. 2015, 140, 414–420. [Google Scholar] [CrossRef]
- Kalkbrenner, A.E.; Windham, G.C.; Serre, M.L.; Akita, Y.; Wang, X.; Hoffman, K.; Thayer, B.P.; Daniels, J.L. Particulate matter exposure, prenatal and postnatal windows of susceptibility, and autism spectrum disorders. Epidemiology 2015, 26, 30–42. [Google Scholar] [CrossRef]
- Chen, G.; Jin, Z.; Li, S.; Jin, X.; Tong, S.; Liu, S.; Yang, Y.; Huang, H.; Guo, Y. Early life exposure to particulate matter air pollution (PM1, PM2.5 and PM10) and autism in Shanghai, China: A case-control study. Environ. Int. 2018, 121, 1121–1127. [Google Scholar] [CrossRef]
- Morales-Suarez-Varela, M.; Peraita-Costa, I.; Llopis-Gonzalez, A. Systematic review of the association between particulate matter exposure and autism spectrum disorders. Environ. Res. 2017, 153, 150–160. [Google Scholar] [CrossRef]
- Chun, H.; Leung, C.; Wen, S.W.; McDonald, J.; Shin, H.H. Maternal exposure to air pollution and risk of autism in children: A systematic review and meta-analysis. Environ. Pollut. 2020, 256, 113307. [Google Scholar] [CrossRef]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Fang, S.; Li, G. The association between particulate matter 2.5 exposure and children with autism spectrum disorder. Int. J. Dev. Neurosci. 2019, 75, 59–63. [Google Scholar] [CrossRef]
- Li, K.; Li, L.; Cui, B.; Gai, Z.; Li, Q.; Wang, S.; Yan, J.; Lin, B.; Tian, L.; Liu, H.; et al. Early Postnatal Exposure to Airborne Fine Particulate Matter Induces Autism-like Phenotypes in Male Rats. Toxicol. Sci. 2018, 162, 189–199. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Meng, G.; Nie, Z.; Zhou, R.; Cheng, W.; Wu, X.; Feng, Y.; Wang, Y. Redox/methylation mediated abnormal DNA methylation as regulators of ambient fine particulate matter-induced neurodevelopment related impairment in human neuronal cells. Sci. Rep. 2016, 6, 33402. [Google Scholar] [CrossRef] [Green Version]
- Jarup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Saghazadeh, A.; Rezaei, N. Systematic review and meta-analysis links autism and toxic metals and highlights the impact of country development status: Higher blood and erythrocyte levels for mercury and lead, and higher hair antimony, cadmium, lead, and mercury. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 340–368. [Google Scholar] [CrossRef]
- Mohamed Fel, B.; Zaky, E.A.; El-Sayed, A.B.; Elhossieny, R.M.; Zahra, S.S.; Salah Eldin, W.; Youssef, W.Y.; Khaled, R.A.; Youssef, A.M. Assessment of Hair Aluminum, Lead, and Mercury in a Sample of Autistic Egyptian Children: Environmental Risk Factors of Heavy Metals in Autism. Behav. Neurol. 2015, 2015, 545674. [Google Scholar] [CrossRef] [Green Version]
- Kardas, F.; Bayram, A.K.; Demirci, E.; Akin, L.; Ozmen, S.; Kendirci, M.; Canpolat, M.; Oztop, D.B.; Narin, F.; Gumus, H.; et al. Increased Serum Phthalates (MEHP, DEHP) and Bisphenol A Concentrations in Children With Autism Spectrum Disorder: The Role of Endocrine Disruptors in Autism Etiopathogenesis. J. Child Neurol. 2016, 31, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.P.; Schluter, M.D.; Steer, R.A.; Guo, L.; Ming, X. Bisphenol A Exposure in Children With Autism Spectrum Disorders. Autism Res. 2015, 8, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeddi, M.Z.; Janani, L.; Memari, A.H.; Akhondzadeh, S.; Yunesian, M. The role of phthalate esters in autism development: A systematic review. Environ. Res. 2016, 151, 493–504. [Google Scholar] [CrossRef]
- Shin, H.M.; Schmidt, R.J.; Tancredi, D.; Barkoski, J.; Ozonoff, S.; Bennett, D.H.; Hertz-Picciotto, I. Prenatal exposure to phthalates and autism spectrum disorder in the MARBLES study. Environ. Health 2018, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Q.; Loganath, A.; Chong, Y.S.; Tan, J.; Obbard, J.P. Persistent organic pollutants and adverse health effects in humans. J Toxicol. Environ. Health A 2006, 69, 1987–2005. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.A.; Sjödin, A.; Yoshida, C.K.; Zerbo, O.; Kharrazi, M.; Windham, G.C. Polychlorinated Biphenyl and Organochlorine Pesticide Concentrations in Maternal Mid-Pregnancy Serum Samples: Association with Autism Spectrum Disorder and Intellectual Disability. Environ. Health Perspect. 2017, 125, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.M.; Woods, R.; Chi, L.H.; Schmidt, R.J.; Pessah, I.N.; Kostyniak, P.J.; LaSalle, J.M. Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder. Environ. Mol. Mutagen 2012, 53, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Granillo, L.; Sethi, S.; Keil, K.P.; Lin, Y.; Ozonoff, S.; Iosif, A.M.; Puschner, B.; Schmidt, R.J. Polychlorinated biphenyls influence on autism spectrum disorder risk in the MARBLES cohort. Environ. Res. 2019, 171, 177–184. [Google Scholar] [CrossRef]
- Nowack, N.; Wittsiepe, J.; Kasper-Sonnenberg, M.; Wilhelm, M.; Scholmerich, A. Influence of Low-Level Prenatal Exposure to PCDD/Fs and PCBs on Empathizing, Systemizing and Autistic Traits: Results from the Duisburg Birth Cohort Study. PLoS ONE 2015, 10, e0129906. [Google Scholar] [CrossRef]
- Guo, Z.; Xie, H.Q.; Zhang, P.; Luo, Y.; Xu, T.; Liu, Y.; Fu, H.; Xu, L.; Valsami-Jones, E.; Boksa, P.; et al. Dioxins as potential risk factors for autism spectrum disorder. Environ. Int. 2018, 121, 906–915. [Google Scholar] [CrossRef]
- Nishijo, M.; Pham, T.T.; Nguyen, A.T.; Tran, N.N.; Nakagawa, H.; Hoang, L.V.; Tran, A.H.; Morikawa, Y.; Ho, M.D.; Kido, T.; et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin in breast milk increases autistic traits of 3-year-old children in Vietnam. Mol. Psychiatry 2014, 19, 1220–1226. [Google Scholar] [CrossRef]
- Kerzee, J.K.; Ramos, K.S. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: Role of the Ahr bHLH/PAS transcription factor. Circ. Res. 2001, 89, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, M.L.; Gottlicher, M.; Gustafsson, J.A.; Poellinger, L. Definition of a novel ligand binding domain of a nuclear bHLH receptor: Co-localization of ligand and hsp90 binding activities within the regulable inactivation domain of the dioxin receptor. EMBO J. 1993, 12, 4169–4179. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The aryl hydrocarbon receptor: A multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Korashy, H.M.; Shayeganpour, A.; Brocks, D.R.; El-Kadi, A.O. Induction of cytochrome P450 1A1 by ketoconazole and itraconazole but not fluconazole in murine and human hepatoma cell lines. Toxicol. Sci. 2007, 97, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korashy, H.M.; El-Kadi, A.O. Modulation of TCDD-mediated induction of cytochrome P450 1A1 by mercury, lead, and copper in human HepG2 cell line. Toxicol. In Vitro 2008, 22, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Pappas, B.; Yang, Y.; Wang, Y.; Kim, K.; Chung, H.J.; Cheung, M.; Ngo, K.; Shinn, A.; Chan, W.K. p23 protects the human aryl hydrocarbon receptor from degradation via a heat shock protein 90-independent mechanism. Biochem. Pharmacol. 2018, 152, 34–44. [Google Scholar] [CrossRef]
- Tsuji, N.; Fukuda, K.; Nagata, Y.; Okada, H.; Haga, A.; Hatakeyama, S.; Yoshida, S.; Okamoto, T.; Hosaka, M.; Sekine, K.; et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 2014, 4, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Uppstad, H.; Ovrebo, S.; Haugen, A.; Mollerup, S. Importance of CYP1A1 and CYP1B1 in bioactivation of benzo[a]pyrene in human lung cell lines. Toxicol. Lett. 2010, 192, 221–228. [Google Scholar] [CrossRef]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef]
- Yi, T.; Wang, J.; Zhu, K.; Tang, Y.; Huang, S.; Shui, X.; Ding, Y.; Chen, C.; Lei, W. Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 6058784. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Meng, Q.; Zhang, Z.; Yi, T.; He, Y.; Zheng, J.; Lei, W. Aryl hydrocarbon receptor pathway: Role, regulation and intervention in atherosclerosis therapy (Review). Mol. Med. Rep. 2019, 20, 4763–4773. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, M.; Malek, G. The Aryl Hydrocarbon Receptor: A Mediator and Potential Therapeutic Target for Ocular and Non-Ocular Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6777. [Google Scholar] [CrossRef]
- Long, M.; Ghisari, M.; Kjeldsen, L.; Wielsoe, M.; Norgaard-Pedersen, B.; Mortensen, E.L.; Abdallah, M.W.; Bonefeld-Jorgensen, E.C. Autism spectrum disorders, endocrine disrupting compounds, and heavy metals in amniotic fluid: A case-control study. Mol. Autism 2019, 10, 1. [Google Scholar] [CrossRef]
- Rybicki, B.A.; Nock, N.L.; Savera, A.T.; Tang, D.; Rundle, A. Polycyclic aromatic hydrocarbon-DNA adduct formation in prostate carcinogenesis. Cancer Lett. 2006, 239, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Mangal, D.; Frey, A.J.; Harvey, R.G.; Blair, I.A.; Penning, T.M. Aryl hydrocarbon receptor facilitates DNA strand breaks and 8-oxo-2′-deoxyguanosine formation by the aldo-keto reductase product benzo[a]pyrene-7,8-dione. J. Biol. Chem. 2009, 284, 29725–29734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ansary, A.; Cannell, J.J.; Bjorklund, G.; Bhat, R.S.; Al Dbass, A.M.; Alfawaz, H.A.; Chirumbolo, S.; Al-Ayadhi, L. In the search for reliable biomarkers for the early diagnosis of autism spectrum disorder: The role of vitamin D. Metab. Brain Dis. 2018, 33, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Mordaunt, C.E.; Park, B.Y.; Bakulski, K.M.; Feinberg, J.I.; Croen, L.A.; Ladd-Acosta, C.; Newschaffer, C.J.; Volk, H.E.; Ozonoff, S.; Hertz-Picciotto, I.; et al. A meta-analysis of two high-risk prospective cohort studies reveals autism-specific transcriptional changes to chromatin, autoimmune, and environmental response genes in umbilical cord blood. Mol. Autism 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.L.; Hsu, C.C.; Gladen, B.C.; Rogan, W.J. In utero PCB/PCDF exposure: Relation of developmental delay to dysmorphology and dose. Neurotoxicol. Teratol. 1991, 13, 195–202. [Google Scholar] [CrossRef]
- Kimura, E.; Kubo, K.I.; Endo, T.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Excessive activation of AhR signaling disrupts neuronal migration in the hippocampal CA1 region in the developing mouse. J. Toxicol. Sci. 2017, 42, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The CHARGE study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemonnier, E.; Grandgeorge, M.; Jacobzone-Lévêque, C.; Bessaguet, C.; Peudenier, S.; Misery, L. Red dermographism in autism spectrum disorders: A clinical sign of cholinergic dysfunction? Res. Autism Spectr. Disord. 2013, 7, 601–605. [Google Scholar] [CrossRef]
- Bali, Y.A.; Kaikai, N.E.; Ba-M’hamed, S.; Bennis, M. Learning and memory impairments associated to acetylcholinesterase inhibition and oxidative stress following glyphosate based-herbicide exposure in mice. Toxicology 2019, 415, 18–25. [Google Scholar] [CrossRef]
- Xu, H.M.; Xie, H.Q.; Tao, W.Q.; Zhou, Z.G.; Li, S.Z.; Zhao, B. Dioxin and dioxin-like compounds suppress acetylcholinesterase activity via transcriptional downregulations in vitro. J. Mol. Neurosci. 2014, 53, 417–423. [Google Scholar] [CrossRef]
- Ahmed, R.G. Perinatal TCDD exposure alters developmental neuroendocrine system. Food Chem. Toxicol. 2011, 49, 1276–1284. [Google Scholar] [CrossRef]
- Xie, H.Q.; Xu, H.M.; Fu, H.L.; Hu, Q.; Tian, W.J.; Pei, X.H.; Zhao, B. AhR-mediated effects of dioxin on neuronal acetylcholinesterase expression in vitro. Environ. Health Perspect. 2013, 121, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhong, H.; Wang, C.; Gao, D.; Zhou, Y.; Zuo, Z. Maternal exposure to the water soluble fraction of crude oil, lead and their mixture induces autism-like behavioral deficits in zebrafish (Danio rerio) larvae. Ecotoxicol. Environ. Saf. 2016, 134P1, 23–30. [Google Scholar] [CrossRef]
- Glazer, L.; Hahn, M.E.; Aluru, N. Delayed effects of developmental exposure to low levels of the aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl (PCB126) on adult zebrafish behavior. Neurotoxicology 2016, 52, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Colter, B.T.; Garber, H.F.; Fleming, S.M.; Fowler, J.P.; Harding, G.D.; Hooven, M.K.; Howes, A.A.; Infante, S.K.; Lang, A.L.; MacDougall, M.C.; et al. Ahr and Cyp1a2 genotypes both affect susceptibility to motor deficits following gestational and lactational exposure to polychlorinated biphenyls. Neurotoxicology 2018, 65, 125–134. [Google Scholar] [CrossRef]
- Klinefelter, K.; Hooven, M.K.; Bates, C.; Colter, B.T.; Dailey, A.; Infante, S.K.; Kania-Korwel, I.; Lehmler, H.J.; Lopez-Juarez, A.; Ludwig, C.P.; et al. Genetic differences in the aryl hydrocarbon receptor and CYP1A2 affect sensitivity to developmental polychlorinated biphenyl exposure in mice: Relevance to studies of human neurological disorders. Mamm. Genome 2018, 29, 112–127. [Google Scholar] [CrossRef]
- Yoon, S.H.; Choi, J.; Lee, W.J.; Do, J.T. Genetic and Epigenetic Etiology Underlying Autism Spectrum Disorder. J. Clin. Med. 2020, 9, 966. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, A.A.; Liu, G.; Kay, S.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell. Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Siniscalco, D.; Cirillo, A.; Bradstreet, J.J.; Antonucci, N. Epigenetic findings in autism: New perspectives for therapy. Int. J. Environ. Res. Public Health 2013, 10, 4261–4273. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.E.; Davies, T.J.; Mc Auley, M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018, 77, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpyleva, S.; Ivanovsky, S.; de Conti, A.; Melnyk, S.; Tryndyak, V.; Beland, F.A.; James, S.J.; Pogribny, I.P. Cerebellar oxidative DNA damage and altered DNA methylation in the BTBR T+tf/J mouse model of autism and similarities with human post mortem cerebellum. PLoS ONE 2014, 9, e113712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, K.P.; Lein, P.J. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenet. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Su, K.-Y.; Li, M.-C.; Lee, N.-W.; Ho, B.-C.; Cheng, C.-L.; Chuang, Y.-C.; Yu, S.-L.; Guo, Y.L. Perinatal polychlorinated biphenyls and polychlorinated dibenzofurans exposure are associated with DNA methylation changes lasting to early adulthood: Findings from Yucheng second generation. Environ. Res. 2019, 170, 481–486. [Google Scholar] [CrossRef]
- Joubert, B.R.; Haberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, O.; Cupul-Uicab, L.A.; et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.; Craig, J.M.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2014, 9, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Mochizuki, K. Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders. Int. J. Environ. Res. Public Health 2016, 13, 504. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Kawaguchi, A.; Miura, R.; Kobayashi, S.; Tran, N.Q.V.; Kobayashi, S.; Miyashita, C.; Araki, A.; Kubota, T.; Yamagata, Z.; et al. Association between DNA methylation in cord blood and maternal smoking: The Hokkaido Study on Environment and Children’s Health. Sci. Rep. 2018, 8, 5654. [Google Scholar] [CrossRef]
- Desaulniers, D.; Xiao, G.H.; Lian, H.; Feng, Y.L.; Zhu, J.; Nakai, J.; Bowers, W.J. Effects of mixtures of polychlorinated biphenyls, methylmercury, and organochlorine pesticides on hepatic DNA methylation in prepubertal female Sprague-Dawley rats. Int. J. Toxicol. 2009, 28, 294–307. [Google Scholar] [CrossRef]
- Desaulniers, D.; Xiao, G.H.; Leingartner, K.; Chu, I.; Musicki, B.; Tsang, B.K. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-o-methyltransferase in prepubertal female Sprague-Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol. Sci. 2005, 86, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Nayyar, T.; Zawia, N.H.; Hood, D.B. Transplacental effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the temporal modulation of Sp1 DNA binding in the developing cerebral cortex and cerebellum. Exp. Toxicol. Pathol. 2002, 53, 461–468. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hannigan, B.; Crews, D. Embryonic PCB exposure alters phenotypic, genetic, and epigenetic profiles in turtle sex determination, a biomarker of environmental contamination. Endocrinology 2014, 155, 4168–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.M.; Goetz, B.M.; Gore, A.C. Dynamic postnatal developmental and sex-specific neuroendocrine effects of prenatal polychlorinated biphenyls in rats. Mol. Endocrinol. 2014, 28, 99–115. [Google Scholar] [CrossRef]
- Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 99. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Karlic, R.; Chung, H.R.; Lasserre, J.; Vlahovicek, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [Green Version]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [Green Version]
- Henningsson, S.; Jonsson, L.; Ljunggren, E.; Westberg, L.; Gillberg, C.; Råstam, M.; Anckarsäter, H.; Nygren, G.; Landén, M.; Thuresson, K.; et al. Possible association between the androgen receptor gene and autism spectrum disorder. Psychoneuroendocrinology 2009, 34, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Geier, M.R.; Geier, D.A. The potential importance of steroids in the treatment of autistic spectrum disorders and other disorders involving mercury toxicity. Med. Hypotheses 2005, 64, 946–954. [Google Scholar] [CrossRef]
- Geier, D.A.; Kern, J.K.; King, P.G.; Sykes, L.K.; Geier, M.R. An evaluation of the role and treatment of elevated male hormones in autism spectrum disorders. Acta Neurobiol. Exp. 2012, 72, 1–17. [Google Scholar]
- Baron-Cohen, S.; Knickmeyer, R.C.; Belmonte, M.K. Sex differences in the brain: Implications for explaining autism. Science 2005, 310, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan-Young, R.M. Hormones, context, and “brain gender”: A review of evidence from congenital adrenal hyperplasia. Soc. Sci. Med. 2012, 74, 1738–1744. [Google Scholar] [CrossRef]
- Casati, L.; Sendra, R.; Poletti, A.; Negri-Cesi, P.; Celotti, F. Androgen receptor activation by polychlorinated biphenyls: Epigenetic effects mediated by the histone demethylase Jarid1b. Epigenetics 2013, 8, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbarian, S.; Huang, H.S. Epigenetic regulation in human brain-focus on histone lysine methylation. Biol. Psychiatry 2009, 65, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Vismara, G.; Simonini, F.; Onesto, E.; Bignamini, M.; Miceli, V.; Martini, L.; Poletti, A. Androgens inhibit androgen receptor promoter activation in motor neurons. Neurobiol. Dis. 2009, 33, 395–404. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Anitha, A.; Thanseem, I. microRNA and Autism. Adv. Exp. Med. Biol. 2015, 888, 71–83. [Google Scholar] [CrossRef]
- Schepici, G.; Cavalli, E.; Bramanti, P.; Mazzon, E. Autism Spectrum Disorder and miRNA: An Overview of Experimental Models. Brain Sci. 2019, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.P.; Singh, U.P.; Guan, H.; Nagarkatti, P.; Nagarkatti, M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS ONE 2012, 7, e45054. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.X.; Nishitani, S.; Iwanaga, R.; Matsuzaki, J.; Kawasaki, C.; Tochigi, M.; Sasaki, T.; Kato, N.; Shinohara, K. Association of Aryl Hydrocarbon Receptor-Related Gene Variants with the Severity of Autism Spectrum Disorders. Front. Psychiatry 2016, 7, 184. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, A.; Warrier, V.; Baron-Cohen, S.; Chakrabarti, B. Genetic variant rs17225178 in the ARNT2 gene is associated with Asperger Syndrome. Mol. Autism 2015, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medhasi, S.; Pasomsub, E.; Vanwong, N.; Ngamsamut, N.; Puangpetch, A.; Chamnanphon, M.; Hongkaew, Y.; Limsila, P.; Pinthong, D.; Sukasem, C. Clinically relevant genetic variants of drug-metabolizing enzyme and transporter genes detected in Thai children and adolescents with autism spectrum disorder. Neuropsychiatr. Dis. Treat. 2016, 12, 843–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, A.L.; Truong, L.; Marvel, S.W.; Reif, D.M.; Garcia, A.; Lu, C.; Simonich, M.T.; Teeguarden, J.G.; Tanguay, R.L. Transgenerational inheritance of neurobehavioral and physiological deficits from developmental exposure to benzo[a]pyrene in zebrafish. Toxicol. Appl. Pharmacol. 2017, 329, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Veatch, O.J.; Pendergast, J.S.; Allen, M.J.; Leu, R.M.; Johnson, C.H.; Elsea, S.H.; Malow, B.A. Genetic variation in melatonin pathway enzymes in children with autism spectrum disorder and comorbid sleep onset delay. J. Autism Dev. Disord. 2015, 45, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Lynch, R.; Diggins, E.L.; Connors, S.L.; Zimmerman, A.W.; Singh, K.; Liu, H.; Talalay, P.; Fahey, J.W. Sulforaphane from Broccoli Reduces Symptoms of Autism: A Follow-up Case Series from a Randomized Double-blind Study. Glob. Adv. Health Med. 2017, 6, 2164957X17735826. [Google Scholar] [CrossRef]
- Santin-Marquez, R.; Alarcon-Aguilar, A.; Lopez-Diazguerrero, N.E.; Chondrogianni, N.; Konigsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef] [Green Version]
- Korashy, H.M.; Brocks, D.R.; El-Kadi, A.O. Induction of the NAD(P)H:quinone oxidoreductase 1 by ketoconazole and itraconazole: A mechanism of cancer chemoprotection. Cancer Lett. 2007, 258, 135–143. [Google Scholar] [CrossRef]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker Exploration in Human Peripheral Blood Mononuclear Cells for Monitoring Sulforaphane Treatment Responses in Autism Spectrum Disorder. Sci. Rep. 2020, 10, 5822. [Google Scholar] [CrossRef] [Green Version]
- Skupinska, K.; Misiewicz-Krzeminska, I.; Stypulkowski, R.; Lubelska, K.; Kasprzycka-Guttman, T. Sulforaphane and its analogues inhibit CYP1A1 and CYP1A2 activity induced by benzo[a]pyrene. J. Biochem. Mol. Toxicol. 2009, 23, 18–28. [Google Scholar] [CrossRef]
- Abdull Razis, A.F.; Hanlon, N.; Soltys, E.; Krizova, V.; Iori, R.; Plant, K.E.; Plant, N.; Ioannides, C. The naturally occurring aliphatic isothiocyanates sulforaphane and erucin are weak agonists but potent non-competitive antagonists of the aryl hydrocarbon receptor. Arch. Toxicol. 2012, 86, 1505–1514. [Google Scholar] [CrossRef] [Green Version]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Napoli, E.; Wong, S.; Giulivi, C. Evidence of reactive oxygen species-mediated damage to mitochondrial DNA in children with typical autism. Mol. Autism 2013, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, G.; Kim, Y. Sulforaphane treatment for autism spectrum disorder: A systematic review. EXCLI J. 2020, 19, 892–903. [Google Scholar] [CrossRef]
- Liu, F.; Horton-Sparks, K.; Hull, V.; Li, R.W.; Martinez-Cerdeno, V. The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 2018, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Gronborg, T.K.; Sorensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Roullet, F.I.; Lai, J.K.; Foster, J.A. In utero exposure to valproic acid and autism—A current review of clinical and animal studies. Neurotoxicol. Teratol. 2013, 36, 47–56. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Matsuo, K.; Yabuki, Y.; Fukunaga, K. 5-aminolevulinic acid inhibits oxidative stress and ameliorates autistic-like behaviors in prenatal valproic acid-exposed rats. Neuropharmacology 2020, 168, 107975. [Google Scholar] [CrossRef]
- You, D.; Wen, X.; Gorczyca, L.; Morris, A.; Richardson, J.R.; Aleksunes, L.M. Increased MDR1 Transporter Expression in Human Brain Endothelial Cells Through Enhanced Histone Acetylation and Activation of Aryl Hydrocarbon Receptor Signaling. Mol. Neurobiol. 2019, 56, 6986–7002. [Google Scholar] [CrossRef] [PubMed]
- Van Breda, S.G.J.; Claessen, S.M.H.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.; Kleinjans, J.C.S. Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology 2018, 393, 160–170. [Google Scholar] [CrossRef]
- Griffiths, A.L.; Schroeder, J.C. Effects of Resveratrol on AhR Activity In Cells Treated with Benzo[A]Pyrene or Indigo. Ga. J. Sci. 2017, 75, 44. [Google Scholar]
- Licznerska, B.; Szaefer, H.; Wierzchowski, M.; Sobierajska, H.; Baer-Dubowska, W. Resveratrol and its methoxy derivatives modulate the expression of estrogen metabolism enzymes in breast epithelial cells by AhR down-regulation. Mol. Cell. Biochem. 2017, 425, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Bambini-Junior, V.; Zanatta, G.; Della Flora Nunes, G.; Mueller de Melo, G.; Michels, M.; Fontes-Dutra, M.; Nogueira Freire, V.; Riesgo, R.; Gottfried, C. Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci. Lett. 2014, 583, 176–181. [Google Scholar] [CrossRef]
- Fontes-Dutra, M.; Santos-Terra, J.; Deckmann, I.; Brum Schwingel, G.; Della-Flora Nunes, G.; Hirsch, M.M.; Bauer-Negrini, G.; Riesgo, R.S.; Bambini-Junior, V.; Hedin-Pereira, C.; et al. Resveratrol Prevents Cellular and Behavioral Sensory Alterations in the Animal Model of Autism Induced by Valproic Acid. Front. Synaptic Neurosci. 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, R.; Kuhad, A. Resveratrol suppresses neuroinflammation in the experimental paradigm of autism spectrum disorders. Neurochem. Int. 2017, 103, 8–23. [Google Scholar] [CrossRef]
- Do, M.T.; Kim, H.G.; Tran, T.T.; Khanal, T.; Choi, J.H.; Chung, Y.C.; Jeong, T.C.; Jeong, H.G. Metformin suppresses CYP1A1 and CYP1B1 expression in breast cancer cells by down-regulating aryl hydrocarbon receptor expression. Toxicol. Appl. Pharmacol. 2014, 280, 138–148. [Google Scholar] [CrossRef]
- Ishola, I.O.; Balogun, A.O.; Adeyemi, O.O. Novel potential of metformin on valproic acid-induced autism spectrum disorder in rats: Involvement of antioxidant defence system. Fundam. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Ghebeh, H.; Alhaider, A.A.; El-Kadi, A.O.; Soshilov, A.A.; Denison, M.S.; Ansari, M.A.; Korashy, H.M. Metformin inhibits 7,12-dimethylbenz[a]anthracene-induced breast carcinogenesis and adduct formation in human breast cells by inhibiting the cytochrome P4501A1/aryl hydrocarbon receptor signaling pathway. Toxicol. Appl. Pharmacol. 2015, 284, 217–226. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, M.; Wagner, K.D. Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacol. Bull. 2016, 46, 18–41. [Google Scholar] [PubMed]
- Fitzpatrick, S.E.; Srivorakiat, L.; Wink, L.K.; Pedapati, E.V.; Erickson, C.A. Aggression in autism spectrum disorder: Presentation and treatment options. Neuropsychiatr. Dis. Treat. 2016, 12, 1525–1538. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.T.; Campbell, M.; Grega, D.M.; Perry, R.; Small, A.M.; Green, W.H. Haloperidol in the treatment of infantile autism: Effects on learning and behavioral symptoms. Am. J. Psychiatry 1984, 141, 1195–1202. [Google Scholar] [CrossRef]
- Goel, R.; Hong, J.S.; Findling, R.L.; Ji, N.Y. An update on pharmacotherapy of autism spectrum disorder in children and adolescents. Int. Rev. Psychiatry 2018, 30, 78–95. [Google Scholar] [CrossRef]
- Ramadoss, P.; Marcus, C.; Perdew, G.H. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2005, 1, 9–21. [Google Scholar] [CrossRef]
- Gunes, A.; Dahl, M.L. Variation in CYP1A2 activity and its clinical implications: Influence of environmental factors and genetic polymorphisms. Pharmacogenomics 2008, 9, 625–637. [Google Scholar] [CrossRef]
- Tyler, M.W.; Zaldivar-Diez, J.; Haggarty, S.J. Classics in Chemical Neuroscience: Haloperidol. ACS Chem. Neurosci. 2017, 8, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Yasui-Furukori, N.; Kondo, T.; Mihara, K.; Inoue, Y.; Kaneko, S. Fluvoxamine dose-dependent interaction with haloperidol and the effects on negative symptoms in schizophrenia. Psychopharmacology 2004, 171, 223–227. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhulkifle, H.; Agouni, A.; Zeidan, A.; Al-Kuwari, M.S.; Parray, A.; Tolefat, M.; Korashy, H.M. Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 9258. https://doi.org/10.3390/ijms22179258
Dhulkifle H, Agouni A, Zeidan A, Al-Kuwari MS, Parray A, Tolefat M, Korashy HM. Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder. International Journal of Molecular Sciences. 2021; 22(17):9258. https://doi.org/10.3390/ijms22179258
Chicago/Turabian StyleDhulkifle, Hevna, Abdelali Agouni, Asad Zeidan, Mohammed Saif Al-Kuwari, Aijaz Parray, Mohamed Tolefat, and Hesham M. Korashy. 2021. "Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder" International Journal of Molecular Sciences 22, no. 17: 9258. https://doi.org/10.3390/ijms22179258