
Epigenetic Clocks Are Not Accelerated in COVID-19 Patients

 , ,
, ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
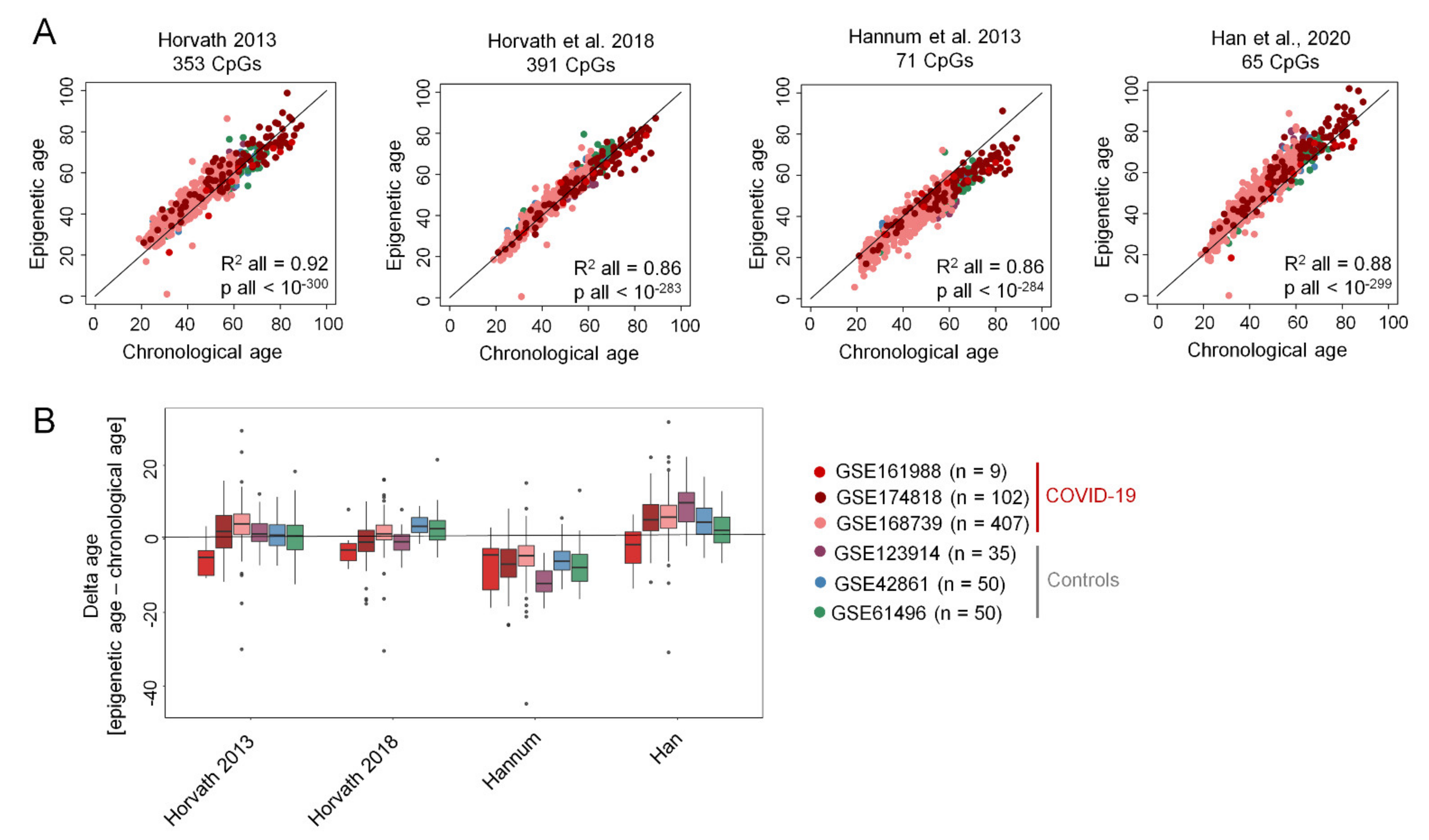
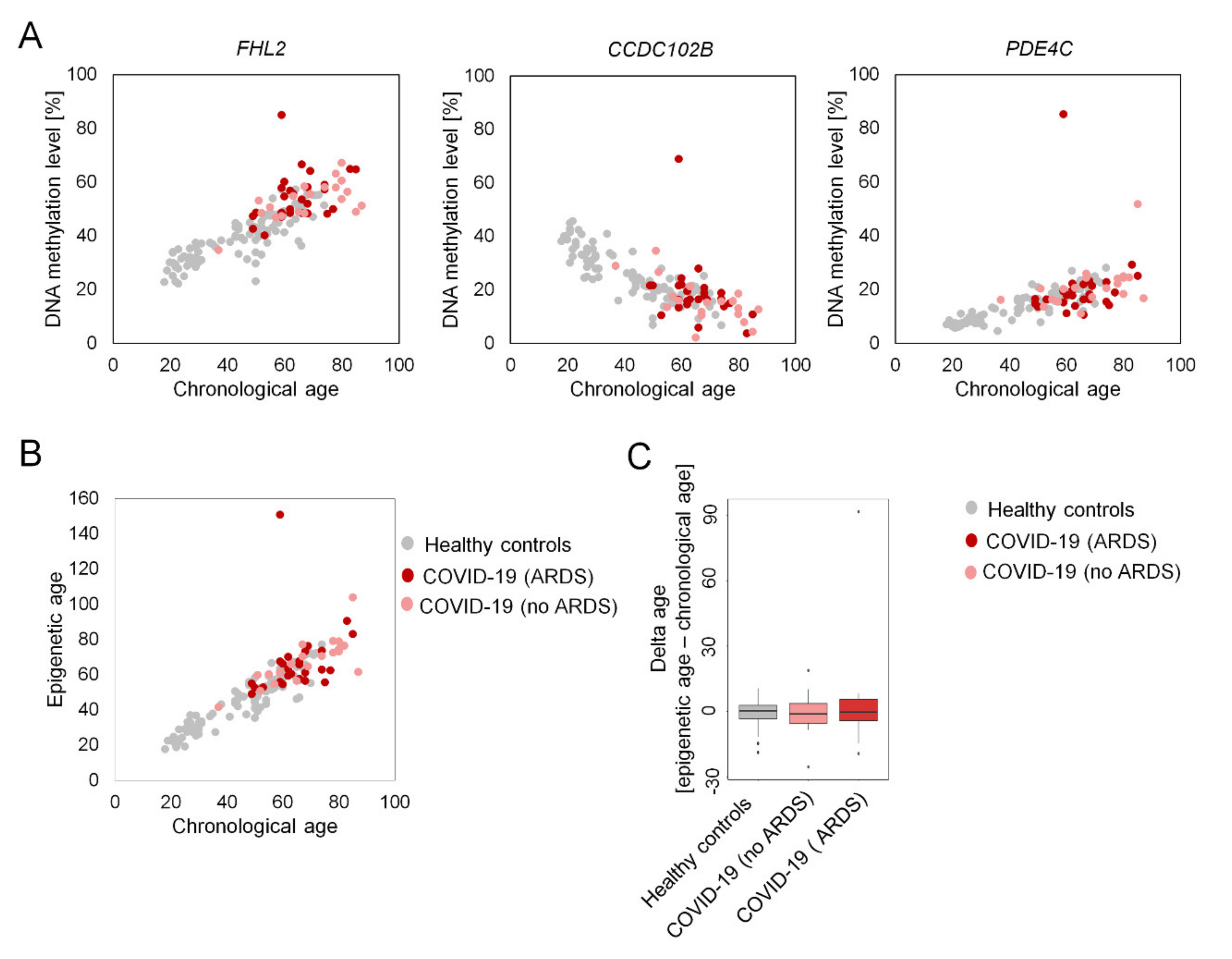
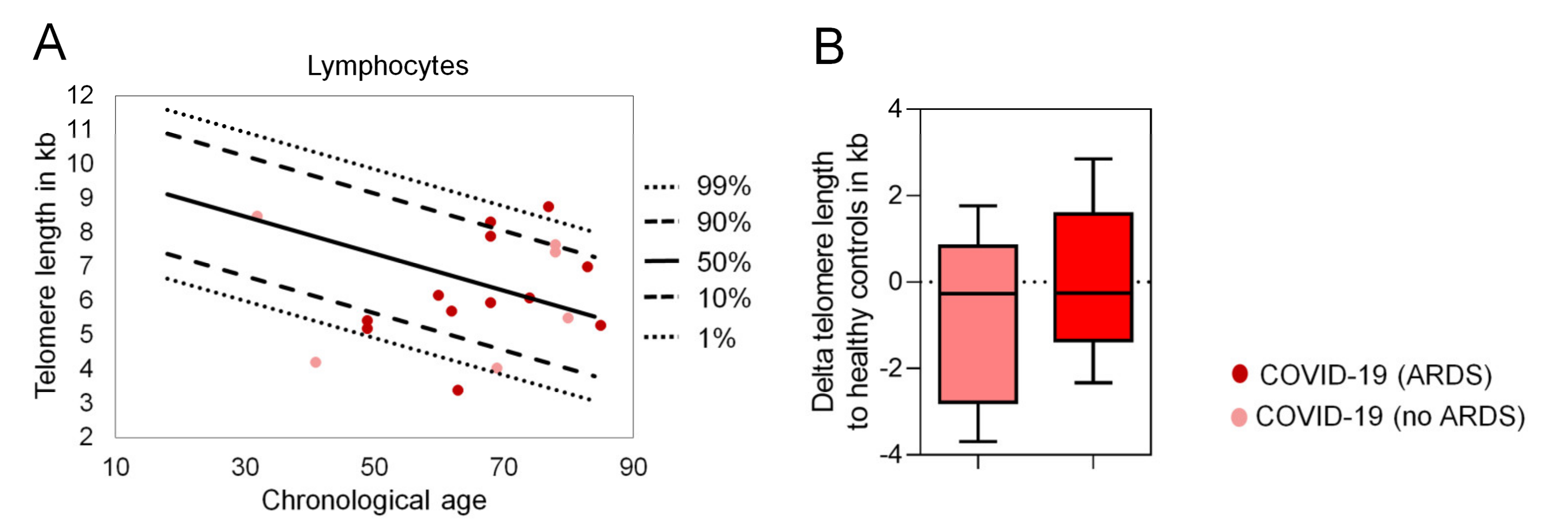
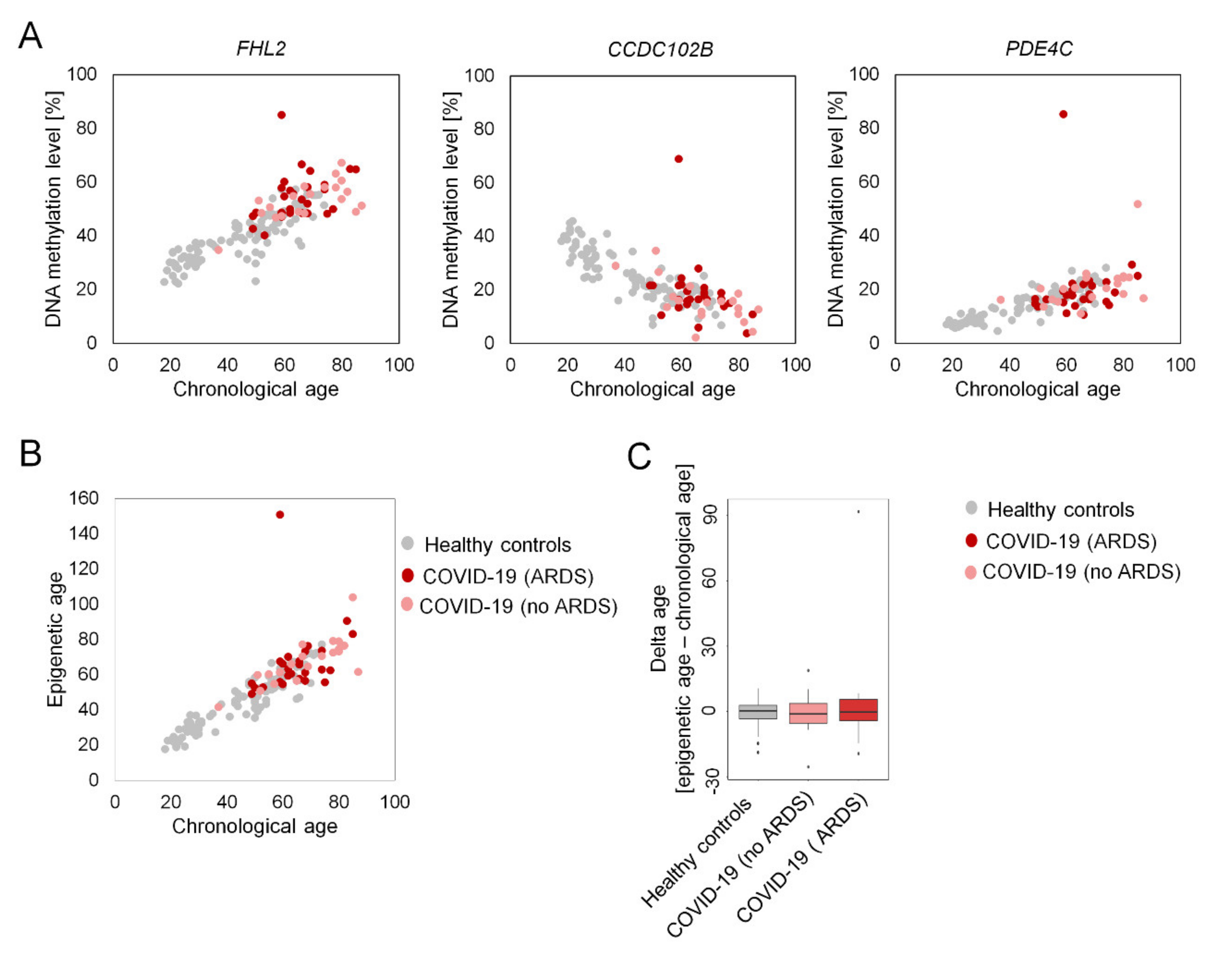
2. Results
3. Discussion
4. Material and Methods
4.1. Blood Samples Used in This Study
4.2. Analysis of DNA Methylation Microarray Data
4.3. Bisulfite Amplicon Sequencing
4.4. Fluorescence In Situ Hybridization (Flow-FISH)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oran, D.P.; Topol, E.J. Prevalence of Asymptomatic SARS-CoV-2 Infection: A Narrative Review. Ann. Intern. Med. 2020, 173, 362–367. [Google Scholar] [CrossRef]
- Levin, A.T.; Hanage, W.P.; Owusu-Boaitey, N.; Cochran, K.B.; Walsh, S.P.; Meyerowitz-Katz, G. Assessing the age specificity of infection fatality rates for COVID-19: Systematic review, meta-analysis, and public policy implications. Eur. J. Epidemiol. 2020, 35, 1123–1138. [Google Scholar] [CrossRef]
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981. [Google Scholar] [CrossRef]
- Polidori, M.C.; Sies, H.; Ferrucci, L.; Benzing, T. COVID-19 mortality as a fingerprint of biological age. Ageing Res. Rev. 2021, 67, 101308. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Custodero, C.; Maggi, S.; Polidori, M.C.; Veronese, N.; Ferrucci, L. A multidimensional approach to frailty in older people. Ageing Res. Rev. 2020, 60, 101047. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef]
- Balnis, J.; Madrid, A.; Hogan, K.J.; Drake, L.A.; Chieng, H.C.; Tiwari, A.; Vincent, C.E.; Chopra, A.; Vincent, P.A.; Robek, M.D.; et al. Blood DNA methylation and COVID-19 outcomes. Clin. Epigenet. 2021, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Castro de Moura, M.; Davalos, V.; Planas-Serra, L.; Alvarez-Errico, D.; Arribas, C.; Ruiz, M.; Aguilera-Albesa, S.; Troya, J.; Valencia-Ramos, J.; Vélez-Santamaria, V.; et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 2021, 66, 103339. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Chen, D.; Bigambo, F.M.; Wei, H.; Wang, X.; Xia, Y. Differences of blood cells, lymphocyte subsets and cytokines in COVID-19 patients with different clinical stages: A network meta-analysis. BMC Infect. Dis. 2021, 21, 156. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Franzen, J.; Stiehl, T.; Gobs, M.; Kuo, C.-C.; Nikolić, M.; Hapala, J.; Koop, B.E.; Strathmann, K.; Ritz-Timme, S.; et al. New targeted approaches for epigenetic age predictions. BMC Biol. 2020, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Froidure, A.; Mahieu, M.; Hoton, D.; Laterre, P.-F.; Yombi, J.C.; Koenig, S.; Ghaye, B.; Defour, J.-P.; Decottignies, A. Short telomeres increase the risk of severe COVID-19. Aging (Albany NY) 2020, 12, 19911–19922. [Google Scholar] [CrossRef]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Mongelli, A.; Barbi, V.; Gottardi Zamperla, M.; Atlante, S.; Forleo, L.; Nesta, M.; Massetti, M.; Pontecorvi, A.; Nanni, S.; Farsetti, A.; et al. Evidence for Biological Age Acceleration and Telomere Shortening in COVID-19 Survivors. Int. J. Mol. Sci. 2021, 22, 6151. [Google Scholar] [CrossRef]
- Capalbo, C.; Aceti, A.; Simmaco, M.; Bonfini, R.; Rocco, M.; Ricci, A.; Napoli, C.; Rocco, M.; Alfonsi, V.; Teggi, A.; et al. The Exponential Phase of the Covid-19 Pandemic in Central Italy: An Integrated Care Pathway. Int. J. Environ. Res. Public Health 2020, 17, 3792. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef]
- Pidsley, R.; Wong, C.C.Y.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.; Deary, I.; Wagner, W. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging (Albany NY) 2016, 8, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.S.V.; Kirschner, M.; Halfmeyer, I.; Estrada, N.; Xicoy, B.; Isfort, S.; Vieri, M.; Zamora, L.; Abels, A.; Bouillon, A.S.; et al. Comparison of flow-FISH and MM-qPCR telomere length assessment techniques for the screening of telomeropathies. Ann. N. Y. Acad. Sci. 2020, 1466, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.; Vieri, M.; Kricheldorf, K.; Ferreira, M.S.V.; Wlodarski, M.W.; Schwarz, M.; Balabanov, S.; Rolles, B.; Isfort, S.; Koschmieder, S.; et al. Androgen derivatives improve blood counts and elongate telomere length in adult cryptic dyskeratosis congenita. Br. J. Haematol. 2020, 193, 669–673. [Google Scholar] [CrossRef]
- Werner, B.; Beier, F.; Hummel, S.; Balabanov, S.; Lassay, L.; Orlikowsky, T.; Dingli, D.; Brümmendorf, T.H.; Traulsen, A. Reconstructing the in vivo dynamics of hematopoietic stem cells from telomere length distributions. eLife 2015, 4, e08687. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzen, J.; Nüchtern, S.; Tharmapalan, V.; Vieri, M.; Nikolić, M.; Han, Y.; Balfanz, P.; Marx, N.; Dreher, M.; Brümmendorf, T.H.; et al. Epigenetic Clocks Are Not Accelerated in COVID-19 Patients. Int. J. Mol. Sci. 2021, 22, 9306. https://doi.org/10.3390/ijms22179306
Franzen J, Nüchtern S, Tharmapalan V, Vieri M, Nikolić M, Han Y, Balfanz P, Marx N, Dreher M, Brümmendorf TH, et al. Epigenetic Clocks Are Not Accelerated in COVID-19 Patients. International Journal of Molecular Sciences. 2021; 22(17):9306. https://doi.org/10.3390/ijms22179306
Chicago/Turabian StyleFranzen, Julia, Selina Nüchtern, Vithurithra Tharmapalan, Margherita Vieri, Miloš Nikolić, Yang Han, Paul Balfanz, Nikolaus Marx, Michael Dreher, Tim H. Brümmendorf, and et al. 2021. "Epigenetic Clocks Are Not Accelerated in COVID-19 Patients" International Journal of Molecular Sciences 22, no. 17: 9306. https://doi.org/10.3390/ijms22179306
APA StyleFranzen, J., Nüchtern, S., Tharmapalan, V., Vieri, M., Nikolić, M., Han, Y., Balfanz, P., Marx, N., Dreher, M., Brümmendorf, T. H., Dahl, E., Beier, F., & Wagner, W. (2021). Epigenetic Clocks Are Not Accelerated in COVID-19 Patients. International Journal of Molecular Sciences, 22(17), 9306. https://doi.org/10.3390/ijms22179306