
Loss of Ift74 Leads to Slow Photoreceptor Degeneration and Ciliogenesis Defects in Zebrafish


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
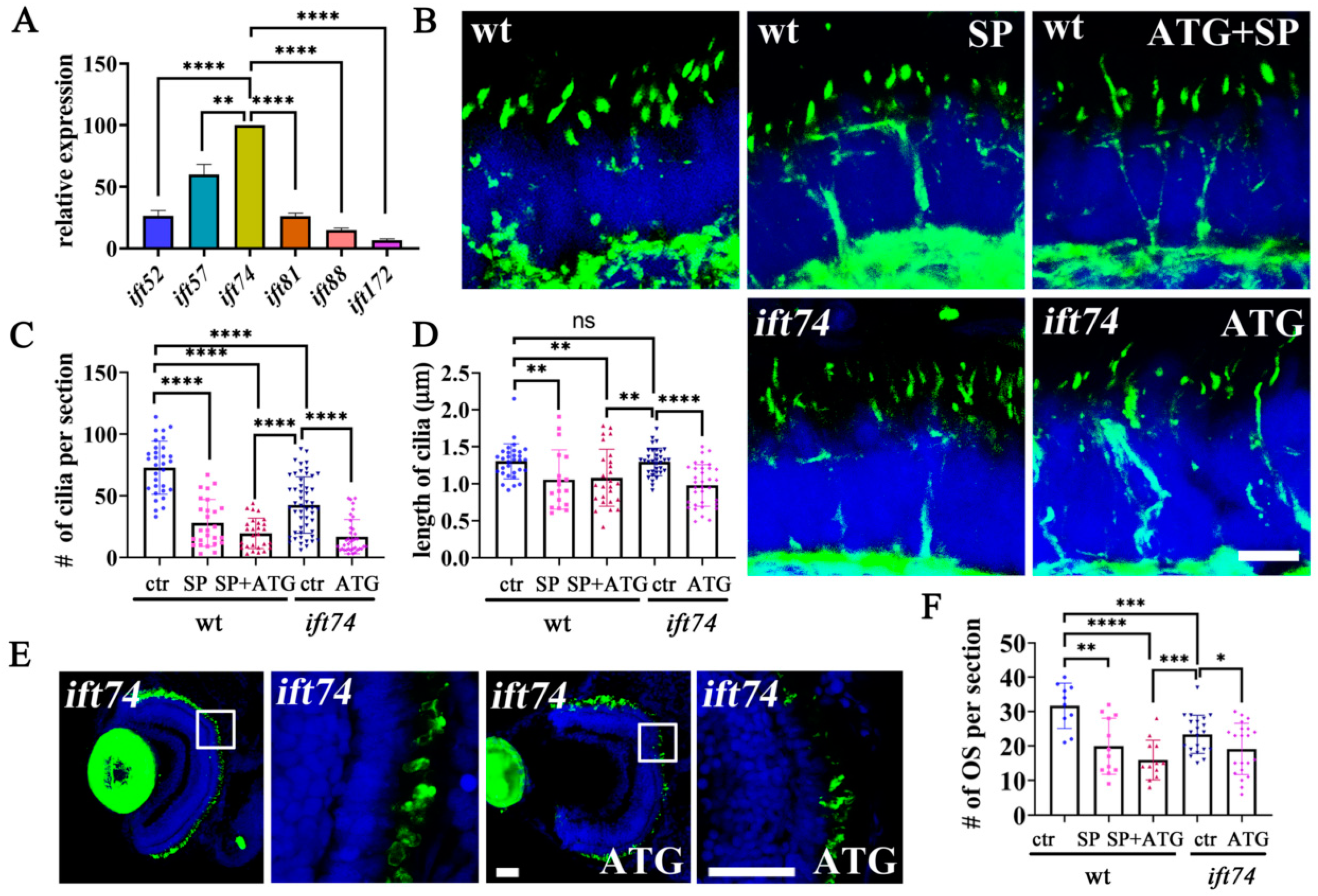
2.1. Zebrafish michelin Locus Encodes ift74, an IFT-B Component
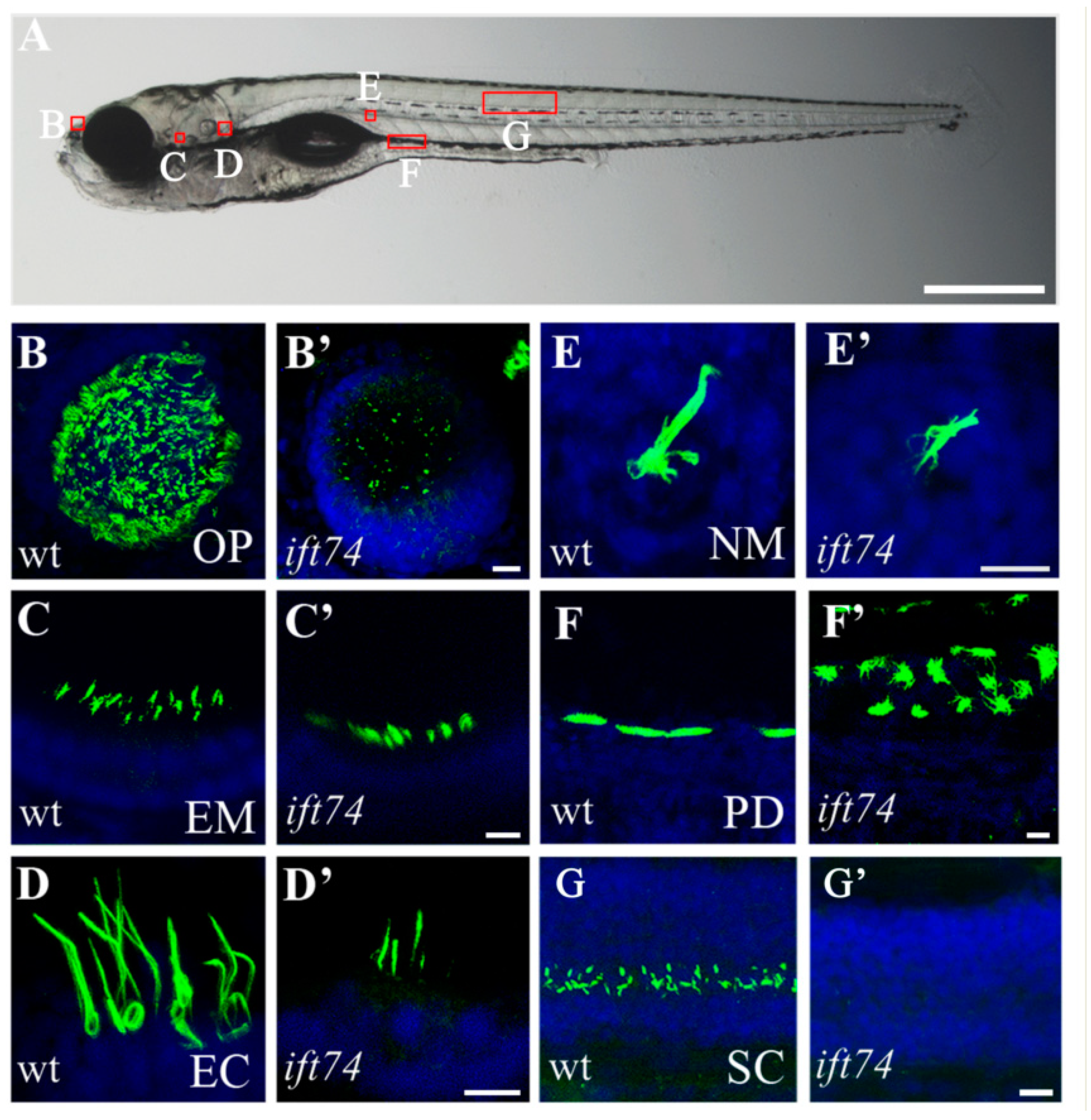
2.2. Ciliary Defects in Multiple Organs of ift74 Mutants
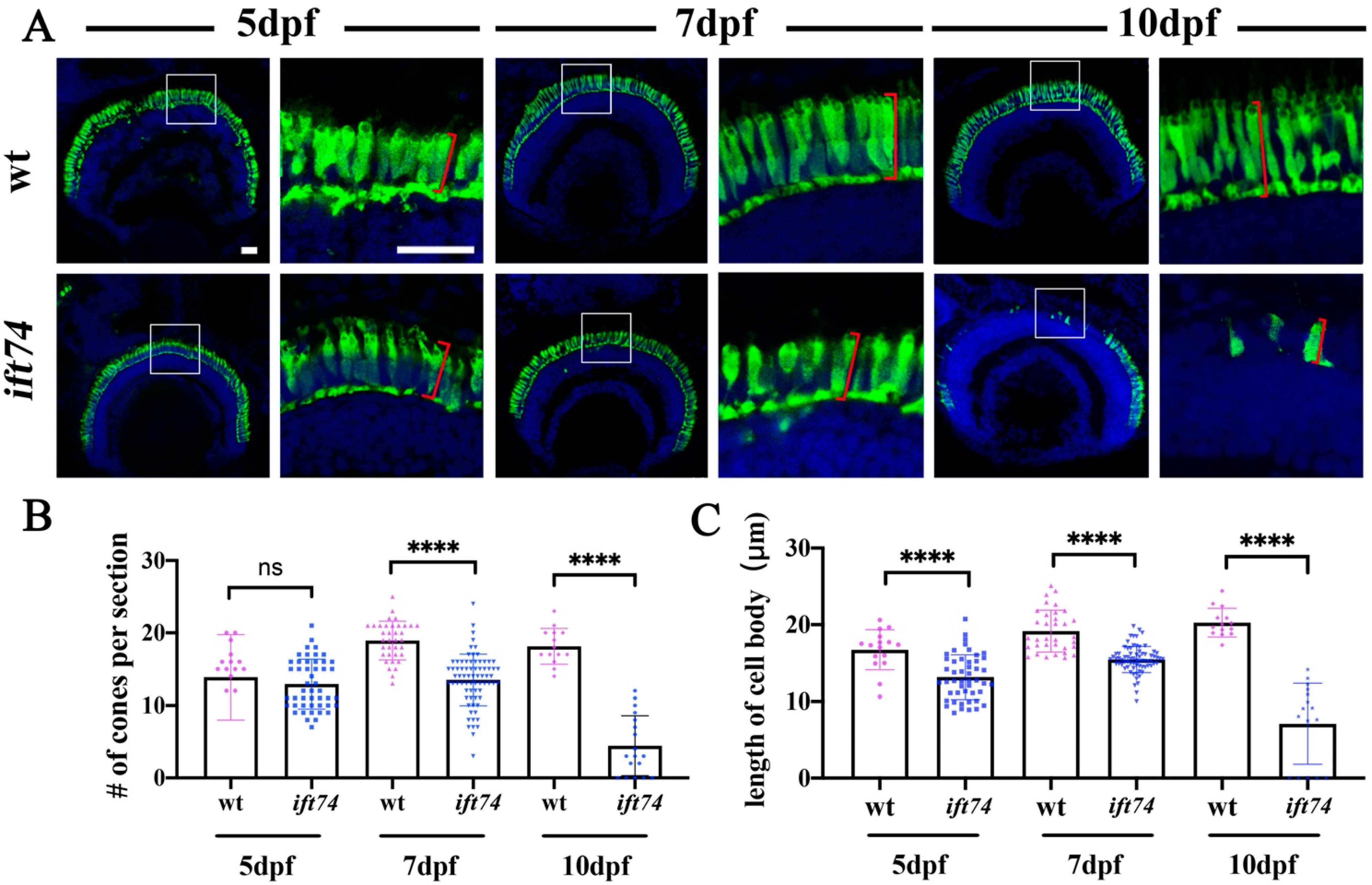
2.3. Slow-Progressing Photoreceptor Cell Degeneration in ift74 Mutant
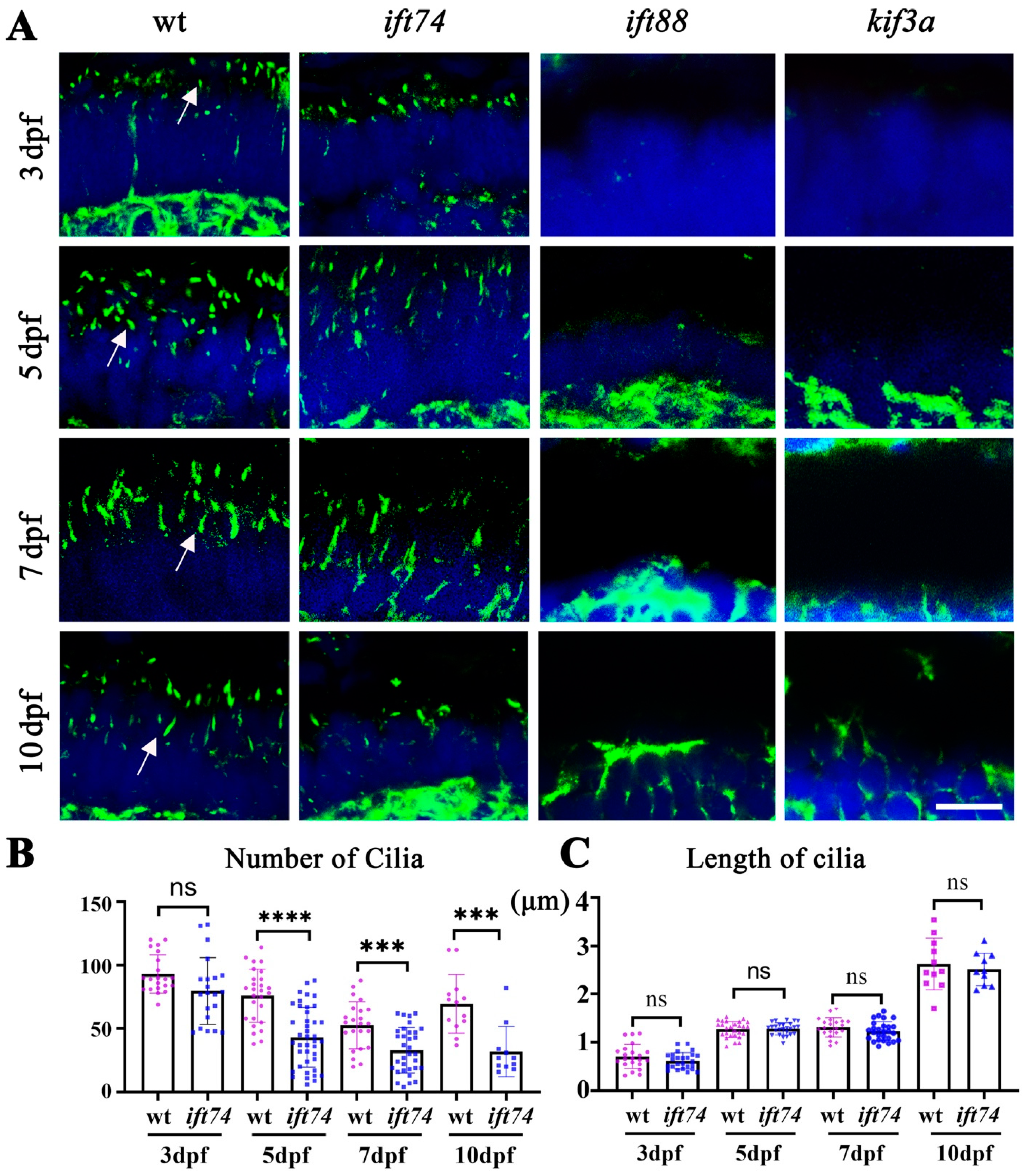
2.4. Ift74 Is Required for the Maintenance of Connecting Cilium
2.5. Defects of Opsin Transport Machinery in ift74 Mutant Retina
2.6. Maternal Ift74 Proteins Contribute to the Early Development of Photoreceptors
3. Discussion
4. Materials and Methods
4.1. Zebrafish Strain
4.2. Genetic Mapping and Cloning
4.3. Whole-Mount In Situ Hybridization and Immunohistochemistry
4.4. Quantitative PCR
4.5. Morpholino Knockdown
4.6. Opsin Transport Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnes, C.L.; Malhotra, H.; Calvert, P.D. Compartmentalization of Photoreceptor Sensory Cilia. Front. Cell Dev. Biol. 2021, 9, 636737. [Google Scholar] [CrossRef]
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Zhang, X.L.; Jia, S.; Yelick, P.C.; Zhao, C.T. Zebrafish as a Model for Human Ciliopathies. J. Genet. Genomics 2016, 43, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G., 3rd; Raleigh, D.R.; Reiter, J.F. How the Ciliary Membrane Is Organized Inside-Out to Communicate Outside-In. Curr. Biol. 2018, 28, R421–R434. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, K.I.; Johnson, C.T.; Jackson, P.K. The primary cilium as a cellular receiver: Organizing ciliary GPCR signaling. Curr. Opin. Cell Biol. 2016, 39, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Jack, B.M.; Wang, H.H.; Kavanaugh, M.A.; Maser, R.L.; Tran, P.V. Intraflagellar Transport Proteins as Regulators of Primary Cilia Length. Front. Cell Dev. Biol. 2021, 9, 661350. [Google Scholar] [CrossRef] [PubMed]
- Lechtreck, K.F. IFT-Cargo Interactions and Protein Transport in Cilia. Trends Biochem. Sci. 2015, 40, 765–778. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Omori, Y.; Brodowska, K.; Kovach, P.; Malicki, J. Kinesin-2 family in vertebrate ciliogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 2388–2393. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Tam, B.M.; Ying, G.; Wu, S.; Hauswirth, W.W.; Frederick, J.M.; Moritz, O.L.; Baehr, W. Kinesin family 17 (osmotic avoidance abnormal-3) is dispensable for photoreceptor morphology and function. FASEB J. 2015, 29, 4866–4880. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Chen, Z.; Yang, K.; Miao, S.; Xu, B.; Kang, Y.; Xie, H.; Zhao, C. The cytoplasmic tail of rhodopsin triggers rapid rod degeneration in kinesin-2 mutants. J. Biol. Chem. 2017, 292, 17375–17386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, V.S.; Jimeno, D.; Khanobdee, K.; Song, X.; Chen, B.; Nusinowitz, S.; Williams, D.S. Dysfunction of heterotrimeric kinesin-2 in rod photoreceptor cells and the role of opsin mislocalization in rapid cell death. Mol. Biol. Cell 2010, 21, 4076–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marszalek, J.R.; Liu, X.; Roberts, E.A.; Chui, D.; Marth, J.D.; Williams, D.S.; Goldstein, L.S. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell 2000, 102, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Perkins, B.D. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009, 49, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Perkins, B.D. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J. Cell Sci. 2008, 121, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, M.; Malicki, J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 2004, 42, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Pazour, G.J.; Baker, S.A.; Deane, J.A.; Cole, D.G.; Dickert, B.L.; Rosenbaum, J.L.; Witman, G.B.; Besharse, J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002, 157, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boubakri, M.; Chaya, T.; Hirata, H.; Kajimura, N.; Kuwahara, R.; Ueno, A.; Malicki, J.; Furukawa, T.; Omori, Y. Loss of ift122, a Retrograde Intraflagellar Transport (IFT) Complex Component, Leads to Slow, Progressive Photoreceptor Degeneration Due to Inefficient Opsin Transport. J. Biol. Chem. 2016, 291, 24465–24474. [Google Scholar] [CrossRef] [Green Version]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A.; et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Lin, Z.; Zhu, T.; Jin, M.; Meng, D.; He, R.; Cao, Z.; Shen, Y.; Lu, C.; Cai, R.; et al. Disrupted intraflagellar transport due to IFT74 variants causes Joubert syndrome. Genet. Med. 2021, 23, 1041–1049. [Google Scholar] [CrossRef]
- Kleinendorst, L.; Alsters, S.I.M.; Abawi, O.; Waisfisz, Q.; Boon, E.M.J.; van den Akker, E.L.T.; van Haelst, M.M. Second case of Bardet-Biedl syndrome caused by biallelic variants in IFT74. Eur. J. Hum. Genet. 2020, 28, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Gao, F.; Gao, S.; Liang, Y.; Long, H.; Lv, Z.; Su, Y.; Ye, N.; Zhang, L.; Zhao, C.; et al. Biodiversity-based development and evolution: The emerging research systems in model and non-model organisms. Sci. China Life Sci. 2021, 64, 1236–1280. [Google Scholar] [CrossRef] [PubMed]
- Larison, K.D.; Bremiller, R. Early onset of phenotype and cell patterning in the embryonic zebrafish retina. Development 1990, 109, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Zhao, C.; Saras, A.; Mukhopadhyay, S.; Kim, W.; Furukawa, T.; Sengupta, P.; Veraksa, A.; Malicki, J. Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat. Cell Biol. 2008, 10, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Malicki, J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 2011, 30, 2532–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takechi, M.; Hamaoka, T.; Kawamura, S. Fluorescence visualization of ultraviolet-sensitive cone photoreceptor development in living zebrafish. FEBS Lett. 2003, 553, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jia, S.; Chen, Z.; Chong, Y.L.; Xie, H.; Feng, D.; Wu, X.; Song, D.Z.; Roy, S.; Zhao, C. Cilia-driven cerebrospinal fluid flow directs expression of urotensin neuropeptides to straighten the vertebrate body axis. Nat. Genet. 2018, 50, 1666–1673. [Google Scholar] [CrossRef]
- Huang, P.; Schier, A.F. Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development 2009, 136, 3089–3098. [Google Scholar] [CrossRef] [Green Version]
- Lores, P.; Kherraf, Z.E.; Amiri-Yekta, A.; Whitfield, M.; Daneshipour, A.; Stouvenel, L.; Cazin, C.; Cavarocchi, E.; Coutton, C.; Llabador, M.A.; et al. A missense mutation in IFT74, encoding for an essential component for intraflagellar transport of Tubulin, causes asthenozoospermia and male infertility without clinical signs of Bardet-Biedl syndrome. Hum. Genet. 2021, 140, 1031–1043. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O'Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome-Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W.; et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, G.M.; Ye, F.; Nager, A.R.; Murphy, J.P.; Lee, J.S.; Aguiar, M.; Breslow, D.K.; Gygi, S.P.; Nachury, M.V. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 2014, 31, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, P.; Xu, J.; Wang, Y.; Zhao, C. Loss of Ift74 Leads to Slow Photoreceptor Degeneration and Ciliogenesis Defects in Zebrafish. Int. J. Mol. Sci. 2021, 22, 9329. https://doi.org/10.3390/ijms22179329
Zhu P, Xu J, Wang Y, Zhao C. Loss of Ift74 Leads to Slow Photoreceptor Degeneration and Ciliogenesis Defects in Zebrafish. International Journal of Molecular Sciences. 2021; 22(17):9329. https://doi.org/10.3390/ijms22179329
Chicago/Turabian StyleZhu, Panpan, Jingjin Xu, Yadong Wang, and Chengtian Zhao. 2021. "Loss of Ift74 Leads to Slow Photoreceptor Degeneration and Ciliogenesis Defects in Zebrafish" International Journal of Molecular Sciences 22, no. 17: 9329. https://doi.org/10.3390/ijms22179329
APA StyleZhu, P., Xu, J., Wang, Y., & Zhao, C. (2021). Loss of Ift74 Leads to Slow Photoreceptor Degeneration and Ciliogenesis Defects in Zebrafish. International Journal of Molecular Sciences, 22(17), 9329. https://doi.org/10.3390/ijms22179329