
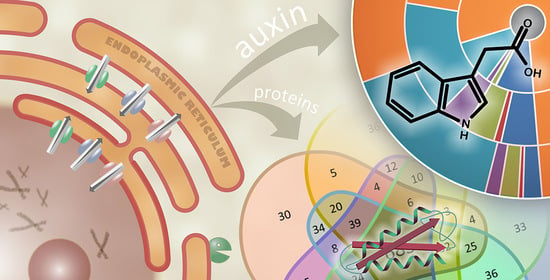
Auxin Metabolome Profiling in the Arabidopsis Endoplasmic Reticulum Using an Optimised Organelle Isolation Protocol

 , , , and
, , , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
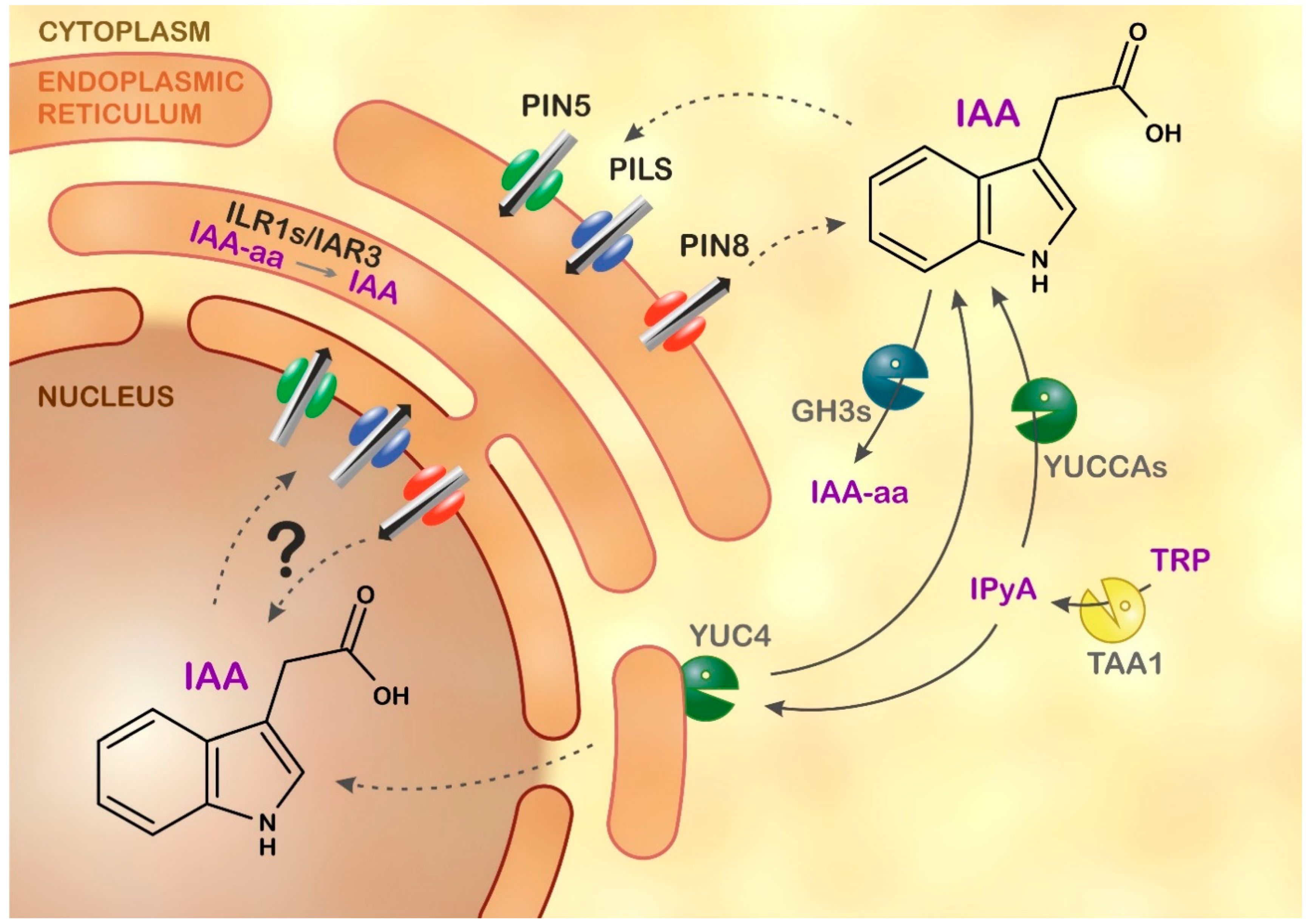
1. Introduction
2. Results
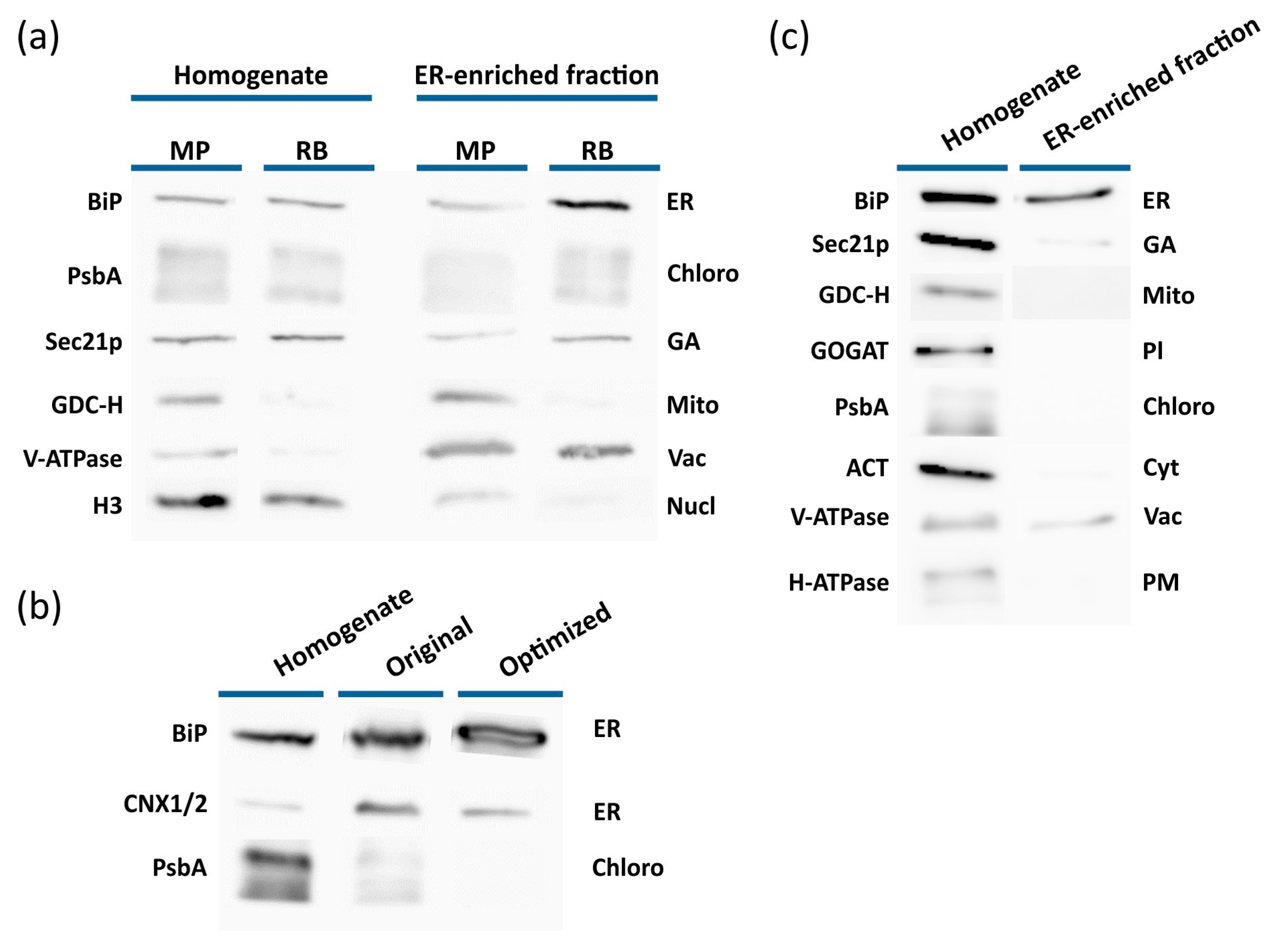
2.1. Isolation of the ER from Arabidopsis Plants
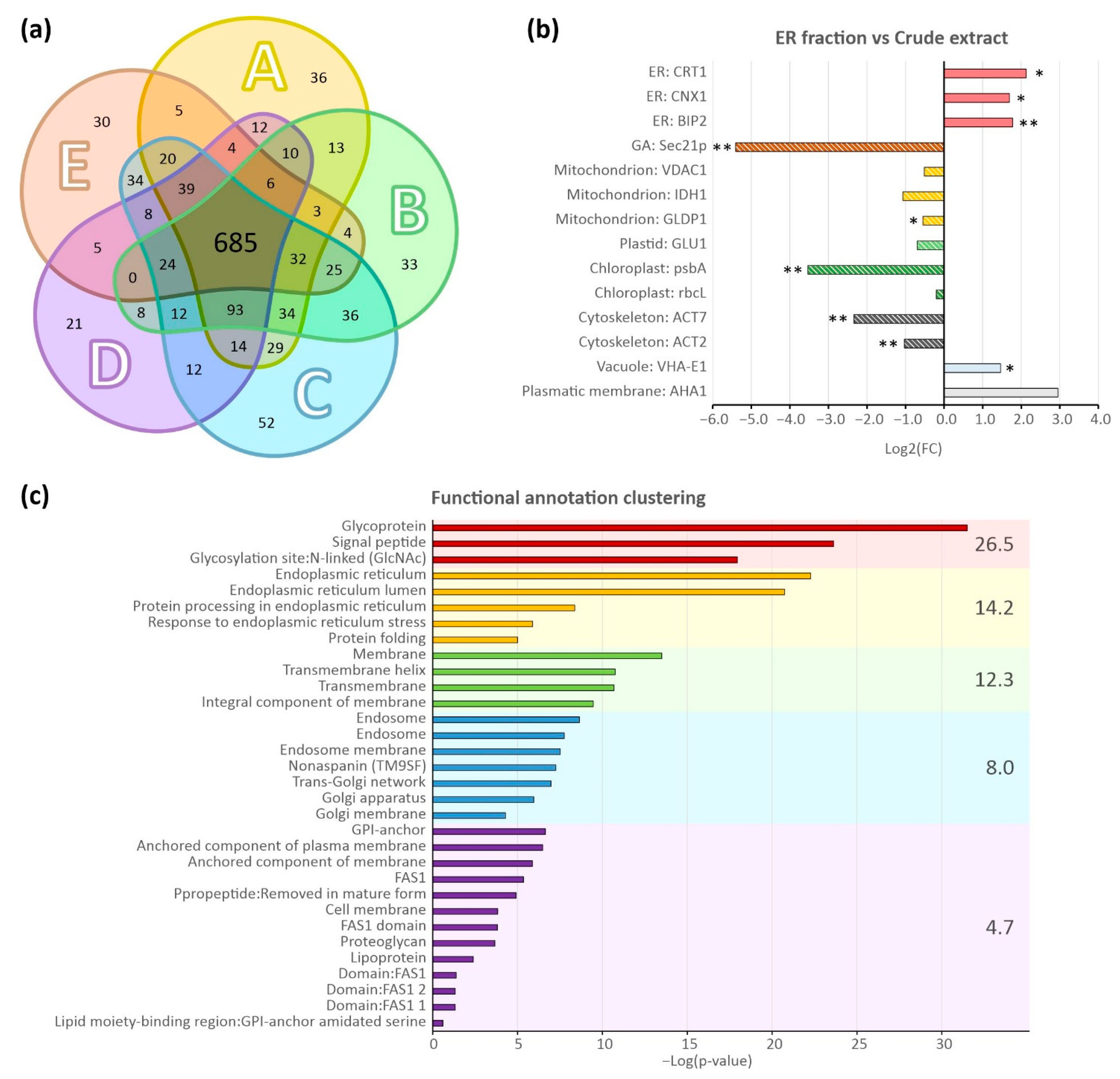
2.2. Confirmation of ER-Enriched Fractions by Proteomic Analysis
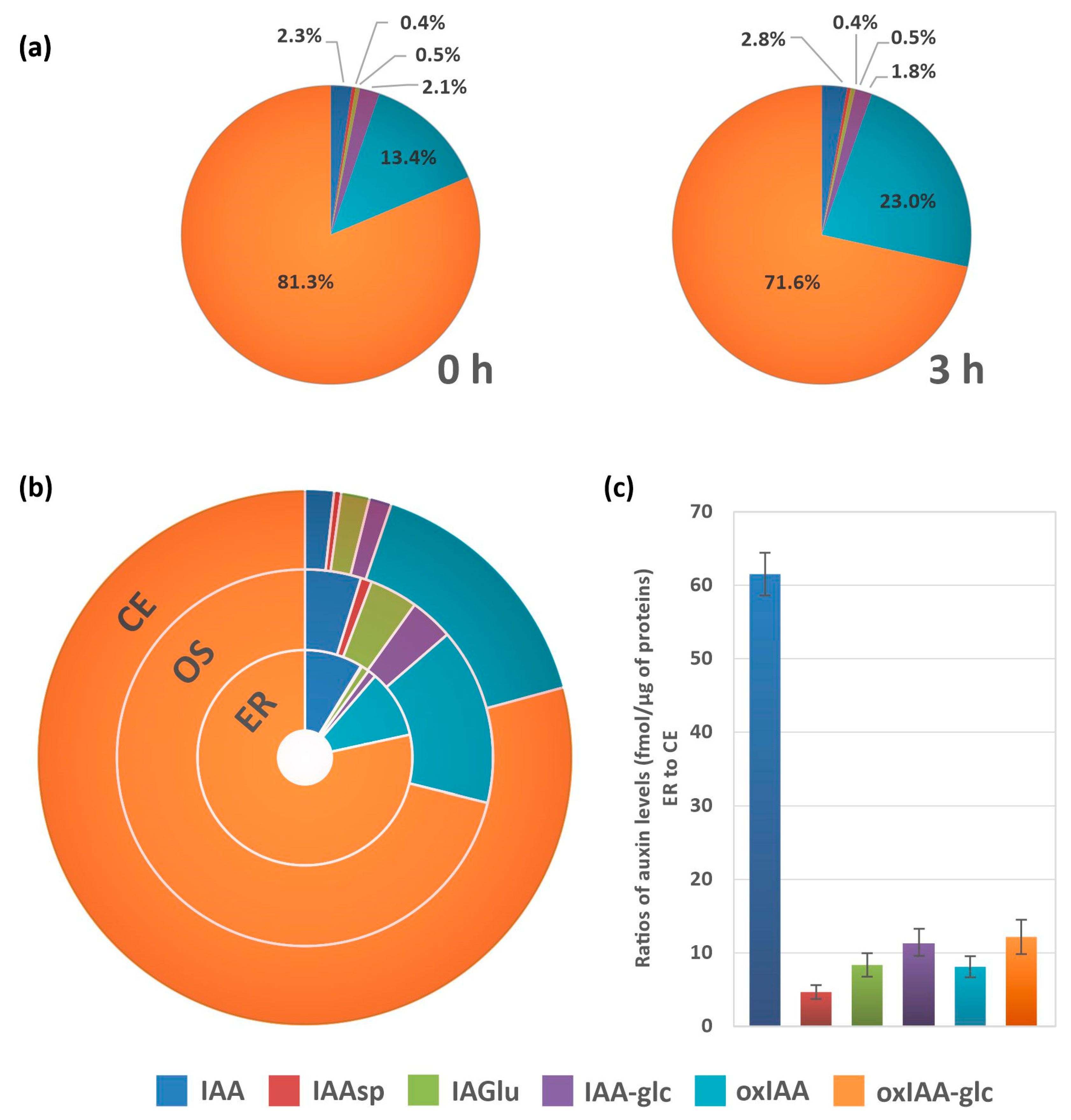
2.3. Auxin Metabolite Determination in ER
3. Discussion
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. Homogenisation
4.3. Optimisation of Initial Centrifugation
4.4. Preparation of ER-Enriched Fraction–Original Protocol
4.5. Preparation of ER-Enriched Fraction-Optimised Protocol
4.6. The SDS-PAGE Western Blot Assay
4.7. Proteomic Analysis
4.8. Optimisation of SPE Protocols
4.9. Control of Auxin Metabolite Profile Stability
4.10. Extraction and Purification of IAA Metabolites
4.11. Quantification of IAA Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Doyle, C.; Qi, X.; Zheng, H. The endoplasmic reticulum: A social network in plant cells. J. Integr. Plant Biol. 2012, 54, 840–850. [Google Scholar] [CrossRef]
- Spang, A. Retrograde traffic from the Golgi to the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013391. [Google Scholar] [CrossRef] [PubMed]
- Hawes, C.; Kiviniemi, P.; Kriechbaumer, V. The endoplasmic reticulum: A dynamic and well-connected organelle. J. Integr. Plant Biol. 2015, 57, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Dashek, W.V. Endoplasmic reticulum. In Plant Cells and their Organelles, 1st ed.; Dashek, W.V., Miglani, G.S., Eds.; Wiley: Hoboken, NJ, USA, 2017; pp. 42–60. [Google Scholar] [CrossRef]
- Mravec, J.; Skůpa, P.; Bailly, A.; Hoyerová, K.; Křeček, P.; Bielach, A.; Petrášek, J.; Zhang, J.; Gaykova, V.; Stierhof, Y.-D.; et al. Subcellular homeostasis of phytohormone auxin is mediated by the ER-localized PIN5 transporter. Nature 2009, 459, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, B.; Moreno, I.; Dupláková, N.; Simon, S.; Carraro, N.; Reemmer, J.; Pěnčík, A.; Chen, X.; Tejos, R.; et al. ER-localized auxin transporter PIN8 regulates auxin homeostasis and male gametophyte development in Arabidopsis. Nat. Commun. 2012, 3, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, C.D.; Dovzhenko, A.; Liu, X.; Woerner, N.; Rensch, T.; Eismann, M.; Eimer, S.; Hegermann, J.; Paponov, I.A.; Ruperti, B.; et al. The endoplasmic reticulum localized PIN8 is a pollen-specific auxin carrier involved in intracellular auxin homeostasis. Plant J. 2012, 71, 860–870. [Google Scholar] [CrossRef]
- Barbez, E.; Kleine-Vehn, J. Divide et Impera—Cellular auxin compartmentalization. Curr. Opin. Plant Biol. 2013, 16, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Middleton, A.M.; Dal Bosco, C.; Chlap, P.; Bensch, R.; Harz, H.; Ren, F.; Bergmann, S.; Wend, S.; Weber, W.; Hayashi, K.-I.; et al. Data-Driven Modeling of Intracellular Auxin Fluxes Indicates a Dominant Role of the ER in Controlling Nuclear Auxin Uptake. Cell Rep. 2018, 22, 3044–3057. [Google Scholar] [CrossRef] [PubMed]
- Caesar, K.; Thamm, A.M.K.; Witthöft, J.; Elgass, K.; Huppenberger, P.; Grefen, C.; Horak, J.; Harter, K. Evidence for the localization of the Arabidopsis cytokinin receptors AHK3 and AHK4 in the endoplasmic reticulum. J. Exp. Bot. 2011, 62, 5571–5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulfetange, K.; Lomin, S.N.; Romanov, G.A.; Stolz, A.; Heyl, A.; Schmülling, T. The cytokinin receptors of Arabidopsis are located mainly to the endoplasmic reticulum. Plant Physiol. 2011, 156, 1808–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-F.; Randlett, M.D.; Findell, J.L.; Schaller, G.E. Localization of the ethylene receptor ETR1 to the endoplasmic reticulum of Arabidopsis. J. Biol. Chem. 2002, 277, 19861–19866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbez, E.; Kubeš, M.; Rolčík, J.; Béziat, C.; Pěnčík, A.; Wang, B.; Rosquete, M.R.; Zhu, J.; Dobrev, P.I.; Lee, Y.; et al. A novel putative auxin carrier family regulates intracellular auxin homeostasis in plants. Nature 2012, 485, 119–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feraru, E.; Feraru, M.I.; Barbez, E.; Waidmann, S.; Sun, L.; Gaidora, A.; Kleine-Vehn, J. PILS6 is a temperature-sensitive regulator of nuclear auxin input and organ growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2019, 116, 3893–3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranocha, P.; Dima, O.; Nagy, R.; Felten, J.; Corratgé-Faillie, C.; Novák, O.; Morreel, K.; Lacombe, B.; Martinez, Y.; Pfrunder, S.; et al. Arabidopsis WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis. Nat. Commun. 2013, 4, 2625. [Google Scholar] [CrossRef] [PubMed]
- Béziat, C.; Barbez, E.; Feraru, M.I.; Lucyshyn, D.; Kleine-Vehn, J. Light triggers PILS-dependent reduction in nuclear auxin signalling for growth transition. Nat. Plants 2017, 3, 17105. [Google Scholar] [CrossRef] [Green Version]
- Ljung, K. Auxin metabolism and homeostasis during plant development. Development 2013, 140, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Casanova-Sáez, R.; Mateo-Bonmatí, E.; Ljung, K. Auxin Metabolism in Plants. Cold Spring Harb. Perspect. Biol. 2021, a039867. [Google Scholar] [CrossRef]
- Stepanova, A.N.; Robertson-Hoyt, J.; Yun, J.; Benavente, L.M.; Xie, D.Y.; Doležal, K.; Schlereth, A.; Jürgens, G.; Alonso, J.M. TAA1-mediated auxin biosynthesis is essential for hormone crosstalk and plant development. Cell 2008, 133, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Ferrer, J.L.; Ljung, K.; Pojer, F.; Hong, F.; Long, J.A.; Li, L.; Moreno, J.E.; Bowman, M.E.; Ivans, L.J.; et al. Rapid synthesis of auxin via a new tryptophan-dependent pathway is required for shade avoidance in plants. Cell 2008, 133, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Sharkhuu, A.; Jin, J.B.; Li, P.; Jeong, J.C.; Baek, D.; Lee, S.Y.; Blakeslee, J.J.; Murphy, A.S.; Bohnert, H.J.; et al. yucca6, a dominant mutation in Arabidopsis, affects auxin accumulation and auxin-related phenotypes. Plant Physiol. 2007, 145, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Kriechbaumer, V.; Botchway, S.W.; Hawes, C. Localization and interactions between Arabidopsis auxin biosynthetic enzymes in the TAA/YUC-dependent pathway. J. Exp. Bot. 2016, 67, 4195–4207. [Google Scholar] [CrossRef] [PubMed]
- Kubeš, M.; Napier, R. Non-canonical auxin signalling: Fast and curious. J. Exp. Bot. 2019, 70, 2609–2614. [Google Scholar] [CrossRef] [PubMed]
- Fendrych, M.; Akhmanova, M.; Merrin, J.; Glanc, M.; Hagihara, S.; Takahashi, K.; Uchida, N.; Torii, K.U.; Friml, J. Rapid and reversible root growth inhibition by TIR1 auxin signalling. Nat. Plants 2018, 4, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Korasick, D.A.; Enders, T.A.; Strader, L.C. Auxin biosynthesis and storage forms. J. Exp. Bot. 2013, 64, 2541–2555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lin, J.E.; Harris, C.; Campos Mastrotti Pereira, F.; Wu, F.; Blakeslee, J.J.; Peer, W.A. DAO1 catalyzes temporal and tissue-specific oxidative inactivation of auxin in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2016, 113, 11010–11015. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.-H.; Ma, X.-M.; Han, P.; Wang, B.; Sun, Y.-G.; Zhang, G.-Z.; Li, Y.-J.; Hou, B.-K. UGT74D1 is a novel auxin glycosyltransferase from Arabidopsis thaliana. PLoS ONE 2013, 8, e61705. [Google Scholar] [CrossRef]
- Staswick, P.E.; Serban, B.; Rowe, M.; Tiryaki, I.; Maldonado, M.T.; Maldonado, M.C.; Suza, W. Characterization of an Arabidopsis Enzyme Family that Conjugates Amino Acids to Indole-3-Acetic Acid. Plant Cell 2005, 17, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Sánchez-García, A.B.; Albacete, A.; González-Bayón, R.; Justamante, M.S.; Ibáñez, S.; Acosta, M.; Pérez-Pérez, J.M. Enhanced Conjugation of Auxin by GH3 Enzymes Leads to Poor Adventitious Rooting in Carnation Stem Cuttings. Front. Plant Sci. 2018, 9, 566. [Google Scholar] [CrossRef] [Green Version]
- Ostin, A.; Kowalczyk, M.; Bhalerao, R.; Sandberg, G. Metabolism of indole-3-acetic acid in Arabidopsis. Plant Physiol. 1998, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Rampey, R.A.; LeClere, S.; Kowalczyk, M.; Ljung, K.; Sandberg, G.; Bartel, B. A family of auxin-conjugate hydrolases that contributes to free indole-3-acetic acid levels during Arabidopsis germination. Plant Physiol. 2004, 135, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Sanchez Carranza, A.P.; Singh, A.; Steinberger, K.; Panigrahi, K.; Palme, K.; Dovzhenko, A.; Dal Bosco, C. Hydrolases of the ILR1-like family of Arabidopsis thaliana modulate auxin response by regulating auxin homeostasis in the endoplasmic reticulum. Sci. Rep. 2016, 6, 24212. [Google Scholar] [CrossRef] [Green Version]
- Di Mambro, R.; Svolacchia, N.; Dello Ioio, R.; Pierdonati, E.; Salvi, E.; Pedrazzini, E.; Vitale, A.; Perilli, S.; Sozzani, R.; Benfey, P.N.; et al. The Lateral Root Cap Acts as an Auxin Sink that Controls Meristem Size. Curr. Biol. 2019, 29, 1199–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porco, S.; Pěnčík, A.; Rashed, A.; Voß, U.; Casanova-Sáez, R.; Bishopp, A.; Golebiowska, A.; Bhosale, R.; Swarup, R.; Swarup, K.; et al. Dioxygenase-encoding AtDAO1 gene controls IAA oxidation and homeostasis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, 11016–11021. [Google Scholar] [CrossRef] [Green Version]
- Kai, K.; Horita, J.; Wakasa, K.; Miyagawa, H. Three oxidative metabolites of indole-3-acetic acid from Arabidopsis thaliana. Phytochemistry 2007, 68, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- Jiskrová, E.; Novák, O.; Pospíšilová, H.; Holubová, K.; Karády, M.; Galuszka, P.; Robert, S.; Frébort, I. Extra- and intracellular distribution of cytokinins in the leaves of monocots and dicots. New Biotechnol. 2016, 33, 735–742. [Google Scholar] [CrossRef]
- Polanská, L.; Vičánková, A.; Nováková, M.; Malbeck, J.; Dobrev, P.I.; Brzobohatý, B.; Vaňková, R.; Macháčková, I. Altered cytokinin metabolism affects cytokinin, auxin, and abscisic acid contents in leaves and chloroplasts, and chloroplast ultrastructure in transgenic tobacco. J. Exp. Bot. 2007, 58, 637–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benková, E.; Witters, E.; Van Dongen, W.; Kolář, J.; Motyka, V.; Brzobohatý, B.; Van Onckelen, H.A.; Macháčková, I. Cytokinins in tobacco and wheat chloroplasts. Occurrence and changes due to light/dark treatment. Plant Physiol. 1999, 121, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Thibivilliers, S.; Anderson, D.; Libault, M. Isolation of Plant Root Nuclei for Single Cell RNA Sequencing. Curr. Protoc. Plant Biol. 2020, 5, e20120. [Google Scholar] [CrossRef]
- Somerville, C.R.; Somerville, S.C.; Ogren, W.L. Isolation of photosynthetically active protoplasts and chloroplastids from Arabidopsis thaliana. Plant Sci. Lett. 1981, 21, 89–96. [Google Scholar] [CrossRef]
- Saxena, P.K.; Fowke, L.C.; King, J. An efficient procedure for isolation of nuclei from plant protoplasts. Protoplasma 1985, 128, 184–189. [Google Scholar] [CrossRef]
- Antoniadi, I.; Skalický, V.; Sun, G.; Ma, W.; Galbraith, D.W.; Novák, O.; Ljung, K. Fluorescence activated cell sorting—A selective tool for plant cell isolation and analysis. Cytom. Part A 2021. [Google Scholar] [CrossRef]
- Muñoz, P.; Norambuena, L.; Orellana, A. Evidence for a UDP-glucose transporter in Golgi apparatus-derived vesicles from pea and its possible role in polysaccharide biosynthesis. Plant Physiol. 1996, 112, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Robert, S.; Zouhar, J.; Carter, C.J.; Raikhel, N. Isolation of intact vacuoles from Arabidopsis rosette leaf–derived protoplasts. Nat. Protoc. 2007, 2, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Keech, O.; Dizengremel, P.; Gardeström, P. Preparation of leaf mitochondria from Arabidopsis thaliana. Physiol. Plant. 2005, 124, 403–409. [Google Scholar] [CrossRef]
- Seigneurin-Berny, D.; Salvi, D.; Dorne, A.-J.; Joyard, J.; Rolland, N. Percoll-purified and photosynthetically active chloroplasts from Arabidopsis thaliana leaves. Plant Physiol. Biochem. 2008, 46, 951–955. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tan, H.T.; Chung, M.C.M. Subcellular fractionation methods and strategies for proteomics. Proteomics 2010, 10, 3935–3956. [Google Scholar] [CrossRef] [PubMed]
- De Araùjo, M.; Huber, L.A.; Stasyk, T. Isolation of endocitic organelles by density gradient centrifugation. Methods Mol. Biol. 2008, 424, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Petrovská, B.; Jeřábková, H.; Chamrád, I.; Vrána, J.; Lenobel, R.; Uřinovská, J.; Šebela, M.; Doležel, J. Proteomic analysis of barley cell nuclei purified by flow sorting. Cytogenet. Genome Res. 2014, 143, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Stefanski, A.; Overbeck, N.; Drews, L.; Reichert, A.S.; Stühler, K.; Weber, A.P.M. Rapid single-step affinity purification of HA-tagged plant mitochondria. Plant Physiol. 2020, 182, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehaus, M.; Straube, H.; Künzler, P.; Rugen, N.; Hegermann, J.; Giavalisco, P.; Eubel, H.; Witte, C.P.; Herde, M. Rapid Affinity Purification of Tagged Plant Mitochondria (Mito-AP) for Metabolome and Proteome Analyses. Plant Physiol. 2020, 182, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Deal, R.B.; Henikoff, S. The INTACT method for cell type–specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Boussardon, C.; Przybyla-Toscano, J.; Carrie, C.; Keech, O. Tissue-Specific Isolation of Arabidopsis/plant Mitochondria—IMTACT (Isolation of Mitochondria Tagged in specific Cell Types). Plant J. 2020, 103, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Novák, O.; Hényková, E.; Sairanen, I.; Kowalczyk, M.; Pospíšil, T.; Ljung, K. Tissue-specific profiling of the Arabidopsis thaliana auxin metabolome. Plant J. 2012, 72, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Pěnčík, A.; Casanova-Sáez, R.; Pilařová, V.; Žukauskaitė, A.; Pinto, R.; Luis Micol, J.; Ljung, K.; Novák, O. Ultra-rapid auxin metabolite profiling for high-throughput mutant screening in Arabidopsis. J. Exp. Bot. 2018, 69, 2569–2579. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Hao, Y.; Sun, A.; Li, T.; Li, W.; Guo, L.; Yan, Y.; Geng, C.; Chen, N.; Zhong, F.; et al. Sample preparation project for the subcellular proteome of mouse liver. Proteomics 2006, 6, 5269–5277. [Google Scholar] [CrossRef]
- Vertommen, A.; Panis, B.; Swennen, R.; Carpentier, S.C. Challenges and solutions for the identification of membrane proteins in non-model plants. J. Proteom. 2011, 74, 1165–1181. [Google Scholar] [CrossRef]
- Parsons, H.T.; Christiansen, K.; Knierim, B.; Carroll, A.; Ito, J.; Batth, T.S.; Smith-Moritz, A.M.; Morrison, S.; McInerney, P.; Hadi, M.Z.; et al. Isolation and Proteomic Characterization of the Arabidopsis Golgi Defines Functional and Novel Components Involved in Plant Cell Wall Biosynthesis. Plant Physiol. 2012, 159, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Viotti, C.; Kruger, F.; Krebs, M.; Neubert, C.; Fink, F.; Lupanga, U.; Scheuring, D.; Boutte, Y.; Frescatada-Rosa, M.; Wolfenstetter, S.; et al. The Endoplasmic Reticulum Is the Main Membrane Source for Biogenesis of the Lytic Vacuole in Arabidopsis. Plant Cell 2013, 25, 3434–3449. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.M.; Castleden, I.R.; Tanz, S.K.; Aryamanesh, N.; Millar, A.H. SUBA4: The interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res. 2017, 45, D1064–D1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biała, W.; Jasiński, M. The Phenylpropanoid Case—It Is Transport that Matters. Front. Plant Sci. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, I.R.; Schwammle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassal, M.; Abukhalaf, M.; Majovsky, P.; Thieme, D.; Herr, T.; Ayash, M.; Tabassum, N.; Al Shweiki, M.R.; Proksch, C.; Hmedat, A.; et al. Reshaping of the Arabidopsis thaliana Proteome Landscape and Co-regulation of Proteins in Development and Immunity. Mol. Plant 2020, 13, 1709–1732. [Google Scholar] [CrossRef] [PubMed]
- Franc, V.; Šebela, M.; Řehulka, P.; Končitíková, R.; Lenobel, R.; Madzak, C.; Kopečný, D. Analysis of N-glycosylation in maize cytokinin oxidase/dehydrogenase 1 using a manual microgradient chromatographic separation coupled offline to MALDI-TOF/TOF mass spectrometry. J. Proteom. 2012, 75, 4027–4037. [Google Scholar] [CrossRef]
- Chamrád, I.; Simerský, R.; Bérešová, L.; Strnad, M.; Šebela, M.; Lenobel, R. Proteomic Identification of a Candidate Sequence of Wheat Cytokinin-Binding Protein 1. J. Plant Growth Regul. 2014, 33, 896–902. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Beck, S.; Michalski, A.; Raether, O.; Lubeck, M.; Kaspar, S.; Goedecke, N.; Baessmann, C.; Hornburg, D.; Meier, F.; Paron, I.; et al. The impact II, a very high-resolution quadrupole time-of-flight instrument (QTOF) for deep shotgun proteomics. Mol. Cell. Proteom. 2015, 14, 2014–2029. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Fariselli, P.; Casadio, R. BaCelLo: A balanced subcellular localization predictor. Bioinformatics 2006, 22, e408–e416. [Google Scholar] [CrossRef] [Green Version]
- Bannai, H.; Tamada, Y.; Maruyama, O.; Nakai, K.; Miyano, S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 2002, 18, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Bodén, M.; Hawkins, J. Prediction of subcellular localization using sequence-biased recurrent networks. Bioinformatics 2005, 21, 2279–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petsalaki, E.I.; Bagos, P.G.; Litou, Z.I.; Hamodrakas, S.J. PredSL: A tool for the N-terminal sequence-based prediction of protein subcellular localization. Genom. Proteom. Bioinform. 2006, 4, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Akutsu, T. Subcellular location prediction of proteins using support vector machines with alignment of block sequences utilizing amino acid composition. BMC Bioinform. 2007, 8, 466. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Vert, J.-P.; Saigo, H.; Ueda, N.; Toh, H.; Akutsu, T. A novel representation of protein sequences for prediction of subcellular location using support vector machines. Protein Sci. 2005, 14, 2804–2813. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Darwin, C.; Darwin, F. The Power of Movement in Plants; John Murray: London, UK, 1880. [Google Scholar]
- Novák, O.; Napier, R.; Ljung, K. Zooming in on Plant Hormone Analysis: Tissue- and Cell-Specific Approaches. Annu. Rev. Plant Biol. 2017, 68, 323–348. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Včelařová, L.; Skalický, V.; Chamrád, I.; Lenobel, R.; Kubeš, M.F.; Pěnčík, A.; Novák, O. Auxin Metabolome Profiling in the Arabidopsis Endoplasmic Reticulum Using an Optimised Organelle Isolation Protocol. Int. J. Mol. Sci. 2021, 22, 9370. https://doi.org/10.3390/ijms22179370
Včelařová L, Skalický V, Chamrád I, Lenobel R, Kubeš MF, Pěnčík A, Novák O. Auxin Metabolome Profiling in the Arabidopsis Endoplasmic Reticulum Using an Optimised Organelle Isolation Protocol. International Journal of Molecular Sciences. 2021; 22(17):9370. https://doi.org/10.3390/ijms22179370
Chicago/Turabian StyleVčelařová, Ludmila, Vladimír Skalický, Ivo Chamrád, René Lenobel, Martin F. Kubeš, Aleš Pěnčík, and Ondřej Novák. 2021. "Auxin Metabolome Profiling in the Arabidopsis Endoplasmic Reticulum Using an Optimised Organelle Isolation Protocol" International Journal of Molecular Sciences 22, no. 17: 9370. https://doi.org/10.3390/ijms22179370
APA StyleVčelařová, L., Skalický, V., Chamrád, I., Lenobel, R., Kubeš, M. F., Pěnčík, A., & Novák, O. (2021). Auxin Metabolome Profiling in the Arabidopsis Endoplasmic Reticulum Using an Optimised Organelle Isolation Protocol. International Journal of Molecular Sciences, 22(17), 9370. https://doi.org/10.3390/ijms22179370